Prevalence of ABCA4 Deep-Intronic Variants and Related Phenotype in An Unsolved “One-Hit” Cohort with Stargardt Disease

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Results
2.1. Genotypic Analysis
2.2. Phenotypic Analysis and Genotype-Phenotype Correlation
2.2.1. c.4253+43G>A
2.2.2. c.4539+2064C>T
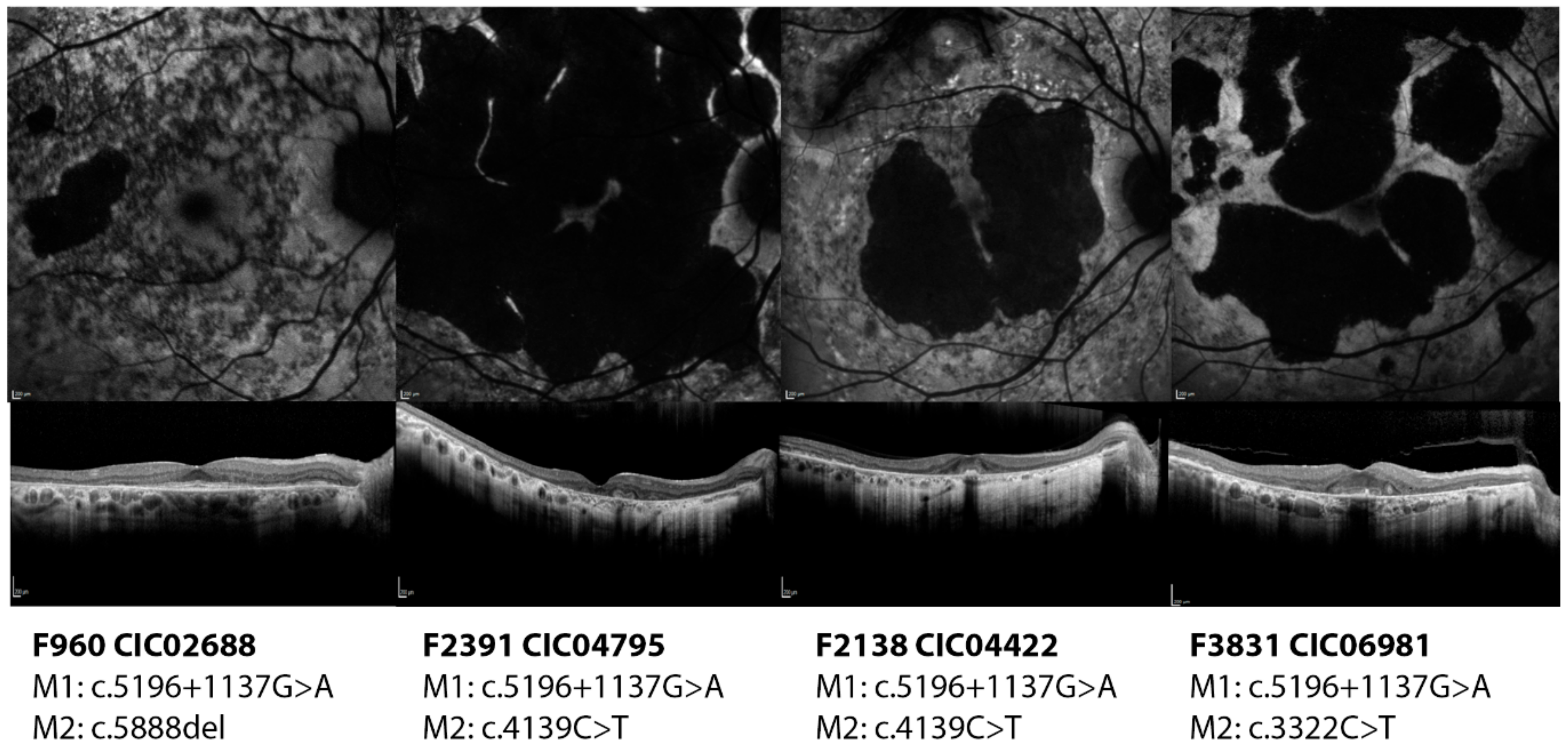
2.2.3. c.5196+1137G>A
2.2.4. Novel Variants
3. Discussion
4. Materials and Methods
4.1. Patients and Preliminary Results
4.2. Literature Review
4.3. Genetic Screening
4.4. Phenotypic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABCA4 | ATP-binding cassette transporter 4 |
| AONs | antisense oligonucleotides |
| BCVA | best corrected visual acuity |
| bp | base pair |
| ECD | Extracellular domain |
| ERG | Electroretinogram |
| ETDRS | Early Treatment Diabetic Retinopathy Study |
| EZ | ellipsoid zone |
| ff-ERG | Full field electroretinogram |
| FP | Fundus Photograph |
| ISCEV | International Society for Clinical of Electrophysiology of Vision |
| MAF | minor allele frequency |
| mf-ERG | Full field electroretinogram |
| NBD | nucleotide binding domain |
| NIR-AF | Near infrared fundus autofluorescence |
| OCT | optical coherence tomography |
| RPE | Retinal pigment epithelium |
| STGD1 | Autosomal recessive Stargardt disease |
| SW-AF | Short wavelength fundus autofluorescence |
References
- Michaelides, M.; Hunt, D.; Moore, A. The genetics of inherited macular dystrophies. J. Med. Genet. 2003, 40, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Allikmets, R.; Singh, N.; Sun, H.; Shroyer, N.F.; Hutchinson, A.; Chidambaram, A.; Gerrard, B.; Baird, L.; Stauffer, D.; Peiffer, A.; et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat. Genet. 1997, 15, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Van Driel, M.A.; Maugeri, A.; Klevering, B.J.; Hoyng, C.B.; Cremers, F.P. ABCR unites what ophthalmologists divide(s). Ophthalmic Genet. 1998, 19, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, S.S.; Bax, N.M.; Zernant, J.; Allikmets, R.; Fritsche, L.G.; den Dunnen, J.T.; Ajmal, M.; Hoyng, C.B.; Cremers, F.P.M. In Silico Functional Meta-Analysis of 5962 ABCA4 Variants in 3928 Retinal Dystrophy Cases. Hum. Mutat. 2017, 38, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Hussain, R.M.; Ciulla, T.A.; Berrocal, A.M.; Gregori, N.Z.; Flynn, H.W.; Lam, B.L. Stargardt macular dystrophy and evolving therapies. Expert Opin. Biol. Ther. 2018, 18, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Lenis, T.L.; Hu, J.; Ng, S.Y.; Jiang, Z.; Sarfare, S.; Lloyd, M.B.; Esposito, N.J.; Samuel, W.; Jaworski, C.; Bok, D.; et al. Expression of ABCA4 in the retinal pigment epithelium and its implications for Stargardt macular degeneration. Proc. Natl. Acad. Sci. USA 2018, 115, E11120–E11127. [Google Scholar] [CrossRef]
- Boulanger-Scemama, E.; El Shamieh, S.; Démontant, V.; Condroyer, C.; Antonio, A.; Michiels, C.; Boyard, F.; Saraiva, J.-P.; Letexier, M.; Souied, E.; et al. Next-generation sequencing applied to a large French cone and cone-rod dystrophy cohort: Mutation spectrum and new genotype-phenotype correlation. Orphanet J. Rare Dis. 2015, 10, 85. [Google Scholar] [CrossRef]
- Klevering, B.J.; Maugeri, A.; Wagner, A.; Go, S.L.; Vink, C.; Cremers, F.P.M.; Hoyng, C.B. Three families displaying the combination of Stargardt’s disease with cone-rod dystrophy or retinitis pigmentosa. Ophthalmology 2004, 111, 546–553. [Google Scholar] [CrossRef]
- Zernant, J.; Schubert, C.; Im, K.M.; Burke, T.; Brown, C.M.; Fishman, G.A.; Tsang, S.H.; Gouras, P.; Dean, M.; Allikmets, R. Analysis of the ABCA4 gene by next-generation sequencing. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8479–8487. [Google Scholar] [CrossRef]
- Zernant, J.; Xie, Y.A.; Ayuso, C.; Riveiro-Alvarez, R.; Lopez-Martinez, M.-A.; Simonelli, F.; Testa, F.; Gorin, M.B.; Strom, S.P.; Bertelsen, M.; et al. Analysis of the ABCA4 genomic locus in Stargardt disease. Hum. Mol. Genet. 2014, 23, 6797–6806. [Google Scholar] [CrossRef]
- Bauwens, M.; De Zaeytijd, J.; Weisschuh, N.; Kohl, S.; Meire, F.; Dahan, K.; Depasse, F.; De Jaegere, S.; De Ravel, T.; De Rademaeker, M.; et al. An augmented ABCA4 screen targeting noncoding regions reveals a deep intronic founder variant in Belgian Stargardt patients. Hum. Mutat. 2015, 36, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Bax, N.M.; Sangermano, R.; Roosing, S.; Thiadens, A.A.H.J.; Hoefsloot, L.H.; van den Born, L.I.; Phan, M.; Klevering, B.J.; Westeneng-van Haaften, C.; Braun, T.A.; et al. Heterozygous deep-intronic variants and deletions in ABCA4 in persons with retinal dystrophies and one exonic ABCA4 variant. Hum. Mutat. 2015, 36, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Sangermano, R.; Bax, N.M.; Bauwens, M.; van den Born, L.I.; De Baere, E.; Garanto, A.; Collin, R.W.J.; Goercharn-Ramlal, A.S.A.; den Engelsman-van Dijk, A.H.A.; Rohrschneider, K.; et al. Photoreceptor Progenitor mRNA Analysis Reveals Exon Skipping Resulting from the ABCA4 c.5461-10T→C Mutation in Stargardt Disease. Ophthalmology 2016, 123, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Sangermano, R.; Khan, M.; Cornelis, S.S.; Richelle, V.; Albert, S.; Garanto, A.; Elmelik, D.; Qamar, R.; Lugtenberg, D.; van den Born, L.I.; et al. ABCA4 midigenes reveal the full splice spectrum of all reported noncanonical splice site variants in Stargardt disease. Genome Res. 2018, 28, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Albert, S.; Garanto, A.; Sangermano, R.; Khan, M.; Bax, N.M.; Hoyng, C.B.; Zernant, J.; Lee, W.; Allikmets, R.; Collin, R.W.J.; et al. Identification and Rescue of Splice Defects Caused by Two Neighboring Deep-Intronic ABCA4 Mutations Underlying Stargardt Disease. Am. J. Hum. Genet. 2018, 102, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Bauwens, M.; Garanto, A.; Sangermano, R.; Naessens, S.; Weisschuh, N.; Zaeytijd, J.D.; Khan, M.; Sadler, F.; Balikova, I.; Cauwenbergh, C.V.; et al. ABCA4-associated disease as a model for missing heritability in autosomal recessive disorders: Novel noncoding splice, cis-regulatory, structural, and recurrent hypomorphic variants. Genet. Med. 2019, 21, 1761–1771. [Google Scholar] [CrossRef] [PubMed]
- Fadaie, Z.; Khan, M.; Del Pozo-Valero, M.; Cornelis, S.S.; Ayuso, C.; Cremers, F.P.M.; Roosing, S. The Abca Study Group, null Identification of splice defects due to noncanonical splice site or deep-intronic variants in ABCA4. Hum. Mutat. 2019. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Cornelis, S.S.; Khan, M.I.; Elmelik, D.; Manders, E.; Bakker, S.; Derks, R.; Neveling, K.; Van de Vorst, M.; Gilissen, C.; et al. Cost-effective molecular inversion probe-based ABCA4 sequencing reveals deep-intronic variants in Stargardt disease. Hum. Mutat. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zernant, J.; Lee, W.; Collison, F.T.; Fishman, G.A.; Sergeev, Y.V.; Schuerch, K.; Sparrow, J.R.; Tsang, S.H.; Allikmets, R. Frequent hypomorphic alleles account for a significant fraction of ABCA4 disease and distinguish it from age-related macular degeneration. J. Med. Genet. 2017, 54, 404–412. [Google Scholar] [CrossRef]
- Lee, W.; Xie, Y.; Zernant, J.; Yuan, B.; Bearelly, S.; Tsang, S.H.; Lupski, J.R.; Allikmets, R. Complex inheritance of ABCA4 disease: Four mutations in a family with multiple macular phenotypes. Hum. Genet. 2016, 135, 9–19. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.A.; Mullins, R.F.; Wagner, A.H.; Andorf, J.L.; Johnston, R.M.; Bakall, B.B.; Deluca, A.P.; Fishman, G.A.; Lam, B.L.; Weleber, R.G.; et al. Non-exomic and synonymous variants in ABCA4 are an important cause of Stargardt disease. Hum. Mol. Genet. 2013, 22, 5136–5145. [Google Scholar] [CrossRef] [PubMed]
- Schulz, H.L.; Grassmann, F.; Kellner, U.; Spital, G.; Rüther, K.; Jägle, H.; Hufendiek, K.; Rating, P.; Huchzermeyer, C.; Baier, M.J.; et al. Mutation Spectrum of the ABCA4 Gene in 335 Stargardt Disease Patients from a Multicenter German Cohort-Impact of Selected Deep Intronic Variants and Common SNPs. Investig. Ophthalmol. Vis. Sci. 2017, 58, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Sangermano, R.; Garanto, A.; Khan, M.; Runhart, E.H.; Bauwens, M.; Bax, N.M.; van den Born, L.I.; Khan, M.I.; Cornelis, S.S.; Verheij, J.B.G.M.; et al. Deep-intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet. Med. 2019, 21, 1751–1760. [Google Scholar] [CrossRef] [PubMed]
- Sergeev, Y.V.; Caruso, R.C.; Meltzer, M.R.; Smaoui, N.; MacDonald, I.M.; Sieving, P.A. Molecular modeling of retinoschisin with functional analysis of pathogenic mutations from human X-linked retinoschisis. Hum. Mol. Genet. 2010, 19, 1302–1313. [Google Scholar] [CrossRef] [PubMed]
- Sergeev, Y.; Pogrebniak, K.; Falsini, B.; Zein, W.; Goetz, K.; Huang, J.; Peeler, C.; Jayasundera, K.; Brooks, B.; Sieving, P. Molecular modeling of functional domain of ABCA4: Towards understanding the genotype-to-phenotype relationships in Stargardt’s disease. Investig. Ophthalmol. Vis. Sci. 2013, 54, 1325. [Google Scholar]
- Fujinami, K.; Sergouniotis, P.I.; Davidson, A.E.; Mackay, D.S.; Tsunoda, K.; Tsubota, K.; Robson, A.G.; Holder, G.E.; Moore, A.T.; Michaelides, M.; et al. The clinical effect of homozygous ABCA4 alleles in 18 patients. Ophthalmology 2013, 120, 2324–2331. [Google Scholar] [CrossRef]
- Cooper, T.A. Use of minigene systems to dissect alternative splicing elements. Methods 2005, 37, 331–340. [Google Scholar] [CrossRef]
- Bickenbach, J.R. Isolation, characterization, and culture of epithelial stem cells. Methods Mol. Biol. 2005, 289, 97–102. [Google Scholar]
- Aukrust, I.; Jansson, R.W.; Bredrup, C.; Rusaas, H.E.; Berland, S.; Jørgensen, A.; Haug, M.G.; Rødahl, E.; Houge, G.; Knappskog, P.M. The intronic ABCA4 c.5461-10T>C variant, frequently seen in patients with Stargardt disease, causes splice defects and reduced ABCA4 protein level. Acta Ophthalmol. 2017, 95, 240–246. [Google Scholar] [CrossRef]
- Millat, G.; Lafont, E.; Nony, S.; Rouvet, I.; Bozon, D. Functional characterization of putative novel splicing mutations in the cardiomyopathy-causing genes. DNA Cell Biol. 2015, 34, 489–496. [Google Scholar] [CrossRef]
- Nguyen, L.S.; Wilkinson, M.F.; Gecz, J. Nonsense-mediated mRNA decay: Inter-individual variability and human disease. Neurosci. Biobehav. Rev. 2014, 46 Pt 2, 175–186. [Google Scholar] [CrossRef]
- Hammond, S.M.; Wood, M.J.A. Genetic therapies for RNA mis-splicing diseases. Trends Genet. 2011, 27, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Garanto, A.; Duijkers, L.; Tomkiewicz, T.Z.; Collin, R.W.J. Antisense Oligonucleotide Screening to Optimize the Rescue of the Splicing Defect Caused by the Recurrent Deep-Intronic ABCA4 Variant c.4539+2001G>A in Stargardt Disease. Genes (Basel) 2019, 10, 452. [Google Scholar] [CrossRef] [PubMed]
- Bonifert, T.; Gonzalez Menendez, I.; Battke, F.; Theurer, Y.; Synofzik, M.; Schöls, L.; Wissinger, B. Antisense Oligonucleotide Mediated Splice Correction of a Deep Intronic Mutation in OPA1. Mol. Ther. Nucleic Acids 2016, 5, e390. [Google Scholar] [CrossRef] [PubMed]
- Slijkerman, R.W.; Vaché, C.; Dona, M.; García-García, G.; Claustres, M.; Hetterschijt, L.; Peters, T.A.; Hartel, B.P.; Pennings, R.J.; Millan, J.M.; et al. Antisense Oligonucleotide-based Splice Correction for USH2A-associated Retinal Degeneration Caused by a Frequent Deep-intronic Mutation. Mol. Ther. Nucleic Acids 2016, 5, e381. [Google Scholar] [CrossRef] [PubMed]
- Collin, R.W.; den Hollander, A.I.; van der Velde-Visser, S.D.; Bennicelli, J.; Bennett, J.; Cremers, F.P. Antisense Oligonucleotide (AON)-based Therapy for Leber Congenital Amaurosis Caused by a Frequent Mutation in CEP290. Mol. Ther. Nucleic Acids 2012, 1, e14. [Google Scholar] [CrossRef] [PubMed]
- Gerard, X.; Perrault, I.; Hanein, S.; Silva, E.; Bigot, K.; Defoort-Delhemmes, S.; Rio, M.; Munnich, A.; Scherman, D.; Kaplan, J.; et al. AON-mediated Exon Skipping Restores Ciliation in Fibroblasts Harboring the Common Leber Congenital Amaurosis CEP290 Mutation. Mol. Ther. Nucleic Acids 2012, 1, e29. [Google Scholar] [CrossRef] [PubMed]
- Parfitt, D.A.; Lane, A.; Ramsden, C.M.; Carr, A.-J.F.; Munro, P.M.; Jovanovic, K.; Schwarz, N.; Kanuga, N.; Muthiah, M.N.; Hull, S.; et al. Identification and Correction of Mechanisms Underlying Inherited Blindness in Human iPSC-Derived Optic Cups. Cell Stem Cell 2016, 18, 769–781. [Google Scholar] [CrossRef]
- Duijkers, L.; van den Born, L.I.; Neidhardt, J.; Bax, N.M.; Pierrache, L.H.M.; Klevering, B.J.; Collin, R.W.J.; Garanto, A. Antisense Oligonucleotide-Based Splicing Correction in Individuals with Leber Congenital Amaurosis due to Compound Heterozygosity for the c.2991+1655A>G Mutation in CEP290. Int. J. Mol. Sci. 2018, 19, 753. [Google Scholar] [CrossRef]
- Garanto, A.; van der Velde-Visser, S.D.; Cremers, F.P.M.; Collin, R.W.J. Antisense Oligonucleotide-Based Splice Correction of a Deep-Intronic Mutation in CHM Underlying Choroideremia. Adv. Exp. Med. Biol. 2018, 1074, 83–89. [Google Scholar] [PubMed]
- Garanto, A.; Chung, D.C.; Duijkers, L.; Corral-Serrano, J.C.; Messchaert, M.; Xiao, R.; Bennett, J.; Vandenberghe, L.H.; Collin, R.W.J. In vitro and in vivo rescue of aberrant splicing in CEP290-associated LCA by antisense oligonucleotide delivery. Hum. Mol. Genet. 2016, 25, 2552–2563. [Google Scholar] [PubMed]
- Gérard, X.; Perrault, I.; Munnich, A.; Kaplan, J.; Rozet, J.-M. Intravitreal Injection of Splice-switching Oligonucleotides to Manipulate Splicing in Retinal Cells. Mol. Ther. Nucleic Acids 2015, 4, e250. [Google Scholar] [CrossRef] [PubMed]
- Murray, S.F.; Jazayeri, A.; Matthes, M.T.; Yasumura, D.; Yang, H.; Peralta, R.; Watt, A.; Freier, S.; Hung, G.; Adamson, P.S.; et al. Allele-Specific Inhibition of Rhodopsin with an Antisense Oligonucleotide Slows Photoreceptor Cell Degeneration. Investig. Ophthalmol. Vis. Sci. 2015, 56, 6362–6375. [Google Scholar] [CrossRef] [PubMed]
- Jaakson, K.; Zernant, J.; Külm, M.; Hutchinson, A.; Tonisson, N.; Glavac, D.; Ravnik-Glavac, M.; Hawlina, M.; Meltzer, M.R.; Caruso, R.C.; et al. Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum. Mutat. 2003, 22, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Nassisi, M.; Mohand-Saïd, S.; Dhaenens, C.-M.; Boyard, F.; Démontant, V.; Andrieu, C.; Antonio, A.; Condroyer, C.; Foussard, M.; Méjécase, C.; et al. Expanding the Mutation Spectrum in ABCA4: Sixty Novel Disease Causing Variants and Their Associated Phenotype in a Large French Stargardt Cohort. Int. J. Mol. Sci. 2018, 19, 2196. [Google Scholar] [CrossRef]
- Zhang, X.H.-F.; Chasin, L.A. Computational definition of sequence motifs governing constitutive exon splicing. Genes Dev. 2004, 18, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef]
- Yeo, G.; Holste, D.; Kreiman, G.; Burge, C.B. Variation in alternative splicing across human tissues. Genome Biol. 2004, 5, R74. [Google Scholar] [CrossRef]
- Pertea, M.; Lin, X.; Salzberg, S.L. GeneSplicer: A new computational method for splice site prediction. Nucleic Acids Res. 2001, 29, 1185–1190. [Google Scholar] [CrossRef]
- Cartegni, L.; Wang, J.; Zhu, Z.; Zhang, M.Q.; Krainer, A.R. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003, 31, 3568–3571. [Google Scholar] [CrossRef] [PubMed]
- Fishman, G.A. Fundus flavimaculatus. A clinical classification. Arch. Ophthalmol. 1976, 94, 2061–2067. [Google Scholar] [CrossRef] [PubMed]
- Duncker, T.; Marsiglia, M.; Lee, W.; Zernant, J.; Tsang, S.H.; Allikmets, R.; Greenstein, V.C.; Sparrow, J.R. Correlations Among Near-Infrared and Short-Wavelength Autofluorescence and Spectral-Domain Optical Coherence Tomography in Recessive Stargardt Disease. Investig. Ophthalmol. Vis. Sci. 2014, 55, 8134–8143. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, K.; Sergouniotis, P.I.; Davidson, A.E.; Wright, G.; Chana, R.K.; Tsunoda, K.; Tsubota, K.; Egan, C.A.; Robson, A.G.; Moore, A.T.; et al. Clinical and molecular analysis of Stargardt disease with preserved foveal structure and function. Am. J. Ophthalmol. 2013, 156, 487–501.e1. [Google Scholar] [CrossRef] [PubMed]
- Hood, D.C.; Bach, M.; Brigell, M.; Keating, D.; Kondo, M.; Lyons, J.S.; Palmowski-Wolfe, A.M. ISCEV guidelines for clinical multifocal electroretinography (2007 edition). Doc. Ophthalmol. 2008, 116, 1–11. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol. 2015, 130, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lois, N.; Holder, G.E.; Bunce, C.; Fitzke, F.W.; Bird, A.C. Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Arch. Ophthalmol. 2001, 119, 359–369. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Allele 1 | Allele 2 | |||||||
|---|---|---|---|---|---|---|---|---|
| Patient ID | Family ID | Exon/Intron | Nucleotide Change | Protein Change | Exon/Intron | Nucleotide Change | Protein Change | |
| CIC00251 | 174 | Index | 42 | c.5882G>A | p.(Gly1961Glu) | IVS30 | c.4539+2064C>T | p.[=,Arg1514Leufs*36] |
| CIC00454 | 174 | Unaffected father | 42 | c.5882G>A | p.(Gly1961Glu) | reference sequence | ||
| CIC00455 | 174 | Unaffected mother | reference sequence | IVS30 | c.4539+2064C>T | p.[=,Arg1514Leufs*36] | ||
| CIC01275 | 765 | Index | 10 | c.1344del | p.(Ile449Metfs*3) | IVS30 | c.4539+2064C>T | p.[=Arg1514Leufs*36] |
| CIC01276 | 765 | Unaffected mother | reference sequence | IVS30 | c.4539+2064C>T | p.[=Arg1514Leufs*36] | ||
| CIC01277 | 765 | Unaffected father | 10 | c.1344del | p.(Ile449Metfs*3) | reference sequence | ||
| CIC02688 | 960 | Index | 42 | c.5888del | p.(Pro1963Argfs*11) | IVS36 | c.5196+1137G>A | p.[=,Met1733Glufs*78] |
| CIC03648 | 1602 | Index | IVS38 | c.5461-10T>C | p.[Thr1821Aspfs*6, Thr1821Valfs*13] | IVS28 | c.4253+43G>A | p.[=,Ile1377Hisfs*3] |
| 40 | c.5603A>T | p.(Asn1868Ile) | ||||||
| CIC03649 | 1602 | Unaffected aunt | IVS38 | c.5461-10T>C | p.[Thr1821Asp*6, Thr1821Valfs*13] | reference sequence | ||
| 40 | c.5603A>T | p.(Asn1868Ile) | ||||||
| CIC04422 | 2138 | Index | 28 | c.4139C>T | p.(Pro1380Leu) | IVS36 | c.5196+1137G>A | p.[=,Met1733Glufs*78] |
| CIC04795 | 2391 | Index | 28 | c.4139C>T | p.(Pro1380Leu) | IVS36 | c.5196+1137G>A | p.[=,Met1733Glufs*78] |
| CIC06528 | 3493 | Index | IVS40 | c.5714+5G>A | p.[=,Glu1863Leufs*33] | IVS30 | c.4539+2064C>T | p.[=,Arg1514Leufs*36] |
| CIC07955 | 3493 | Affected cousin | IVS40 | c.5714+5G>A | p.[=,Glu1863Leufs*33] | IVS30 | c.4539+2064C>T | p.[=,Arg1514Leufs*36] |
| CIC06981 | 3831 | Index | 22 | c.3322C>T | p.(Arg1108Cys) | IVS36 | c.5196+1137G>A | p.[=,Met1733Glufs*78] |
| CIC08281 | 4645 | Index | 43 | c.5914G>A | p.(Gly1972Arg) | IVS28 | c.4253+43G>A | p.[=,Ile1377Hisfs*3] |
| CIC07459 | 4128 | Index | 13 | c.1804C>T | p.(Arg602Trp) | IVS36 | c.5196+1137G>A | p.[=,Met1733Glufs*78] |
| CIC07460 | 4128 | Unaffected mother | reference sequence | IVS36 | c.5196+1137G>A | p.[=,Met1733Glufs*78] | ||
| CIC07461 | 4128 | Unaffected father | 13 | c.1804C>T | p.(Arg602Trp) | reference sequence | ||
| CIC08968 | 4128 | Unaffected sister | reference sequence | reference sequence | ||||
| CIC08809 | 5010 | Index | 6 | c.688T>G | p.(Cys230Gly) | IVS30 | c.4539+2064C>T | p.[=,Arg1514Leufs*36] |
| CIC09117 | 5207 | Index | 6 | c.686T>C | p.(Leu229Pro) | IVS28 | c.4253+43G>A | p.[=,Ile1377Hisfs*3] |
| CIC09118 | 5207 | Unaffected mother | 6 | c.686T>C | p.(Leu229Pro) | reference sequence | ||
| CIC09119 | 5207 | Unaffected father | IVS28 | c.4253+43G>A | p.[=,Ile1377Hisfs*3] | IVS28 | c.4253+43G>A | p.[=,Ile1377Hisfs*3] |
| CIC09817 | 5639 | Index | 40 | c.5603A>T | p.(Asn1868Ile) | IVS28 | c.4253+43G>A | p.[=,Ile1377Hisfs*3] |
| CIC10544 | 6093 | Index | 13 | c.1804C>T | p.(Arg602Trp) | IVS36 | c.5196+1137G>A | p.[=,Met1733Glufs*78] |
| # Subject | Location | Variant (CCDS747.1) | SNP ID | Nucleotide Conservation | MAF (Allele Count/Allele Total/Number of Homozygous) | SSF | MaxEntScan | NNSplice | GeneSplicer | ESE Finder |
|---|---|---|---|---|---|---|---|---|---|---|
| CIC03520 | IVS6 | c.768+353T>C | rs79372932 | Not conserved | 0.003281 (103/31392/0) | No changes | No changes | Mild activation of an acceptor site | Mild activation of an acceptor site | No changes |
| CIC03956 | IVS6 | c.768+508A>G | rs77933221 | Moderately Conserved | 0.007579 (238/31404/2) | Strong inactivation of a donor site and activation of an acceptor site | Moderate inactivation of a donor site and activation of an acceptor site | No changes | No changes | Formations of binding sites for SF2/ASF |
| CIC08792 | IVS6 | c.769-775C>T | - | Not conserved | Absent | No changes | No changes | No changes | No changes | No changes |
| CIC03956 | IVS7 | c.859-364C>T | rs544917926 | Not conserved | 0.003154 (99/31388/2) | No changes | No changes | No changes | No changes | No changes |
| CIC01916 | IVS7 | c.859-256A>G | rs538160992 | Not Conserved | 0.00009553 (3/31404/0) | No changes | No changes | No changes | No changes | No changes |
| CIC01916 | IVS7 | c.859-245_859-243delinsTGA | - | C and T highly conserved, A not conserved | Absent | Strong activation of a donor site | Strong activation of a donor site | Strong activation of a donor site | Strong activation of a donor site | Formations of binding sites for SF2/ASF and Srp40 |
| CIC10548 | IVS7 | c.859-241A>C | rs56378813 | Highly conserved | 0.0009871 (31/31404/0) | No changes | No changes | No changes | No changes | No changes |
| CIC01916 | IVS7 | c.859-235T>C | - | Not Conserved | Absent | No changes | No changes | No changes | No changes | No changes |
| CIC00952 CIC00973 CIC01413 CIC02688 CIC09897 CIC10529 CIC10548 CIC10577 | IVS13 | c.1938-703C>T | - | Highly conserved | Absent | No changes | No changes | No changes | No changes | No changes |
| CIC06173 | IVS14 | c.2160+462A>C | - | Not conserved | Absent | No changes | No changes | No changes | No changes | No changes |
| CIC09897 | IVS30 | c.4539+1168C>G | - | Moderately Conserved | Absent | No changes | No changes | No changes | No changes | Formations of binding sites for SF2/ASF |
| CIC06353 | IVS44 | c.6148-489C>T | rs894440427 | Not conserved | 0.0003821 (12/31408/0) | No changes | No changes | No changes | Mild inactivation of donor site and activation of acceptor site | No changes |
| Patient CIC# | Aoo (years) | Aoe (years) | Duration (years) | Sex | Active Smoking | History | Symptoms at Onset | BCVA RE/LE | Color Vision (axis) | Binocular Perimetry | Fundus grading | AF | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Group | Atrophy RPE | Peripapillary Sparing | SD-OCT Foveal Sparing | ERG | ||||||||||||
| 00251 | 10 | 44 | 34 | M | No | Myopic | Decreased VA | 20/200 / 20/200 | Normal | Central scotoma 20° | III | II | Present | Atrophy | No | I |
| 01275 | 11 | 16 | 5 | M | No | Myopic | Decreased VA photophobia | 20/200 / 20/250 | Deutan | Central scotoma 20° | II | II | Present | Flecks | No | III |
| 02688 | - | 63 | - | F | - | - | - | - | - | - | III | II | Present | Yes | Yes | II |
| 03648 | 16 | 26 | 10 | F | No | Negative | Decreased VA | 20/160 / 20/160 | Normal | Central scotoma 5° | III | II | Present | Flecks | No | II |
| 10544 | - | 49 | - | F | - | Negative | - | 20/32 / 20/25 | Normal | - | II | II | Present | Yes | Yes | I |
| 04795 | - | 52 | - | F | - | - | Decreased VA photophobia | 20/500 / 20/200 | Multiple | Central scotoma 20° | III | II | Present | Atrophy | Yes | II |
| 06528 | 26 | 56 | 30 | M | No | Negative | Decreased VA | 20/500 / 20/500 | Deutan | Central scotoma 50° | IV | II | Present | Atrophy | No | III |
| 06981 | 61 | 72 | 11 | M | No | Negative | Decreased VA | 20/50 / 20/50 | Normal | Paracentral scotoma 30° | IV | II | Present | Flecks | Yes | I |
| 08281 | 25 | 45 | 20 | F | No | Right eye amblyopic | Decreased VA | 20/125 / 20/125 | Normal | Central scotoma 10° | I | IV | Present | Yes | No | I |
| 07459 | 15 | 22 | 7 | F | No | Negative | Decreased VA | 20/200 / 20/160 | Tetartan | Central scotoma 20° | II | II | Absent | Flecks | No | II |
| 08809 | 14 | 38 | 24 | F | No | Negative | Decreased VA | 20/320 / 20/250 | Protan | Central scotoma 30° | IV | II | Present | Yes | No | II |
| 09117 | 41 | 47 | 6 | F | No | Negative | Decreased VA photophobia | 20/50 / 20/200 | Deutan | Paracentral scotoma 10° | I | II | Present | Yes | Yes | I |
| 09817 | 50 | 66 | 16 | F | Yes | Negative | Decreased VA | 20/160 / 20/32 | Protan Tritan | Paracentral scotoma 40° | III | II | Present | Yes | Yes | II |
| 04422 | 56 | 72 | 16 | M | Past | Negative | Decreased VA night blindness | 20/50 / 20/32 | Tritan | Paracentral scotoma 20° | IV | II | Present | Yes | Yes | III |
| 01916 | 21 | 40 | 19 | M | No | Negative | Decreased VA | 20/500 / 20/200 | Normal | Central scotoma 30° | IV | II | Present | Yes | No | I |
| 03956 | - | - | - | F | - | - | - | - | - | - | - | - | - | - | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nassisi, M.; Mohand-Saïd, S.; Andrieu, C.; Antonio, A.; Condroyer, C.; Méjécase, C.; Varin, J.; Wohlschlegel, J.; Dhaenens, C.-M.; Sahel, J.-A.; et al. Prevalence of ABCA4 Deep-Intronic Variants and Related Phenotype in An Unsolved “One-Hit” Cohort with Stargardt Disease. Int. J. Mol. Sci. 2019, 20, 5053. https://doi.org/10.3390/ijms20205053
Nassisi M, Mohand-Saïd S, Andrieu C, Antonio A, Condroyer C, Méjécase C, Varin J, Wohlschlegel J, Dhaenens C-M, Sahel J-A, et al. Prevalence of ABCA4 Deep-Intronic Variants and Related Phenotype in An Unsolved “One-Hit” Cohort with Stargardt Disease. International Journal of Molecular Sciences. 2019; 20(20):5053. https://doi.org/10.3390/ijms20205053
Chicago/Turabian StyleNassisi, Marco, Saddek Mohand-Saïd, Camille Andrieu, Aline Antonio, Christel Condroyer, Cécile Méjécase, Juliette Varin, Juliette Wohlschlegel, Claire-Marie Dhaenens, José-Alain Sahel, and et al. 2019. "Prevalence of ABCA4 Deep-Intronic Variants and Related Phenotype in An Unsolved “One-Hit” Cohort with Stargardt Disease" International Journal of Molecular Sciences 20, no. 20: 5053. https://doi.org/10.3390/ijms20205053
APA StyleNassisi, M., Mohand-Saïd, S., Andrieu, C., Antonio, A., Condroyer, C., Méjécase, C., Varin, J., Wohlschlegel, J., Dhaenens, C.-M., Sahel, J.-A., Zeitz, C., & Audo, I. (2019). Prevalence of ABCA4 Deep-Intronic Variants and Related Phenotype in An Unsolved “One-Hit” Cohort with Stargardt Disease. International Journal of Molecular Sciences, 20(20), 5053. https://doi.org/10.3390/ijms20205053