KSIMC: Predicting Kinase–Substrate Interactions Based on Matrix Completion


Abstract
:1. Introduction
2. Experiments and Results
2.1. Evaluation Metrics
2.2. Comparison with Network-Based Method
2.3. Comparison with Different Predictors by De Novo Test
2.4. Case Studies
3. Materials and Methods
3.1. Data Resources
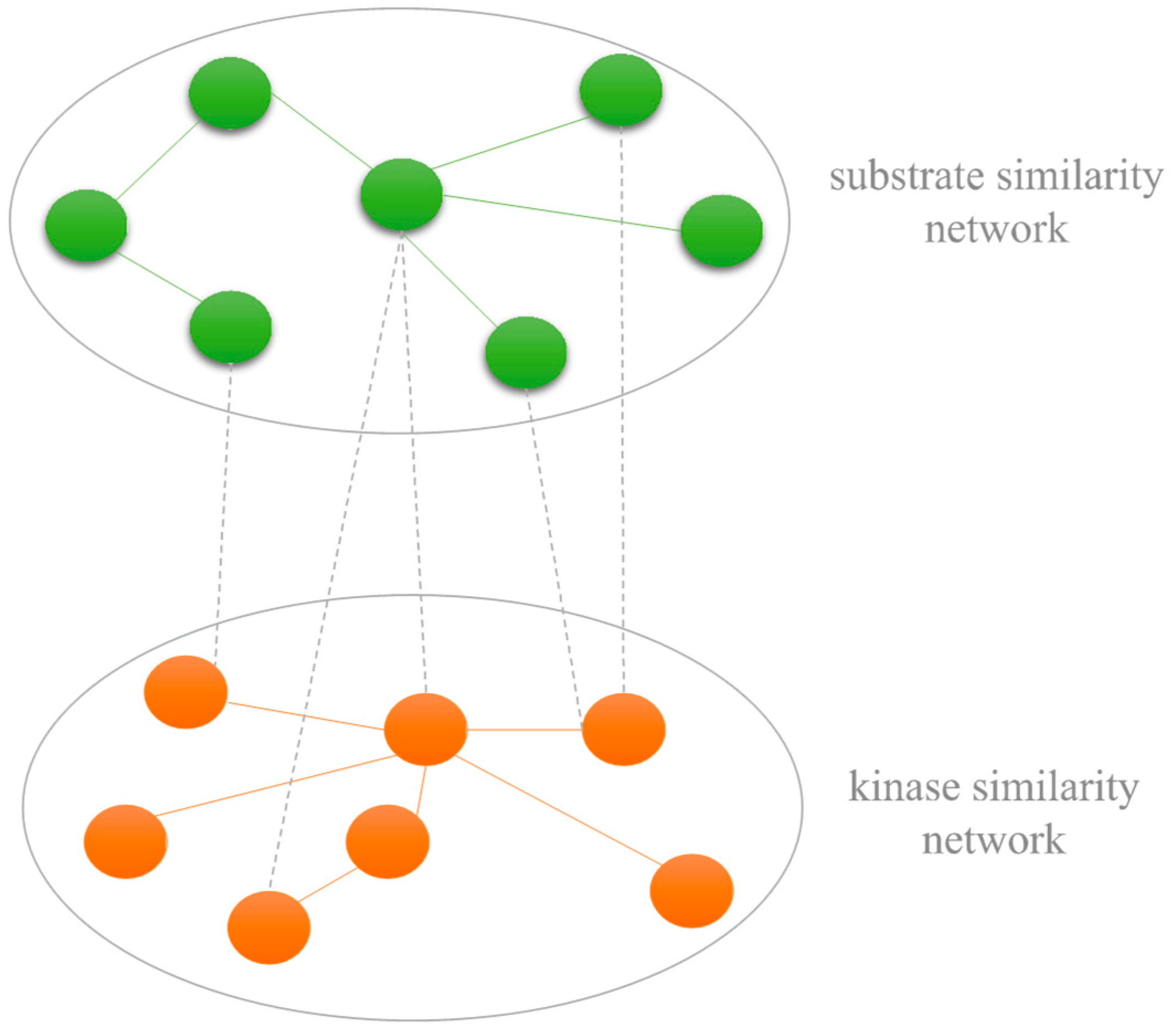
3.2. Kinase-Kinase and Substrate-Substrate Similarity Measure
3.3. Adjust the Kinase-Substrate Interaction Network
3.4. Construction of Kinase-Substrate Heterogenous Network
3.5. Predicting Kinase-Substrate Interactions by Using Matrix Completion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| KSIMC | Predicting kinase-substrate interactions based on matrix completion |
| AUC | Area Under roc Curve |
| ROC | Receiver Operating Characteristic Curve |
| TP | True Positive |
| FP | False Positive |
| TN | True Negative |
| FN | False Negative |
| TPR | True Positive Rate |
| FPR | False Positive Rate |
References
- Cohen, P. The origins of protein phosphorylation. Nat. Cell Biol. 2002, 4, E127–E130. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef]
- Zheng, R.; Li, M.; Chen, X.; Wu, F.; Pan, Y.; Wang, J. BiXGBoost: A scalable, flexible boosting based method for reconstructing gene regulatory networks. Bioinformatics 2018, bty908. [Google Scholar] [CrossRef] [PubMed]
- Grabiec, A.M.; Korchynskyi, O.; Tak, P.P.; Reedquist, K.A. Histone deacetylase inhibitors suppress rheumatoid arthritis fibroblast-like synoviocyte and macrophage IL-6 production by accelerating mRNA decay. Ann. Rheum. Dis. 2012, 71, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. The role of protein phosphorylation in human health and disease. Eur. J. Biochem. 2001, 268, 5001–5010. [Google Scholar] [CrossRef] [PubMed]
- Lan, W.; Wang, J.; Li, M.; Peng, W.; Wu, F. Computational approaches for prioritizing candidate disease genes based on PPI networks. Tsinghua Sci. Technol. 2015, 20, 500–512. [Google Scholar] [CrossRef]
- Zheng, R.; Li, M.; Li, Y.; Wu, F.; Wang, J. MGT-SM: A Method for Constructing Cellular Signal Transduction Networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017. [Google Scholar] [CrossRef]
- Lan, W.; Wang, J.; Li, M.; Liu, J.; Li, Y.; Wu, F.; Pan, Y. Predicting drug–target interaction using positive-unlabeled learning. Neurocomputing 2016, 206, 50–57. [Google Scholar] [CrossRef]
- Salinas, M.; Wang, J.; de Sagarra, M.R.; Martín, D.; Rojo, A.I.; Martin-Perez, J.; de Montellano, P.R.O.; Cuadrado, A. Protein kinase Akt/PKB phosphorylates heme oxygenase-1 in vitro and in vivo. FEBS Lett. 2004, 578, 90–94. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Xie, Z.; Zhu, H.; Qian, J. Understanding protein phosphorylation on a systems level. Brief. Funct. Genom. 2010, 9, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Dinkel, H.; Chica, C.; Via, A.; Gould, C.M.; Jensen, L.J.; Gibson, T.J.; Diella, F. Phospho. ELM: A database of phosphorylation sites—Update 2011. Nucleic Acids Res. 2010, 39, D261–D267. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Kornhauser, J.M.; Tkachev, S.; Zhang, B.; Skrzypek, E.; Murray, B.; Latham, V.; Sullivan, M. PhosphoSitePlus: A comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2011, 40, D261–D270. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Chen, Q.; Liu, Z.; Zheng, R.; Liu, J.; Wang, J.; Lan, W. KSIBW: Predicting Kinase-Substrate Interactions Based on Bi-random Walk. In Bioinformatics Research and Applications: 14th International Symposium, ISBRA 2018, Beijing, China, 8–11 June 2018; Springer International Publishing: Cham, Switzerland, 2018; pp. 151–162. [Google Scholar]
- Chen, Q.; Wang, Y.; Chen, B.; Zhang, C.; Wang, L.; Li, J. Using Propensity Scores to Predict the Kinases of Unannotated Phosphopeptides. Knowl.-Based Syst. 2017, 135, 60–76. [Google Scholar] [CrossRef]
- Linding, R.; Jensen, L.J.; Ostheimer, G.J.; van Vugt, M.A.; Jørgensen, C.; Miron, I.M.; Diella, F.; Colwill, K.; Taylor, L.; Elder, K. Systematic discovery of in vivo phosphorylation networks. Cell 2007, 129, 1415–1426. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.H.; Van Leemput, K.; Verschoren, A.; Laukens, K. Prediction of kinase-specific phosphorylation sites using conditional random fields. Bioinformatics 2008, 24, 2857–2864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.-F.; Xue, Y.; Chen, G.-L.; Yao, X. GPS: A novel group-based phosphorylation predicting and scoring method. Biochem. Biophys. Res. Commun. 2004, 325, 1443–1448. [Google Scholar] [CrossRef]
- Zou, L.; Wang, M.; Shen, Y.; Liao, J.; Li, A.; Wang, M. PKIS: Computational identification of protein kinases for experimentally discovered protein phosphorylation sites. BMC Bioinform. 2013, 14, 247. [Google Scholar] [CrossRef]
- Torii, M.; Liu, H.; Hu, Z.-Z. Support vector machine-based mucin-type o-linked glycosylation site prediction using enhanced sequence feature encoding. AMIA Annu. Symp. Proc. 2009, 2009, 640–644. [Google Scholar]
- Patrick, R.; Lê Cao, K.-A.; Kobe, B.; Bodén, M. PhosphoPICK: Modelling cellular context to map kinase-substrate phosphorylation events. Bioinformatics 2014, 31, 382–389. [Google Scholar] [CrossRef]
- Fan, W.; Xu, X.; Shen, Y.; Feng, H.; Li, A.; Wang, M. Prediction of protein kinase-specific phosphorylation sites in hierarchical structure using functional information and random forest. Amino Acids 2014, 46, 1069–1078. [Google Scholar] [CrossRef]
- Li, A.; Xu, X.; Zhang, H.; Wang, M. Kinase identification with supervised laplacian regularized least squares. PLoS ONE 2015, 10, e0139676. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Wang, H.; Wang, J.; Leier, A.; Marquez-Lago, T.; Yang, B.; Zhang, Z.; Akutsu, T.; Webb, G.I.; Daly, R.J. PhosphoPredict: A bioinformatics tool for prediction of human kinase-specific phosphorylation substrates and sites by integrating heterogeneous feature selection. Sci Rep. 2017, 7, 6862. [Google Scholar] [CrossRef] [PubMed]
- Gnad, F.; Gunawardena, J.; Mann, M. PHOSIDA 2011: The posttranslational modification database. Nucleic Acids Res. 2011, 39, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Ye, M.; Liu, Z.; Cheng, H.; Jiang, X.; Han, G.; Songyang, Z.; Tan, Y.; Wang, H.; Ren, J. Systematic analysis of protein phosphorylation networks from phosphoproteomic data. Mol. Cell. Proteom. 2012, 11, 1070–1083. [Google Scholar] [CrossRef] [PubMed]
- Damle, N.P.; Mohanty, D. Deciphering kinase–substrate relationships by analysis of domain-specific phosphorylation network. Bioinformatics 2014, 30, 1730–1738. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, M.; Xu, X. Prediction of kinase–substrate relations based on heterogeneous networks. J. Bioinform. Comput. Biol. 2015, 13, 1542003. [Google Scholar] [CrossRef] [PubMed]
- Qin, G.-M.; Li, R.-Y.; Zhao, X.-M. PhosD: Inferring kinase–substrate interactions based on protein domains. Bioinformatics 2016, 33, 1197–1204. [Google Scholar] [CrossRef]
- Shi, C.; Kong, X.; Huang, Y.; Philip, S.Y.; Wu, B. HeteSim: A General Framework for Relevance Measure in Heterogeneous Networks. IEEE Trans. Knowl. Data Eng. 2014, 26. [Google Scholar] [CrossRef]
- Walker, M.P.; DiAugustine, R.P.; Zeringue, E.; Bunger, M.K.; Schmitt, M.; Archer, T.K.; Richards, R.G. An Insulin-Like Growth Factor-1 (IGF-1)/Insulin Receptor Substrate-1 (IRS-1) Pathway Stimulates a Mitotic Kinase (cdk1) in the Uterine Epithelium During the Proliferative Response to Estradiol. J. Endocrinol. 2010, 207, 225–235. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Chen, Q.F.; Lan, W.; Wang, J.X. Mining featured patterns of MiRNA interaction based on sequence and structure similarity. IEEE/ACM Trans. Comput. Biol. Bioinform. 2013, 10, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Lan, W.; Chen, Q.F.; Li, T.S.; Yuan, C.G.; Mann, S.; Chen, B.S. Identification of important positions within miRNAs by integrating sequential and structural features. Curr. Protein Pept. Sci. 2014, 15, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, B.K. Sparse approximate solutions to linear systems. SIAM J. Comput. 1995, 24, 227–234. [Google Scholar] [CrossRef]
- Cai, J.-F.; Candès, E.J.; Shen, Z. A singular value thresholding algorithm for matrix completion. SIAM J. Optim. 2010, 20, 1956–1982. [Google Scholar] [CrossRef]
- Luo, H.; Li, M.; Wang, S.; Liu, Q.; Li, Y.; Wang, J. Computational drug repositioning using low-rank matrix approximation and randomized algorithms. Bioinformatics 2018, 34, 1904–1912. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yu, W. A Fast Implementation of Singular Value Thresholding Algorithm using Recycling Rank Revealing Randomized Singular Value Decomposition. arXiv, 2017; arXiv:1704.05528. [Google Scholar]
- Lan, W.; Wang, J.; Li, M.; Liu, J.; Wu, F.; Pan, Y. Predicting microRNA-disease associations based on improved microRNA and disease similarities. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 15, 1774–1782. [Google Scholar] [CrossRef]
- Liu, J.; Li, M.; Lan, W.; Wu, F.; Pan, Y.; Wang, J. Classification of alzheimer’s disease using whole brain hierarchical network. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 15, 624–632. [Google Scholar] [CrossRef]
- Lan, W.; Li, M.; Zhao, K.; Liu, J.; Wu, F.; Pan, Y.; Wang, J. LDAP: A web server for lncRNA-disease association prediction. Bioinformatics 2017, 33, 458–460. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Top | Substrate | Predicted Kinase | Evidence |
|---|---|---|---|
| 1 | IRS1 | CDK1 | PMID: 20798132 |
| 2 | IRS1 | MAPK1 | PhosphoNET |
| 3 | IRS1 | PRKCA | PhosphoNET |
| 4 | IRS1 | ABL1 | Unknown |
| 5 | IRS1 | CSNK2A1 | Unknown |
| 6 | IRS1 | MAPK8 | PhosphoNET |
| 7 | IRS1 | PRKCE | PhosphoNET |
| 8 | IRS1 | GSK3B | Unknown |
| 9 | IRS1 | PRKG1 | Unknown |
| 10 | IRS1 | RPS6KA3 | Unknown |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gan, J.; Qiu, J.; Deng, C.; Lan, W.; Chen, Q.; Hu, Y. KSIMC: Predicting Kinase–Substrate Interactions Based on Matrix Completion. Int. J. Mol. Sci. 2019, 20, 302. https://doi.org/10.3390/ijms20020302
Gan J, Qiu J, Deng C, Lan W, Chen Q, Hu Y. KSIMC: Predicting Kinase–Substrate Interactions Based on Matrix Completion. International Journal of Molecular Sciences. 2019; 20(2):302. https://doi.org/10.3390/ijms20020302
Chicago/Turabian StyleGan, Jingzhong, Jie Qiu, Canshang Deng, Wei Lan, Qingfeng Chen, and Yanling Hu. 2019. "KSIMC: Predicting Kinase–Substrate Interactions Based on Matrix Completion" International Journal of Molecular Sciences 20, no. 2: 302. https://doi.org/10.3390/ijms20020302
APA StyleGan, J., Qiu, J., Deng, C., Lan, W., Chen, Q., & Hu, Y. (2019). KSIMC: Predicting Kinase–Substrate Interactions Based on Matrix Completion. International Journal of Molecular Sciences, 20(2), 302. https://doi.org/10.3390/ijms20020302