Specific Features for the Competent Binding of Substrates at the FMN Adenylyltransferase Site of FAD Synthase from Corynebacterium ammoniagenes



Abstract
1. Introduction
2. Results
2.1. Occurrence of P56, P58, and L98 Residues in Different Prokaryotic FADSs
2.2. Mutations at P56, P58 and L98 Produce Subtle Changes in the Environment of Aromatic Residues
2.3. P56, P58 and L98 Contribute to Maintain the FMNAT and FADpp Activities of CaFADS and to Modulate Its RFK Activity
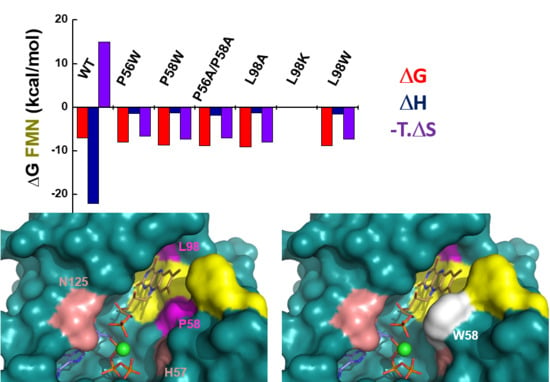
2.4. P56, P58 and L98 Contribute to the Binding of Flavinic Substrates/Products at the FMNAT Site of CaFADS
3. Discussion
4. Materials and Methods
4.1. Biological Material
4.2. Spectroscopic Analysis
4.3. Qualitative Detection of RFK, FMNAT and FADpp Activities
4.4. Quantitative Determination of Steady-State Kinetics Parameters for the RFK Activity
4.5. Isothermal Titration Calorimetry (ITC)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP | Adenosine 5′-triphosphate |
| CD | Circular dichroism |
| FAD | Flavin adenine dinucleotide, Riboflavin 5′-adenosine diphosphate |
| FADpp | FAD pyrophosphorylase |
| FADS | FAD synthase |
| FMN | Flavin mononucleotide, Riboflavin 5′-phosphate |
| FMNAT | ATP:FMN adenylyl transferase |
| HPLC | High performance chromatography |
| ITC | Isothermal titration calorimetry |
| MD | Molecular dynamics |
| NT | Nucleotidyl transferase |
| PIPES | 1,4-Piperazinediethanesulfonic acid |
| RF | Riboflavin |
| RFK | ATP:riboflavin kinase |
| TLC | Thin-layer chromatography |
| UV | Ultraviolet |
| WT | Wilde type |
References
- Serrano, A.; Ferreira, P.; Martínez-Júlvez, M.; Medina, M. The prokaryotic FAD synthetase family: A potential drug target. Curr. Pharm. Des. 2013, 19, 2637–2648. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, S.; Zhou, Q.; Mseeh, F.; Grishin, N.V.; Osterman, A.L.; Zhang, H. Crystal structure of human riboflavin kinase reveals a beta barrel fold and a novel active site arch. Structure 2003, 11, 265–273. [Google Scholar] [CrossRef]
- Herguedas, B.; Martinez-Julvez, M.; Frago, S.; Medina, M.; Hermoso, J.A. Oligomeric state in the crystal structure of modular FAD synthetase provides insights into its sequential catalysis in prokaryotes. J. Mol. Biol. 2010, 400, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Herguedas, B.; Lans, I.; Sebastián, M.; Hermoso, J.A.; Martínez-Júlvez, M.; Medina, M. Structural insights into the synthesis of FMN in prokaryotic organisms. Acta Cryst. D Biol. Cryst. 2015, 71, 2526–2542. [Google Scholar] [CrossRef]
- Bauer, S.; Kemter, K.; Bacher, A.; Huber, R.; Fischer, M.; Steinbacher, S. Crystal structure of Schizosaccharomyces pombe riboflavin kinase reveals a novel ATP and riboflavin-binding fold. J. Mol. Biol. 2003, 326, 1463–1473. [Google Scholar] [CrossRef]
- Barile, M.; Brizio, C.; Valenti, D.; De Virgilio, C.; Passarella, S. The riboflavin/FAD cycle in rat liver mitochondria. Eur. J. Biochem. 2000, 267, 4888–4900. [Google Scholar] [CrossRef]
- Barile, M.; Giancaspero, T.A.; Brizio, C.; Panebianco, C.; Indiveri, C.; Galluccio, M.; Vergani, L.; Eberini, I.; Gianazza, E. Biosynthesis of flavin cofactors in man: Implications in health and disease. Curr. Pharm. Des. 2013, 19, 2649–2675. [Google Scholar] [CrossRef]
- Barile, M.; Giancaspero, T.A.; Leone, P.; Galluccio, M.; Indiveri, C. Riboflavin transport and metabolism in humans. J. Inherit. Metab. Dis. 2016, 39, 545–557. [Google Scholar] [CrossRef]
- Leulliot, N.; Blondeau, K.; Keller, J.; Ulryck, N.; Quevillon-Cheruel, S.; van Tilbeurgh, H. Crystal structure of yeast FAD synthetase (Fad1) in complex with FAD. J. Mol. Biol. 2010, 398, 641–646. [Google Scholar] [CrossRef]
- Sebastián, M.; Anoz-Carbonell, E.; Gracia, B.; Cossio, P.; Aínsa, J.A.; Lans, I.; Medina, M. Discovery of antimicrobial compounds targeting bacterial type FAD synthetases. J. Enzyme Inhib. Med. Chem. 2018, 33, 241–254. [Google Scholar] [CrossRef]
- Efimov, I.; Kuusk, V.; Zhang, X.; McIntire, W.S. Proposed steady-state kinetic mechanism for Corynebacterium ammoniagenes FAD synthetase produced by Escherichia coli. Biochemistry 1998, 37, 9716–9723. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.; Frago, S.; Velázquez-Campoy, A.; Medina, M. Role of key residues at the flavin mononucleotide (FMN):adenylyltransferase catalytic site of the bifunctional riboflavin kinase/flavin adenine dinucleotide (FAD) Synthetase from Corynebacterium ammoniagenes. Int. J. Mol. Sci. 2012, 13, 14492–14517. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.; Sebastian, M.; Arilla-Luna, S.; Baquedano, S.; Pallares, M.C.; Lostao, A.; Herguedas, B.; Velazquez-Campoy, A.; Martinez-Julvez, M.; Medina, M. Quaternary organization in a bifunctional prokaryotic FAD synthetase: Involvement of an arginine at its adenylyltransferase module on the riboflavin kinase activity. Biochim. Biophys. Acta 2015. [Google Scholar] [CrossRef] [PubMed]
- Marcuello, C.; Arilla-Luna, S.; Medina, M.; Lostao, A. Detection of a quaternary organization into dimer of trimers of Corynebacterium ammoniagenes FAD synthetase at the single-molecule level and at the in cell level. Biochim. Biophys. Acta 2013, 1834, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.; Sebastián, M.; Arilla-Luna, S.; Baquedano, S.; Herguedas, B.; Velázquez-Campoy, A.; Martínez-Júlvez, M.; Medina, M. The trimer interface in the quaternary structure of the bifunctional prokaryotic FAD synthetase from Corynebacterium ammoniagenes. Sci. Rep. 2017, 7, 404. [Google Scholar] [CrossRef] [PubMed]
- Sebastián, M.; Arilla-Luna, S.; Bellalou, J.; Yruela, I.; Medina, M. The Biosynthesis of Flavin Cofactors in Listeria monocytogenes. J. Mol. Biol. 2019, 431, 2762–2776. [Google Scholar] [CrossRef]
- Sebastián, M.; Lira-Navarrete, E.; Serrano, A.; Marcuello, C.; Velázquez-Campoy, A.; Lostao, A.; Hurtado-Guerrero, R.; Medina, M.; Martínez-Júlvez, M. The FAD synthetase from the human pathogen Streptococcus pneumoniae: A bifunctional enzyme exhibiting activity-dependent redox requirements. Sci. Rep. 2017, 7, 7609. [Google Scholar] [CrossRef]
- Sebastián, M.; Serrano, A.; Velázquez-Campoy, A.; Medina, M. Kinetics and thermodynamics of the protein-ligand interactions in the riboflavin kinase activity of the FAD synthetase from Corynebacterium ammoniagenes. Sci. Rep. 2017, 7, 7281. [Google Scholar] [CrossRef]
- Matern, A.; Pedrolli, D.; Großhennig, S.; Johansson, J.; Mack, M. Uptake and Metabolism of Antibiotics Roseoflavin and 8-Demethyl-8-Aminoriboflavin in Riboflavin-Auxotrophic Listeria monocytogenes. J. Bacteriol. 2016, 198, 3233–3243. [Google Scholar] [CrossRef]
- Lans, I.; Seco, J.; Serrano, A.; Burbano, R.; Cossio, P.; Daza, M.C.; Medina, M. The Dimer-of-Trimers Assembly Prevents Catalysis at the Transferase Site of Prokaryotic FAD Synthase. Biophys. J. 2018, 115, 988–995. [Google Scholar] [CrossRef]
- Frago, S.; Martínez-Júlvez, M.; Serrano, A.; Medina, M. Structural analysis of FAD synthetase from Corynebacterium ammoniagenes. BMC Microbiol. 2008, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Krupa, A.; Sandhya, K.; Srinivasan, N.; Jonnalagadda, S. A conserved domain in prokaryotic bifunctional FAD synthetases can potentially catalyze nucleotide transfer. Trends Biochem. Sci. 2003, 28, 9–12. [Google Scholar] [CrossRef]
- Huerta, C.; Borek, D.; Machius, M.; Grishin, N.V.; Zhang, H. Structure and mechanism of a eukaryotic FMN adenylyltransferase. J. Mol. Biol. 2009, 389, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Frago, S.; Velázquez-Campoy, A.; Medina, M. The puzzle of ligand binding to Corynebacterium ammoniagenes FAD synthetase. J. Biol. Chem. 2009, 284, 6610–6619. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.; Frago, S.; Herguedas, B.; Martinez-Julvez, M.; Velazquez-Campoy, A.; Medina, M. Key residues at the riboflavin kinase catalytic site of the bifunctional riboflavin kinase/FMN adenylyltransferase from Corynebacterium ammoniagenes. Cell Biochem. Biophys. 2013, 65, 57–68. [Google Scholar] [CrossRef]
- Olland, A.M.; Underwood, K.W.; Czerwinski, R.M.; Lo, M.C.; Aulabaugh, A.; Bard, J.; Stahl, M.L.; Somers, W.S.; Sullivan, F.X.; Chopra, R. Identification, characterization, and crystal structure of Bacillus subtilis nicotinic acid mononucleotide adenylyltransferase. J. Biol. Chem. 2002, 277, 3698–3707. [Google Scholar] [CrossRef]
- Gill, S.C.; von Hippel, P.H. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989, 182, 319–326. [Google Scholar] [CrossRef]
- Leskovac, V. Comprehensive Enzyme Kinetics; Kluwer Adacemic/Plenum Publishers: New York, NY, USA, 2003. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CaFADS | kcat2,3 (min−1) | KmRF 2,3 (µM) | Ki2,3 (µM) | kcat/KmRF (min−1·µM−1) | kcat3,4 (min−1) | KmATP 4 (µM) | kcat/KmATP 3,4 (min−1·µM−1) |
|---|---|---|---|---|---|---|---|
| WT 1 | 408 ± 200 | 12 ± 3 | 4.9 ± 3.9 | 35 ± 21 | 155 ± 5 | 28 ± 4 | 5.5 ± 0.8 |
| P56W | 45 ± 5 | 0.4 ± 0.1 | 6.5 ± 1.9 | 125 ± 33 | 54 ± 2 | 47 ± 6 | 1.2 ± 0.2 |
| P58W | 98 ± 19 | 1.3 ± 0.7 | 39 ± 21 | 78 ± 43 | 60 ± 2 | 52 ± 7 | 1.2 ± 0.2 |
| P56A/P58A | 78 ± 12 | 0.9 ± 0.4 | 54 ± 29 | 87 ± 45 | 67 ± 4 | 46 ± 9 | 1.4 ± 0.3 |
| L98A | 138 ± 26 | 1.5 ± 0.7 | 32 ± 15 | 94 ± 48 | 110 ± 3 | 30 ± 4 | 3.6 ± 0.5 |
| L98K | 248 ± 34 | 4.0 ± 1.1 | 41 ± 13 | 63 ± 19 | 84 ± 3 | 30 ± 5 | 2.8 ± 0.4 |
| L98W | 140 ± 21 | 1.6 ± 0.6 | 46 ± 20 | 85 ± 35 | 70 ± 4 | 39 ± 8 | 1.8 ± 0.4 |
| CaFADS | KdFMN (μM) | KdFAD (μM) | KdATP (μM) |
|---|---|---|---|
| 10 mM MgCl2 | + | + | - |
| WT | 7.1 ± 0.4 | 1.1 ± 0.1 | 48 ± 7 |
| P56W | 1.7 ± 0.1 | 8.3 ± 0.8 | 44 ± 5 |
| P58W | 0.50 ± 0.03 | 2.6 ± 0.2 | 16 ± 1 |
| P56A/P58A | 0.35 ± 0.02 | 1.9 ± 0.1 | 21 ± 3 |
| L98A | 0.23 ± 0.03 | n.d. | 7 ± 5 |
| L98K | n.d. | n.d. | 82 ± 21 |
| L98W | 0.42 ± 0.02 | 3.8 ± 0.1 | 47 ± 7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arilla-Luna, S.; Serrano, A.; Medina, M. Specific Features for the Competent Binding of Substrates at the FMN Adenylyltransferase Site of FAD Synthase from Corynebacterium ammoniagenes. Int. J. Mol. Sci. 2019, 20, 5083. https://doi.org/10.3390/ijms20205083
Arilla-Luna S, Serrano A, Medina M. Specific Features for the Competent Binding of Substrates at the FMN Adenylyltransferase Site of FAD Synthase from Corynebacterium ammoniagenes. International Journal of Molecular Sciences. 2019; 20(20):5083. https://doi.org/10.3390/ijms20205083
Chicago/Turabian StyleArilla-Luna, Sonia, Ana Serrano, and Milagros Medina. 2019. "Specific Features for the Competent Binding of Substrates at the FMN Adenylyltransferase Site of FAD Synthase from Corynebacterium ammoniagenes" International Journal of Molecular Sciences 20, no. 20: 5083. https://doi.org/10.3390/ijms20205083
APA StyleArilla-Luna, S., Serrano, A., & Medina, M. (2019). Specific Features for the Competent Binding of Substrates at the FMN Adenylyltransferase Site of FAD Synthase from Corynebacterium ammoniagenes. International Journal of Molecular Sciences, 20(20), 5083. https://doi.org/10.3390/ijms20205083