Abstract
Zn2+ deficiency in the human population is frequent in underdeveloped countries. Worldwide, approximatively 2 billion people consume Zn2+-deficient diets, accounting for 1–4% of deaths each year, mainly in infants with a compromised immune system. Depending on the severity of Zn2+ deficiency, clinical symptoms are associated with impaired wound healing, alopecia, diarrhea, poor growth, dysfunction of the immune and nervous system with congenital abnormalities and bleeding disorders. Poor nutritional Zn2+ status in patients with metastatic squamous cell carcinoma or with advanced non-Hodgkin lymphoma, was accompanied by cutaneous bleeding and platelet dysfunction. Forcing Zn2+ uptake in the gut using different nutritional supplementation of Zn2+ could ameliorate many of these pathological symptoms in humans. Feeding adult rodents with a low Zn2+ diet caused poor platelet aggregation and increased bleeding tendency, thereby attracting great scientific interest in investigating the role of Zn2+ in hemostasis. Storage protein metallothionein maintains or releases Zn2+ in the cytoplasm, and the dynamic change of this cytoplasmic Zn2+ pool is regulated by the redox status of the cell. An increase of labile Zn2+ pool can be toxic for the cells, and therefore cytoplasmic Zn2+ levels are tightly regulated by several Zn2+ transporters located on the cell surface and also on the intracellular membrane of Zn2+ storage organelles, such as secretory vesicles, endoplasmic reticulum or Golgi apparatus. Although Zn2+ is a critical cofactor for more than 2000 transcription factors and 300 enzymes, regulating cell differentiation, proliferation, and basic metabolic functions of the cells, the molecular mechanisms of Zn2+ transport and the physiological role of Zn2+ store in megakaryocyte and platelet function remain elusive. In this review, we summarize the contribution of extracellular or intracellular Zn2+ to megakaryocyte and platelet function and discuss the consequences of dysregulated Zn2+ homeostasis in platelet-related diseases by focusing on thrombosis, ischemic stroke and storage pool diseases.
1. Introduction
Zinc (Zn2+) is the second most important trace metal in the body. Zn2+ enters into the organism through dietary consummation and plays a critical role in many biological processes including development, proliferation, differentiation, cell metabolism, insulin synthesis and secretion, immune function, regulation of DNA synthesis and genomic stability [1,2]. In humans, daily Zn2+ ingestion can point to 14–30 mg/kg. The body absorbs 20–40% of Zn2+ in the gut, mainly in the jejunum through the enterocytes, and residual Zn2+ is excreted. Zn2+ is mainly stored in skeletal muscle (60%) and bone (30%), and only a small fraction of body Zn2+ is circulating in the blood at a concentration of 10–20 µM [3]. The major part of blood plasma-resident Zn2+ is covalently linked to albumin (75–85%) and β2-macroglobulin (10–20%) and a minor part (2%) can bind to amino acids, organic anions, histidine, cysteine and citrate. Therefore, only a small fraction of Zn2+ exists as free labile form (0.1–2 µM) which can be taken up by blood cells, also including endothelial cells and platelets [4,5,6]. Most of the cells can uptake Zn2+ through receptor-mediated endocytosis or other Zn2+ transport mechanisms [7,8].
Zn2+ has an important catalytic function by stabilizing negative charges in biochemical reactions in metabolic enzymes. Cellular Zn2+ uptake can increase the activity of several enzymes, such as mitogen-activated protein kinases (MAPKs), caspases and metalloproteinases (MMPs), [1,9,10,11,12]. Many of secretory cells, such as pancreatic β-cells, neutrophils and platelets, can uptake, store and release a significant amount of Zn2+, regulating diverse Zn2+-dependent intracellular and extracellular pathways [3,8,13,14,15,16,17,18,19]. Zn2+ is also responsible for maintaining the protein structure in zinc-finger transcription factors and LIM domain proteins, thus Zn2+ can be coordinated by oxygen, sulfur and nitrogen atoms of the polypeptide chain [20].
2. Zn2+ Homeostasis in Megakaryocytes and Platelets
The amount of metabolically active Zn2+ is very limited in the cell due to its cytotoxicity, therefore the majority of Zn2+ is associated to cytoplasmic proteins or transported and stored in intracellular organelles [1,3]. In mammalian cells, Zn2+ is mainly located in the nucleus, cytoplasm, endoplasmic reticulum, Golgi apparatus, vesicles and secretory granules. In the cytoplasm, Zn2+ is sequestered by metallothioneins (MTs), which are small cysteine-rich proteins, complexing up to seven Zn2+ ions [3]. Depending on the type and activated state of cells, approximatively 5–15% of cytoplasmic Zn2+ can bind to MTs [3]. When the sulfur group of MTs is oxidized, Zn2+ cannot bind MTs, thereby depleting MT-dependent Zn2+ store in the cytoplasm. Conversely, enhanced Zn2+ uptake induces MT gene expression by metal transcription factor 1 (MTF-1), thereby increasing the Zn2+ storage capacity of MTs inside the cell [3]. In human platelets, several MTs were detected and the amount of MT is approximatively 40 µg/1010 cells [21], and their storage function is regulated by reactive oxygen species (ROS), [22]. Incubation of platelets with MTs, decreasing the metabolically active labile pool of Zn2+, or injection of MTs into mice inhibits aggregation responses to collagen, probably due to the reduced phospholipase C (PLC)-mediated Ca2+ store mobilization and thromboxane A2 (TxA2) synthesis [23]. In addition, this led to the increased production of cyclic guanosine monophosphate (cGMP), thereby inducing antithrombotic effects in mesenteric venules [23]. Similarly, chelation of free ionic pool of Zn2+ with N,N,N′,N′-tetrakis 2-pyridinylmethyl-1,2-ethanediamine (TPEN) in the cytoplasm inhibits platelet aggregation and tyrosine phosphorylation [24], suggesting an important role of Zn2+ as a second messenger in platelet signaling.
The phospholipid membrane of eukaryotic cells is impermeable for Zn2+, therefore transport and storage of Zn2+ is ensured by Zn2+ transporters [2,8]. Resting platelets can uptake Zn2+ from the blood plasma and store it, indicating the existence of active Zn2+ transport and storage mechanisms [15,25,26]. Zn2+ concentration in blood serum was found to be higher than in blood plasma [27,28], suggesting that activated platelets could release a significant amount of Zn2+ during blood clotting. Later studies showed that incubation of platelets with extracellular Zn2+ increased the cytoplasmic and granular Zn2+ concentrations [29]. Hence, approximatively 50–80 pieces of α-granule fulfill the body of a single platelet [30,31,32], which possibly stores a significant amount of Zn2+. Therefore, it was proposed that the largest Zn2+ store could be located in platelet α-granules [26,29]. In addition, Zn2+ has a high affinity to bind fibrinogen, albumin, histidine-rich glycoprotein (HRG) and factor XIII, which are also accumulated in α-granules of platelets, suggesting that Zn2+ store exists as a protein-bound form in this type of granules [33]. Using inductively coupled plasma mass spectrometry (ICP-MS), Gorodetsky et al., reported that approximately 40% of Zn2+ is stored in granules, while 60% of Zn2+ is stored in other intracellular organelles and also in the cytoplasm [33]. Recently, the labile Zn2+ pool was visualized by our group and quantified in human and mouse platelets using a Zn2+ specific fluorescence dye FluoZin3 [26]. The specificity of the dye was characterized with Zn2+ supplementation or chelation of Zn2+ with ZnCl2 or TPEN, respectively. Using confocal microscopy, FluoZin3 staining showed several stained foci in the platelet cytoplasm, which became markedly reduced when platelets were spread on a fibrinogen-coated surface [26]. In addition, the rapid loss of intracellular FluoZin3 staining measured by flow cytometry, correlated to Zn2+ efflux in activated platelets [26]. Although α-granules are indicated as a major Zn2+ store in platelets [26,29], granule-located Zn2+ transporters and their functions in platelet signaling have not been investigated.
In mammalian cells, Zn2+ transporters are encoded by 24 different solute-linked carrier genes (Slc30a/Slc39a) and grouped into two protein families called ZIP and ZnT [16,34]. The Slc39a family was named ZIP (Zrt-, Irt-related protein), after the first two known members Irt1 of Arabidopsis thaliana and Zrt1 of Saccharomyces cerevisiae. So far, the exact structure of mammalian ZIP proteins has not been established, but 14 members of human and mouse ZIP isoforms and their genes have been isolated and characterized [16,17,35]. ZIP family members mediate Zn2+ influx from the extracellular place into cells, or from the intracellular Zn2+ store into the cytoplasm, thereby increasing cytoplasmic Zn2+ concentrations. Some ZIP isoforms are not strictly specific for Zn2+, and other metals such as iron (Fe), copper (Cu), cadmium (Cd), or manganese (Mn) could be also transported, depending on the pathophysiological status of the cell [2,8]. Recently, we showed mRNA expression levels of ZIP family members using in vitro-grown primary mouse MKs and quantitative real-time polymerase chain reactions (qRT-PCRs). Several isoforms of ZIP were predominantly expressed, including ZIP1, ZIP4, ZIP6, ZIP7, ZIP9 and ZIP10, while other ZIP isoforms showed limited mRNA expression, and ZIP2 was not detected [26]. Studies in mouse models showed the relevance of ZIP transporters in the regulation of systemic or cellular Zn2+ homeostasis [16,17,36], but the function of these transporters have been investigated neither in MKs nor in platelets. The members of Slc30a gene family, also called ZnT, are selective Zn2+ transporters located in the cell surface and on the membrane of intracellular organelles. So far 10 isoforms of ZnT have been identified in mammalians. ZnTs classically form homodimers and reduce Zn2+ levels using a H+/Zn2+ antiport mechanism. ZnT isoforms regulate Zn2+ efflux from the cytosol to the extracellular place or into intracellular organelles decreasing cytoplasmic Zn2+ concentration in mammalian cells [37]. Our studies showed that among ZnT isoforms, ZnT1, ZnT5, ZnT6, ZnT7 and ZnT9 are moderately or highly expressed in mouse MKs, while other ZnTs were weakly or not expressed [26].
Hematopoietic Zn2+-finger gene (Hzf) is expressed mostly in megakaryocyte lineage [38]. Interestingly, modification of the zinc-finger domain of Hzf leads to impaired platelet formation and α-granule biogenesis [38]. Using FluoZin3, we have proposed that the intracellular Zn2+ store is located in α-granules of MKs [26], and therefore we speculate that Hzf function may be affected by the impaired granular Zn2+ store (Figure 1). In addition, Zn2+ also regulates the function of transcription factors GATA, which plays an essential role in megakaryopoiesis [39]. Although these studies indicate on potential Zn2+ homeostasis in MKs is very limited, the actual Zn2+ status regulates megakaryopoiesis and platelet biogenesis in mammalians needs further investigation.
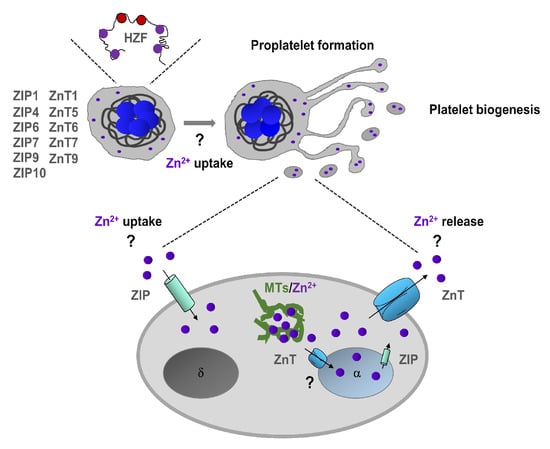
Figure 1.
Zn2+ homeostasis in megakaryopoiesis and platelet biogenesis. Megakaryocytes express Zn2+-finger gene (Hzf), which regulates platelet formation and α-granule biogenesis. In addition, megakaryocytes express several ZIP (Zrt-, Irt-related protein)/Znt transporters which can regulate uptake and storage in α-granules. Extracellular and granule-resident Zn2+ may also regulate proplatelet formation and platelet biogenesis. In platelets, intracellular Zn2+ is mainly stored in α-granules, but it can also be sequestered by metallothioneins (MTs) in the cytoplasm. ZIP/Znt transporters are involved in Zn2+ homeostasis by regulating either efflux or influx of Zn2+ or both.
3. Zn2+-Dependent Regulation of Platelet Function
The first link between Zn2+ and platelet function derives from the studies on rats submitted to experimental Zn2+ deficiency [40,41,42]. In these studies, Zn2+ deficiency resulted in prolonged bleeding time [40,42] and reduced platelet aggregation [41]. In other studies, using a similar experimental model, Zn2+ deficiency could impair platelet reactivity to agonists, such as adenosine diphosphate (ADP) and thrombin [43,44]. In addition, Zn2+ deficiency in rats is associated with impaired Ca2+ uptake of platelets, which can be normalized by in vitro incubation of blood with glutathione [45,46].
Bleeding abnormality associated with Zn2+ deficiency was also observed in human patients. In 1982, Gordon et al., showed that human volunteers submitted to experimental Zn2+ deficiency displayed defective platelet aggregation response to ADP and arachidonate, and Zn2+ supplementation could normalize these defects [47]. Later, ecchymosis, abnormally prolonged bleeding and platelet aggregation responses were described in two cancer patients, one with squamous cell carcinoma and another with non-Hodgkin’s lymphoma, with concurrent nutritional Zn2+ deficiency [48].
Recently, it has been proposed that intracellular chelation of Zn2+ in platelets can inhibit tyrosine phosphorylation cascades, thereby attenuating platelet reactivity and aggregation responses in vitro [24]. On the other hand, it has been shown that increased levels of dietary Zn2+ in rats correlates with enhanced platelet responses to suboptimal doses of collagen, ADP, thrombin and epinephrine, suggesting the link between actual Zn2+ status and platelet reactivity [49]. Zn2+-mediated platelet aggregation is enhanced [49], because fibrinogen binding to the integrin αIIbβ3 was enhanced and consequently increasing the binding capacity of platelets to other blood cells and also to the fibrin clot.
Receptor-mediated activation of phospholipase C (PLC) is essential to increase the activity of diacylglycerol (DAG)-sensitive Ca2+ channel transient receptor potential cation channel subfamily C member 6 (TRPC6), and to induce Ca2+ store depletion through inositol 1,4,5-trisphosphate (IP3)-mediated activation of IP3-receptor (IP3R), which further modulates store-operated Ca2+ entry (SOCE) by activation of the stromal interaction molecule 1 (STIM1)–Orai1 complex in platelets [50]. Interestingly, Zn2+ status of the cells is connected to Ca2+ homeostasis and regulates the actual cytoplasmic Ca2+ levels as well, thereby indirectly influencing platelet reactivity. Indeed, when rats were fed with a low Zn2+ diet, increased basal Ca2+ concentration at resting stage was observed, but surprisingly, Ca2+ influx was significantly reduced without any change on Ca2+ store depletion, indicating that platelet Zn2+ deficiency impairs Ca2+ channel function, independently of SOCE mechanism [46]. However, in sharp contrast, 50% reduction of IP3 production was observed in platelets isolated from Zn2+-deficient rats, when Zn2+ concentration was at least 70% lower in the blood plasma than controls [51], suggesting that depending on the severity of Zn2+ deficiency, Zn2+ could also influence Ca2+ store depletion and SOCE. However, the molecular mechanisms of Zn2+-dependent SOCE and the physiological crosstalk between Zn2+-dependent signaling and Ca2+ channels has not been established. Interestingly, protein kinase C (PKC), the downstream effector of PLC, directly binds Zn2+ [52]. Subcellular localization of PKC is modified by increased levels of Zn2+ in the cytoplasm; accumulation of activated PKC was detected close to the plasma membrane of platelets. On the other hand, a PKC antagonist GF109203X could strongly inhibit Zn2+-mediated platelet aggregation responses [24], indicating a complex role of PKC in Zn2+ and Ca2+-mediated signaling pathways.
Zn2+ binds to sulfhydryl groups and protects several signaling molecules and channels from oxidation, thereby regulating ROS-mediated signaling of the cells [53]. Reduced intracellular Zn2+ levels can change the biochemical properties of signaling molecules and the activity of cation channels by increasing oxidation of free sulfhydryl groups, thereby modifying the structure of these proteins. Interestingly, TRPC6 is directly activated by ROS [54], while transient receptor potential melastatin channel 2 (TRPM2) is indirectly activated, which further enhances the channel activity in MKs [55]. Although there are other TRPM channels, transient receptor potential melastatin channel 7 (TRPM7) and the Orai1 channel are also redox-sensitive, and their channel activity is inhibited by ROS [56,57]. In addition, it has been shown that TRPC6 and TRPM7 can transport both Zn2+ and Ca2+ into the cells [58,59,60], but their functions in Zn2+ homeostasis have not been investigated in MKs and platelets. Therefore, future studies are necessary to elucidate the function of these channels in the regulation of Zn2+ and Ca2+ homeostasis, which are probably connected by PKC and ROS in MKs and platelets.
4. Zn2+-Dependent Hemostasis and Fibrin Clot Formation
Platelets cannot synthesize fibrinogen, it is produced and released by liver hepatocytes into the blood plasma and taken up and stored in α-granules of circulating platelets [61]. Fibrinogen is composed of two pairs of three disulfide-linked polypeptide chains [62,63]. These chains are symmetrically folded and composed of a central E-domain with two lateral D-globular domains. Thrombin activation is an essential step of the hemostatic process, regulating the coagulant activity, and converting the soluble fibrinogen to insoluble fibrin fibers thereby stabilizing the growing blood clot [64]. Fibrin assembly is initiated after thrombin-induced removal of fibrinopeptide A from plasma fibrinogen. Several steps of thrombin proteolysis on fibrinogen create an active fibrin monomer, which is polymerized subsequently to fibrin protofibrils. Lateral assembly and covalent linkage of hundreds of protofibrils could form the fibrin fiber [62,65]. Irregular assembly of fibrin fibers finally forms the fibrin network within the blood clot. The fibrin clot is composed of different diameters and lengths of fibrin fibers. Depending on the ratio between thin and thick fibers, which is regulated by thrombin, Zn2+, Ca2+ and other factors, fibrin clot structure and mechanical stability can be different [62,66]. When fibrin clot is mainly composed of thin and densely packed fibers, fibrin clot is less porous and stiffer, thereby increasing their potential for rupture and embolism. When thick fibers are dominant within the fibrin clot, it is less stiff, more porous; therefore, a fibrin clot can change easily the size and form, depending on the actual shear stress [62]. Several studies showed the multiple effects of Zn2+ and Ca2+ on fibrin clot formation because fibrinogen contains several binding sites for both cations [66,67]. Binding of fibrinogen to Ca2+ enhances the kinetics of fibrin clot formation and also modifies the structure of the fibrin network; more thick fibers were polymerized [68]. Although it is well established that Ca2+ plays an important role in different steps of fibrin clot formation, Ca2+-dependent mechanism behind this process has been published controversial [69,70].
Initial studies showed that Zn2+ can inhibit amidolytic activity of thrombin [71,72] and diminishes thrombin-mediated fibrinopeptide A release [73]. Studies by Marx also demonstrated that Zn2+ can bind fibrinogen and fibrin with Kd values ranging from 8 to 18 µM and with a ratio of six Zn2+ atoms per fibrin/fibrinogen molecule [66]. Interestingly, binding sites of Zn2+ are mapped in the lateral D-globular domains, but not in the central E-domain and sites are distinct from those of Ca2+ [74,75,76], indicating an alternative regulatory mechanism of fibrin formation. Similar to Ca2+, Zn2+ also accelerates the rate of thrombin-induced fibrin clot formation, whereas this effect was not observed in the presence of magnesium (Mg2+) or manganese (Mn2+) [66,67], suggesting that modulation of thrombin-induced fibrin formation is specific to Zn2+ and Ca2+. It has been shown that the lateral association of fibrin monomers to protofibrils is enhanced by Zn2+ to a greater extent than Ca2+, and Zn2+ induces a fibrin clot that has reduced fiber stiffness, compared to Ca2+-induced fibrin clot [66,75,77,78]. Interestingly, on the one hand Zn2+ attenuates the activity of thrombin but on the other hand can still enhance fibrin clot formation. This may indicate that Zn2+ enhances clot formation likely through a mechanism potentiating tridimensional assembly of fibrin network.
Recently, we demonstrated that lack of α-granule content strongly influences the structure of the fibrin network in vitro [26]. Releasate of wild-type and Nbeal2-/- platelets were observed by scanning electron microscopy (SEM) and ultrastructure of fibrin clot was obviously different and fibrin structure was partially modified in the presence of extracellular Zn2+, indicating an important role of α-granule-resident Zn2+ and fibrinogen release during fibrin clot formation [26].
Fibrin formation is enhanced by the release of extrinsic factors from the damaged vessel wall or intrinsic factors from blood cells and platelets [62,68]. The intrinsic pathway of coagulation is initiated by activation of factor XII which further modulates the kininogen/kallikrein system, referred to as the plasma contact system. Polyphosphate accumulation on the surface (also called polyanionic surface) of endothelial cells and platelets triggers the activation of factor XII which further activates its substrate factor XI, thereby activating fibrin formation [79,80]. On the other hand, factor XII also induces the pro-inflammatory cascade through the liberation of the inflammatory peptide hormone bradykinin by kallikrein-mediated cleavage of kininogen [80,81]. Accumulation of bradykinin on the endothelial cell surface increases vascular permeability, promoting adhesion of inflammatory immune cells [82,83,84]. Zn2+ is also known to regulate several steps of the coagulation cascade to activate plasma factors and the contact system [25,85]. Zn2+ directly binds to factor XII, inducing a conformational change, thereby improving its susceptibility for enzymatic activation [86]. Moreover in the presence of Zn2+, factor XI binds to glycocalicin (the soluble extracellular region of GPIbα), thereby affecting the thrombin generation pathway [87]. The activated form of factor XI enhances factor IX activity and the formation of the tenase complex, thereby further promoting thrombin activation and fibrin formation, [25,83]. Consequently, Zn2+ deficiency could strongly impair the coagulation cascade and fibrin formation leading to prolonged bleeding times, as already demonstrated in rodent models and human patients [15]. Together these studies suggest that Zn2+ may contribute to hemostasis through several mechanisms modulating platelet aggregation, coagulation and fibrin network formation (Figure 2).
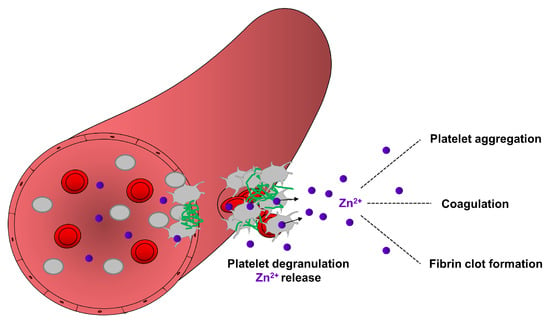
Figure 2.
Contribution of platelet-resident Zn2+ in the regulation of hemostasis. During hemostasis, a process which stops bleeding, platelets accumulate in the site of injury and became activated. Activated platelets release Zn2+ from their granules, which may enhance the aggregation of circulating platelets, modulate the activity of proteins of coagulation and fibrinolytic pathways. Zn2+ can bind to fibrinogen and fibrin and modulate the ultrastructure and mechanical strength of fibrin, thereby supporting fibrin clot formation during hemostasis.
5. Zn2+ in Thrombosis
Platelet accumulation and activation at the sites of vascular injury lead to the release of granular located Zn2+ from platelets to the microenvironment of the vascular network [25]. Several indirect experimental pieces of evidence support the role of platelet Zn2+ in thrombosis, but the molecular mechanisms of how platelet-resident Zn2+ may modulate this process has not been established. It is possible that activated platelets can increase the local concentration of Zn2+ during thrombosis, enhancing fibrin deposition and subsequent recruitment of additional circulating platelets, thereby potentiating thrombus growth. Zn2+ released by platelets may act as a co-factor for initiating the assembly of the contact system on the polyanionic surfaces and also on the procoagulant surface of platelets and endothelial cells during thrombosis [15,25,88,89], (Figure 3).
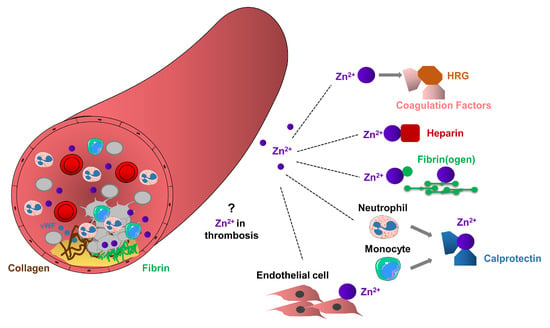
Figure 3.
Potential role of Zn2+ in arterial thrombosis. Arterial thrombosis occurs in atherosclerotic plaque rupture, as a result of clot formation, leading to platelet aggregation, thrombus formation and vessel occlusion. Platelets through glycoproteins, interact with collagen or von Willebrand Factor (vWF), change their shape and adhere to the site of injury. Adherent platelets become activated and release their granule content leading to the recruitment of additional circulating platelets. Extracellular Zn2+ or Zn2+ released from platelet granules may enhance thrombosis by mediating interactions between HRG and coagulation factors, Zn2+ sequesters heparin, or interact with fibrinogen and fibrin, thereby enhancing fibrin deposition and stimulating recruitment of circulating platelets and subsequent thrombus growth. Calprotectin released from neutrophils and monocytes may interact with Zn2+ and consequently enhance prothrombotic events. Procoagulant endothelial cells may provide favorable surface to the accumulation of Zn2+, thereby enhancing thrombosis.
HRG is important regulator in the coagulation cascade since they can directly bind Zn2+, heparin, plasminogen, fibrinogen and other factors in the blood plasma [90]. In addition, interactions between HRG and many of these proteins are stimulated by Zn2+ [25]. High levels of HRG are associated with the clinical symptoms of various cardiovascular disorders, while congenital deficiency of HRG is connected to thrombophilia in humans [91,92]. Interestingly, free fatty acids (FFAs) allosterically disrupt Zn2+ binding to albumin, indicating that increased FFA content could modulate the dissociation of Zn2+ from albumin, thereby increasing the concentrations of metabolically active Zn2+ pool in the blood [93,94]. Consequently, high levels of FFA in patients with cancer, diabetes or obesity may shift the Zn2+ pool from albumin to HRG, thereby increasing the risk of thrombosis [93]. In addition, Zn2+ may regulate the adhesion of immune cells through the ability to bind the plasma HRG [90].
Calprotectin (a complex of S100A8 and S100A9) is one of the major Ca2+ and Zn2+ binding proteins. Calprotectin can sequester Zn2+, reducing the metabolically active labile Zn2+ pool in the blood, thereby limiting Zn2+ uptake by microbes, as a process termed nutritional immunity [95]. Under inflammatory conditions, calprotectin is released from activated neutrophils and monocytes to the bloodstream [95]. Therefore, the permanent increase of plasma concentrations of calprotectin can induce hyperzincaemia in human patients, characterized by recurrent infections, hepatosplenomegaly, anemia and chronic systemic inflammation, and in addition, presenting dermal ulcers, cutaneous inflammation and spontaneous hematomas [96]. Moreover, in patients with coronary artery diseases, high levels of calprotectin were associated with a reduced effect of aspirin, as indicated by increased platelet aggregation [97,98]. Although platelets can store and release calprotectin [98,99], the function of platelet-resident calprotectin in hyperzincaemia induced by chronic inflammation has not been investigated.
6. Zn2+ in Ischemic Stroke
Aberrant Zn2+ accumulation in the brain was observed in various neurological disorders, including traumatic brain injury, stroke and seizure [100,101]. Ischemic stroke is known as thrombo-inflammatory diseases involving the recruitment of platelets and immune cells to the site of ischemic vascular injury, damaging the permeability of the blood-brain barrier (BBB), thereby triggering neuronal cell death [50]. BBB and the blood-cerebrospinal fluid barriers maintain Zn2+ transport in the brain. In animal models of transient global and forebrain ischemia, accumulation of Zn2+ was observed in neuronal tissue at a late step of ischemic insult, before the onset of cellular death [102]. After 3 h of mild transient focal ischemia, Zn2+ accumulation in cortical neurons was also obvious, thus accelerating the development of cerebral infarction [102]. In addition, aberrant excess of Zn2+ into the synaptic cleft of ischemic neurons was found to induce toxicity, therefore Zn2+ can be an independent risk factor for ischemic stroke [102]. Consequently, administration of the Zn2+ chelator, di-calcium ethylene diamine tetra-acetic acid (Ca2EDTA) in rats subjected to experimental ischemia resulted in a neuroprotective effect. Ca2EDTA could inhibit neuronal damage by reducing the brain infarct size, improves neurological function and prevents apoptosis in ischemic neurons [102]. Furthermore, a selective Zn2+ and Ca2+ chelator DP-b99 also has neuroprotective properties in animal models and also in patients with acute ischemic stroke [103,104,105]. It has also been shown that copper/zinc superoxide dismutase could attenuate neuronal death and hippocampal injury after transient focal and global ischemia [106,107]. In rats, systemic administration of Zn2+ protects hippocampus from neuronal damage during the reperfusion phase of transient cerebral ischemia [108]. Patients with ischemic stroke frequently have a lower dietary Zn2+ intake (50% of the recommended amount) during hospitalization, and after normalization of Zn2+ intake, patients have better recovery of neurological deficits [109]. These conflicting results suggest that Zn2+ has multiple roles in the early and late phases of stroke development, and the observed positive or negative effects described here are strongly dependent on the in vivo experimental conditions.
In neurons, 10% of total Zn2+ content is metabolically active Zn2+ which is mainly located in presynaptic vesicles of glutamatergic nerve terminals and acts as a second messenger on the postsynaptic neuron during degranulation [110]. Following ischemic brain insult, Zn2+ is released together with glutamate into the synaptic cleft, and depending on the activating stimulus, Zn2+ concentration can be increased up to 100 µM [111]. In pathological conditions, glutamate-induced Ca2+ overload is a major cause of neuronal death, but besides this process, abnormal accumulation of Zn2+ can also act on the postsynaptic membrane in a synergistic mode with Ca2+. Indeed, N-methyl-D-aspartate receptors (NMDARs), voltage-sensitive Ca2+ channels (VSCCs), or a cation-permeable form of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors on the postsynaptic membrane can regulate both Zn2+ and Ca2+ uptake mechanisms [112]. Consequently, pharmacological inhibition of glutamate receptor 2 (GluR2)-lacking AMPA receptor reduces the Zn2+ level in the cytoplasm and, therefore, neurons become protective in the ischemic brain [113].
Neuroprotective strategies of ischemic stroke were strongly focused on the blockade of aberrant Ca2+ influx-mediated by the intracellular Ca2+ store and SOCE [50,114,115]. Inhibition of neuronal SOCE in mouse models of a stroke, regulated by stromal interaction molecule (STIM) 2 and ORAI2, was proposed to be an alternative route to avoid Ca2+ overload in ischemic neurons [116]. Interestingly, glutamate-induced Ca2+ overload leads to acidification of the cytoplasm, which modulates the MT-resident Zn2+ store, thereby further increasing the concentration of the labile Zn2+ pool, inducing cytotoxicity of ischemic neurons [117]. Consequently, blockade of SOCE in ischemic neurons may inhibit both the Ca2+ overload and aberrant Zn2+ response from the intracellular Zn2+ store regulated by MTs.
Beside Ca2+ and Mg2+, TRPM7 channel can also transport Zn2+ and regulate systemic and cellular Zn2+ homeostasis in mice [118]. Several in vivo studies demonstrated that TRPM7 function contributes to ischemic stroke [119], although the involvement of its channel and kinase activity in the regulation of Zn2+ homeostasis under ischemic conditions is still under investigation. Recently, we demonstrated that the kinase activity of TRPM7 is a key modulator of SOCE in platelets, and this signaling pathway was confirmed in other cell types, suggesting an interplay between the kinase domain of TRPM7 and STIM1 [120]. Consequently, deletion of TRPM7 kinase activity in mice significantly reduces SOCE in platelets, and protected mice from ischemic brain insults, indicating that the increased kinase function, rather than its channel function of TRPM7, plays an important role in ischemic stroke [120]. Furthermore, impaired channel activity of TRPM7 induces macrothrombocytopenia in mice and humans [121], implicating that long-term inhibition of TRPM7 channel activity in disease condition may alter platelet biogenesis and function. Therefore, an alternative strategy to develop safe drugs against TRPM7 function in stroke would be a selective targeting of the kinase activity of TRPM7, thereby inhibiting thrombo-inflammation and protecting BBB and neurons from ischemic insults.
So far, the function of Zn2+ transporters in stroke development has not been established, and only a few experimental studies determined the expression profile of Zn2+ transporters. Tsuda and colleagues examined the induction of ZnT1 mRNA expression in the CA1 subfield of the hippocampus following global ischemia [122]. It was postulated that in response to the increased levels of intracellular Zn2+, vulnerable neurons would upregulate ZnT1, to facilitate Zn2+ efflux in the plasma membrane. While ZnT1 mRNA expression was enhanced as soon as 12 h post-ischemia, without subsequent ZnT1 protein expression, cellular demise ensured by three days post-ischemia [122]. The expression levels of ZnT1 and ZnT6 transporters were increased in the mouse model of hippocampal damage-induced by hypobaric hypoxia. Moreover, chelation of Zn2+ with Ca2EDTA decreased the expression of ZIP6 transporter and attenuated the hypobaric hypoxia-induced oxidative stress, inflammation and apoptosis [123].
The present therapy for acute ischemic stroke is limited to thrombolysis with the recombinant tissue plasminogen activator (rtPA) and mechanical recanalization [124]. Therefore, a better understanding of the molecular interplay between Zn2+ and Ca2+ homeostasis in ischemic stroke is needed for the development of more effective therapies. Future studies are needed to address the regulatory mechanisms, whether platelet-resident Zn2+ may directly regulate stroke or other thrombo-inflammatory diseases.
7. Dysregulated Zn2+ Homeostasis in Storage Pool Diseases
Secretory granule release is critical for platelet activation and hemostasis. Human platelets characterized by the lack or defective function of secretory granules show impaired aggregation responses, but depending on a genetic disorder, only exhibits minor to moderate bleeding [125]. Storage pool disease (SPD) comprises two groups of genetic disorders in which the function of platelet α and/or δ granules is defective [126]. In humans and mice, mutations in the Nbeal2 gene results in gray platelet syndrome (GPS), a rare inherited bleeding disorder characterized by thrombocytopenia, lack of α-granules within blood platelets and progressive development of bone marrow fibrosis [127,128,129,130]. NBEAL2 function was proposed to regulate the sorting and packaging processes of α-granules in MKs [31], only large empty vacuoles could be detected in the platelet cytoplasm of GPS patients, α-granule content is very limited or missing, and the function of residual granule-like structures is defective [131]. Consequently, most of the α-granule-resident proteins are degraded in the lysosomes, although P-selectin (as a marker of α-granules) could be detected on the platelet surface [131]. The lack of α-granules in GPS platelets strongly reduces fibrinogen content, but interestingly, fibrinogen uptake is not defective, thus a limited amount of fibrinogen can be stored and secreted by GPS platelets [132]. It is important to note that δ-granule biogenesis and function are normal in GPS patients, therefore ADP, serotonin and cations can be released upon platelet activation, maintaining the feedforward loop of platelet activation. Nevertheless, the lack of α-granules and degradation of α-granule-resident proteins, including plasma factors and fibrinogen, possibly impair fibrin clot formation in GPS patients. Recently, we investigated whether defective granule biogenesis or secretion may impair the platelet Zn2+ store and release, thereby influencing fibrin clot formation [26]. For this study, a turbidity assay was performed to quantify the thrombin-induced fibrin formation in wild-type mice and also in a mouse model of GPS (Nbeal2-/- mice). We found that turbidity was lower in wild-type platelet-releasate, which was further decreased in the presence of extracellular Zn2+. In Nbeal2-/- mice turbidity was similar to resting level in platelet releasate after thrombin activation, but it was significantly reduced in the presence of Zn2+ [26]. However, extracellular Zn2+ cannot fully restore fibrin formation to the wild-type level, due to the strongly reduced fibrinogen content and release from α-granules of Nbeal2-/- platelets [26]. Using microfluidics, we measured the kinetics of fibrin clot formation and calculated the accumulation rate of fluorescently-labeled platelets and fibrin fibers. Zn2+ supplementation did not modify the kinetics of fibrin formation in wild-type blood, suggesting that extracellular Zn2+ seems to be not a limiting factor of blood plasma in healthy conditions [26]. However, in disease conditions, platelet-dependent fibrin clot formation under flow was impaired in whole blood samples from Nbeal2-/- mice, and the kinetics of fibrin formation was accelerated by Zn2+ supplementation [26]. These alterations may also point to an impaired Zn2+ transport into the available secretory granules, dysregulating cytoplasmic Zn2+ levels in platelets. Taken these results together, we propose that platelets from GPS mouse model and human patients have a strongly reduced Zn2+ store, which influences platelet Zn2+ homeostasis and fibrin clot formation. Limited Zn2+ efflux during degranulation of GPS platelets, in addition to reduced fibrinogen release, could also inhibit fibrin formation and the activation of coagulation cascade. However, further investigation is necessary to understand the exact role of platelet Zn2+ release in these pathological conditions.
Prolonged bleeding time is frequently measured in SPD patients, which is usually ascribed with reduced ADP and serotonin content and release. However, the genetic background of SPD disease with δ-granule deficiency is still under debate. Hermansky–Pudlak Syndrome (HPS) is phenotypically associated with malformation or reduced numbers of platelet δ-granules and a bleeding diathesis [125,131], but α-granule content and function are unaffected, and therefore platelet activation is only mildly or moderately defective. Depending on the genetic mutation, HPS patients are divided into several subtypes, HPS-1 is the most common form characterized by hypopigmentation and albinism. In clinical treatment, antifibrinolytic agents have been administrated to these patients to prevent bleeding complications [126]. Using FluoZin3 measurement, our preliminary data showed impaired basal Zn2+ levels in HPS and SPD patients, indicating defective Zn2+ homeostasis in the granular store of these patients [26]. However, we assume that platelet responses to Zn2+ and also fibrin formation could be different, depending on the genetic background of the HPS/SPD patients and the severity of granule release. In the mouse model, platelets lacking the putative vesicle docking protein Munc13–4 (Unc13d-/-) have a completely abolished δ-granule secretion [133]. The lack of the feedforward loop induced by second wave mediators (ADP, serotonin) located in δ-granule strongly impairs α-granule release in Unc13d-/-platelets as well, which resulted in platelet dysfunction and severely prolonged tail-bleeding time [133], probably due to the combined defects of ADP/serotonin release from δ-granules and α-granule-resident proteins vWF, fibrinogen and coagulation factors. Using flow chamber assay, we showed that platelet-dependent fibrin clot formation was impaired in whole blood samples of mutant mice, and the kinetics of fibrin formation was accelerated by Zn2+ supplementation [26].
8. Conclusions
Taking into account these findings from the limited number of patients and mouse models with granule storage deficiency or granule release defects, it worth postulating that Zn2+ homeostasis could be strongly impaired in patients with platelet granular abnormalities. Consequently, reduced α-granule content or blockade of α-granule secretion can impair the coagulation pathway and fibrin clot formation in humans, which can be more critical in patients with combined defects of both α and δ-granules or with thrombocytopenia. In future, Zn2+ supplementation could be used in mouse models of GPS and SPD, to manipulate the size and structure of fibrin fibers and study fibrin clot formation under static and shear conditions, to allow for a better understanding of Zn2+-dependent clot stability and bleeding tendency in these pathological conditions. In addition, future studies are needed to identify the subcellular localization of platelet-specific Zn2+ transporters as a basis for understanding the molecular mechanisms of platelet Zn2+ uptake, storage and release and contribution of ZIP/ZnT isoforms to Zn2+ homeostasis. Whether the expression profiles of ZIP/ZnT isoforms are changing during megakaryopoiesis in normal and disease conditions, and which ZIP/ZnT isoforms regulate basic metabolic pathways between the Zn2+ store and platelet cytoplasm, which are impaired in GPS/SPD platelets, still needs to be further investigated at mRNA and protein levels. Future studies using mouse models with ZIP/ZnT deficiency in megakaryocyte/platelet lineage are needed to investigate the role of Zn2+ transport and storage in megakaryocytes and platelet-related diseases, including thrombosis and ischemic stroke.
Author Contributions
E.M.-B. and A.B. wrote the manuscript.
Funding
This work was supported by the funding from Deutsche Forschungsgemeinschaft (DFG, German Research Foundation—Projektnummer 374031971–TRR 240/A09) to A.B.
Acknowledgments
E.M.-B. is supported by EFFRE (Europäischer Fonds für regionale Entwicklung)-Bayern.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Roohani, N.; Hurrell, R.; Kelishadi, R.; Schulin, R. Zinc and its importance for human health: An integrative review. J. Res. Med. Sci. 2013, 18, 144–157. [Google Scholar] [PubMed]
- Fukada, T.; Kambe, T. Zinc signaling in physiology and pathogenesis. Int. J. Mol. Sci. 2018. [Google Scholar] [CrossRef]
- Kambe, T.; Tsuji, T.; Hashimoto, A.; Itsumura, N. The Physiological, Biochemical, and Molecular Roles of Zinc Transporters in Zinc Homeostasis and Metabolism. Physiol. Rev. 2015, 95, 749–784. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Stewart, A.J.; Sadler, P.J.; Pinheiro, T.J.; Blindauer, C.A. Albumin as a zinc carrier: Properties of its high-affinity zinc-binding site. Biochem. Soc. Trans. 2008, 36, 1317–1321. [Google Scholar] [CrossRef]
- Chilvers, D.C.; Dawson, J.B.; Bahreyni-Toosi, M.-H.; Hodgkinson, A. Identification and determination of copper-and zinc-protein complexes in blood plasma after chromatographic separation on DEAE-Sepharose CL-6B. Analyst 1984, 109, 871–876. [Google Scholar] [CrossRef]
- Foote, J.W.; Delves, H.T. Distribution of zinc amongst human serum proteins determined by affinity chromatography and atomic-absorption spectrophotometry. Analyst 1983, 108, 492–504. [Google Scholar] [CrossRef]
- Rowe, D.J.; Bobilya, D.J. Albumin facilitates zinc acquisition by endothelial cells. Proc. Soc. Exp. Biol. Med. 2000, 224, 178–186. [Google Scholar] [CrossRef]
- Hojyo, S.; Fukada, T. Zinc transporters and signaling in physiology and pathogenesis. Arch. Biochem. Biophys. 2016, 611, 43–50. [Google Scholar] [CrossRef]
- Haas, C.E.; Rodionov, D.A.; Kropat, J.; Malasarn, D.; Merchant, S.S.; de Crecy-Lagard, V. A subset of the diverse COG0523 family of putative metal chaperones is linked to zinc homeostasis in all kingdoms of life. BMC Genom. 2009, 10, 470. [Google Scholar] [CrossRef]
- Nimmanon, T.; Ziliotto, S.; Morris, S.; Flanagan, L.; Taylor, K.M. Phosphorylation of zinc channel ZIP7 drives MAPK, PI3K and mTOR growth and proliferation signalling. Metallomics 2017, 9, 471–481. [Google Scholar] [CrossRef]
- Truong-Tran, A.Q.; Carter, J.; Ruffin, R.E.; Zalewski, P.D. The role of zinc in caspase activation and apoptotic cell death. Biometals 2001, 14, 315–330. [Google Scholar] [CrossRef]
- Jacobsen, J.A.; Major Jourden, J.L.; Miller, M.T.; Cohen, S.M. To bind zinc or not to bind zinc: An examination of innovative approaches to improved metalloproteinase inhibition. Biochim. Biophys. Acta. 2010, 1803, 72–94. [Google Scholar] [CrossRef] [PubMed]
- Hojyo, S.; Fukada, T. Roles of Zinc Signaling in the Immune System. J. Immunol. Res. 2016, 2016, 6762343. [Google Scholar] [CrossRef] [PubMed]
- Gammoh, N.Z.; Rink, L. Zinc in Infection and Inflammation. Nutrients 2017, 9, 624. [Google Scholar] [CrossRef]
- Taylor, K.A.; Pugh, N. The contribution of zinc to platelet behaviour during haemostasis and thrombosis. Metallomics 2016, 8, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Bin, B.H.; Seo, J.; Kim, S.T. Function, Structure, and Transport Aspects of ZIP and ZnT Zinc Transporters in Immune Cells. J. Immunol. Res. 2018, 2018, 9365747. [Google Scholar] [CrossRef]
- Norouzi, S.; Adulcikas, J.; Sohal, S.S.; Myers, S. Zinc transporters and insulin resistance: Therapeutic implications for type 2 diabetes and metabolic disease. J. Biomed. Sci. 2017, 24, 87. [Google Scholar] [CrossRef]
- Maret, W. Zinc in Pancreatic Islet Biology, Insulin Sensitivity, and Diabetes. Prev. Nutr. Food Sci. 2017, 22, 1–8. [Google Scholar] [CrossRef]
- Prasad, A.S.; Cossack, Z.T. Neutrophil zinc: An indicator of zinc status in man. Trans. Assoc. Am. Phys. 1982, 95, 165–176. [Google Scholar]
- Bird, A.J.; McCall, K.; Kramer, M.; Blankman, E.; Winge, D.R.; Eide, D.J. Zinc fingers can act as Zn2+ sensors to regulate transcriptional activation domain function. EMBO J. 2003, 22, 5137–5146. [Google Scholar] [CrossRef]
- Sugiura, T.; Nakamura, H. Metallothionein in platelets. Int. Arch. Allergy Immunol. 1994, 103, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Ruttkay-Nedecky, B.; Nejdl, L.; Gumulec, J.; Zitka, O.; Masarik, M.; Eckschlager, T.; Stiborova, M.; Adam, V.; Kizek, R. The role of metallothionein in oxidative stress. Int. J. Mol. Sci. 2013, 14, 6044–6066. [Google Scholar] [CrossRef] [PubMed]
- Sheu, J.R.; Hsiao, G.; Shen, M.Y.; Wang, Y.; Lin, K.H.; Lin, C.H.; Chou, D.S. Inhibitory mechanisms of metallothionein on platelet aggregation in in vitro and platelet plug formation in in vivo experiments. Exp. Biol. Med. 2003, 228, 1321–1328. [Google Scholar] [CrossRef]
- Watson, B.R.; White, N.A.; Taylor, K.A.; Howes, J.-M.; Malcor, J.-D.M.; Bihan, D.; Sage, S.O.; Farndale, R.W.; Pugh, N. Zinc is a transmembrane agonist that induces platelet activation in a tyrosine phosphorylation-dependent manner. Metallomics 2016, 8, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.T.; Fredenburgh, J.C.; Weitz, J.I. Zinc: An important cofactor in haemostasis and thrombosis. Thromb. Haemost. 2013, 109, 421–430. [Google Scholar] [CrossRef]
- Gotru, S.K.; van Geffen, J.P.; Nagy, M.; Mammadova-Bach, E.; Eilenberger, J.; Volz, J.; Manukjan, G.; Schulze, H.; Wagner, L.; Eber, S.; et al. Defective Zn(2+) homeostasis in mouse and human platelets with α- and δ-storage pool diseases. Sci. Rep. 2019, 9, 8333. [Google Scholar] [CrossRef]
- Kasperek, K.; Kiem, J.; Iyengar, G.V.; Feinendegen, L.E. Concentration differences between serum and plasma of the elements cobalt, iron, mercury, rubidium, selenium and zinc determined by neutron activation analysis. Sci. Total Environ. 1981, 17, 133–143. [Google Scholar] [CrossRef]
- Chen, W.J.; Zhao, C.Y.; Zheng, T.L. Comparison of zinc contents in human serum and plasma. Clin. Chim. Acta 1986, 155, 185–187. [Google Scholar]
- Marx, G.; Korner, G.; Mou, X.; Gorodetsky, R. Packaging zinc, fibrinogen, and factor XIII in platelet α-granules. J. Cell. Physiol. 1993, 156, 437–442. [Google Scholar] [CrossRef]
- Blair, P.; Flaumenhaft, R. Platelet α-granules: Basic biology and clinical correlates. Blood Rev. 2009, 23, 177–189. [Google Scholar] [CrossRef]
- Kahr, W.H.A.; Lo, R.W.; Li, L.; Pluthero, F.G.; Christensen, H.; Ni, R.; Vaezzadeh, N.; Hawkins, C.E.; Weyrich, A.S.; Di Paola, J.; et al. Abnormal megakaryocyte development and platelet function in Nbeal2−/− mice. Blood 2013, 122, 3349–3358. [Google Scholar] [CrossRef] [PubMed]
- Wijten, P.; Van Holten, T.; Woo, L.L.; Bleijerveld, O.B.; Roest, M.; Heck, A.J.; Scholten, A. High precision platelet releasate definition by quantitative reversed protein profiling—Brief report. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Gorodetsky, R.; Mou, X.; Blankenfeld, A.; Marx, G. Platelet multielemental composition, lability, and subcellular localization. Am. J. Hematol. 1993, 42, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Liuzzi, J.P.; Cousins, R.J. Mammalian zinc transporters. Annu. Rev. Nutr. 2004, 24, 151–172. [Google Scholar] [CrossRef]
- Jeong, J.; Eide, D.J. The SLC39 family of zinc transporters. Mol. Asp. Med. 2013, 34, 612–619. [Google Scholar] [CrossRef]
- Bafaro, E.; Liu, Y.; Xu, Y.; Dempski, R.E. The emerging role of zinc transporters in cellular homeostasis and cancer. Signal Transduct. Target. Ther. 2017, 2, 17029. [Google Scholar] [CrossRef]
- Kambe, T. Molecular architecture and function of ZnT transporters. Curr. Top. Membr. 2012, 69, 199–220. [Google Scholar]
- Kimura, Y.; Hart, A.; Hirashima, M.; Wang, C.; Holmyard, D.; Pittman, J.; Pang, X.-L.; Jackson, C.W.; Bernstein, A. Zinc finger protein, Hzf, is required for megakaryocyte development and hemostasis. J. Exp. Med. 2002, 195, 941–952. [Google Scholar] [CrossRef]
- Bresnick, E.H.; Katsumura, K.R.; Lee, H.Y.; Johnson, K.D.; Perkins, A.S. Master regulatory GATA transcription factors: Mechanistic principles and emerging links to hematologic malignancies. Nucl. Acids Res. 2012, 40, 5819–5831. [Google Scholar] [CrossRef]
- Apgar, J. Effect of zinc deficiency on parturition in the rat. Am. J. Physiol. 1968, 215, 160–163. [Google Scholar] [CrossRef]
- Gordon, P.R.; O’Dell, B.L. Rat platelet aggregation impaired by short-term zinc deficiency. J. Nutr. 1980, 110, 2125–2129. [Google Scholar] [CrossRef] [PubMed]
- O’Dell, B.L.; Reynolds, G.; Reeves, P.G. Analogous effects of zinc deficiency and aspirin toxicity in the pregnant rat. J. Nutr. 1977, 107, 1222–1228. [Google Scholar] [CrossRef] [PubMed]
- Faure, P.; Durand, P.; Blache, D.; Favier, A.; Roussel, A.M. Influence of a long-term zinc-deficient diet on rat platelet function and fatty acid composition. Biometals 1995, 8, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; O’Dell, B.L. Zinc deficiency in rats decreases thrombin-stimulated platelet aggregation by lowering protein kinase C activity secondary to impaired calcium uptake. J. Nutr. Biochem. 1995, 6, 661–666. [Google Scholar] [CrossRef]
- O’Dell, B.L.; Emery, M.; Xia, J.; Browning, J.D. In vitro addition of glutathione to blood from zinc-deficient rats corrects platelet defects: Impaired aggregation and calcium uptakE. Nutr. Biochem. 1997, 8, 346–350. [Google Scholar] [CrossRef]
- O’Dell, B.L.; Emery, M. Compromised zinc status in rats adversely affects calcium metabolism in platelets. J. Nutr. 1991, 121, 1763–1768. [Google Scholar] [CrossRef]
- Gordon, P.R.; Woodruff, C.W.; Anderson, H.L.; O’Dell, B.L. Effect of acute zinc deprivation on plasma zinc and platelet aggregation in adult males. Am. J. Clin. Nutr. 1982, 35, 113–119. [Google Scholar] [CrossRef]
- Stefanini, M. Cutaneous bleeding related to zinc deficiency in two cases of advanced cancer. Cancer 1999, 86, 866–870. [Google Scholar] [CrossRef]
- Marx, G.; Krugliak, J.; Shaklai, M. Nutritional zinc increases platelet reactivity. Am. J. Hematol. 1991, 38, 161–165. [Google Scholar] [CrossRef]
- Mammadova-Bach, E.; Nagy, M.; Heemskerk, J.W.M.; Nieswandt, B.; Braun, A. Store-operated calcium entry in thrombosis and thrombo-inflammation. Cell Calcium 2019, 77, 39–48. [Google Scholar] [CrossRef]
- Roth, H.P.; Moser, C.; Kirchgessner, M. Subcellular distribution of protein kinase C (pKC) in erythrocytes and concentration of D-myo-inositol-1,4,5-trisphosphate (IP3) in platelets and monocytes of force-fed zinc-deficient rats. Biol. Trace Elem. Res. 1996, 53, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Forbes, I.J.; Zalewski, P.D.; Giannakis, C.; Petkoff, H.S.; Cowled, P.A. Interaction between protein kinase C and regulatory ligand is enhanced by a chelatable pool of cellular zinc. Biochim. Biophys. Acta. 1990, 1053, 113–117. [Google Scholar] [CrossRef]
- Marreiro, D.D.; Cruz, K.J.; Morais, J.B.; Beserra, J.B.; Severo, J.S.; de Oliveira, A.R. Zinc and Oxidative Stress: Current Mechanisms. Antioxidants 2017, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.; Ding, M.; Ding, Y.; Sours-Brothers, S.; Luchowski, R.; Gryczynski, Z.; Yorio, T.; Ma, H.; Ma, R. Canonical transient receptor potential 6 (TRPC6), a redox-regulated cation channel. J. Biol. Chem. 2010, 285, 23466–23476. [Google Scholar] [CrossRef] [PubMed]
- Carter, R.N.; Tolhurst, G.; Walmsley, G.; Vizuete-Forster, M.; Miller, N.; Mahaut-Smith, M.P. Molecular and electrophysiological characterization of transient receptor potential ion channels in the primary murine megakaryocyte. J Physiol. 2006, 576, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Murayama, T.; Tashiro, M.; Sakurai, T.; Konishi, M. Mg2+- and ATP-dependent inhibition of transient receptor potential melastatin 7 by oxidative stress. Free Radic. Biol. Med. 2014, 72, 257–266. [Google Scholar] [CrossRef]
- Huang, C.; Miller, R.T.; Freter, C.E. Signaling regulation and role of filamin A cleavage in Ca2+-stimulated migration of androgen receptor-deficient prostate cancer cells. Oncotarget 2017, 8, 3840–3853. [Google Scholar]
- Gees, M.; Colsoul, B.; Nilius, B. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a003962. [Google Scholar] [CrossRef]
- Monteilh-Zoller, M.K.; Hermosura, M.C.; Nadler, M.J.; Scharenberg, A.M.; Penner, R.; Fleig, A. TRPM7 provides an ion channel mechanism for cellular entry of trace metal ions. J. Gen. Physiol. 2003, 121, 49–60. [Google Scholar] [CrossRef]
- Chevallet, M.; Jarvis, L.; Harel, A.; Luche, S.; Degot, S.; Chapuis, V.; Boulay, G.; Rabilloud, T.; Bouron, A. Functional consequences of the over-expression of TRPC6 channels in HEK cells: Impact on the homeostasis of zinc. Metallomics 2014, 6, 1269–1276. [Google Scholar] [CrossRef]
- Tennent, G.A.; Brennan, S.O.; Stangou, A.J.; O’Grady, J.; Hawkins, P.N.; Pepys, M.B. Human plasma fibrinogen is synthesized in the liver. Blood 2007, 109, 1971–1974. [Google Scholar] [CrossRef] [PubMed]
- Weisel, J.W.; Litvinov, R.I. Fibrin Formation, Structure and Properties. Subcell. Biochem. 2017, 82, 405–456. [Google Scholar] [PubMed]
- Zavyalova, E.G.; Protopopova, A.D.; Kopylov, A.M.; Yaminsky, I.V. Investigation of early stages of fibrin association. Langmuir 2011, 27, 4922–4927. [Google Scholar] [CrossRef] [PubMed]
- Wolberg, A.S. Thrombin generation and fibrin clot structure. Blood Rev. 2007, 21, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Mosesson, M.W. Fibrinogen and fibrin structure and functions. J. Thromb. Haemost. 2005, 3, 1894–1904. [Google Scholar] [CrossRef] [PubMed]
- Marx, G. Zinc binding to fibrinogen and fibrin. Arch. Biochem. Biophys. 1988, 266, 285–288. [Google Scholar] [CrossRef]
- Kanaide, H.; Uranishi, T.; Nakamura, M. Effects of divalent cations on the conversion of fibrinogen to fibrin and fibrin polymerization. Am. J. Hematol. 1982, 13, 229–237. [Google Scholar] [CrossRef]
- Weisel, J.W.; Litvinov, R.I. Mechanisms of fibrin polymerization and clinical implications. Blood 2013, 121, 1712–1719. [Google Scholar] [CrossRef]
- Ryan, E.A.; Mockros, L.F.; Weisel, J.W.; Lorand, L. Structural origins of fibrin clot rheology. Biophys. J. 1999, 77, 2813–2826. [Google Scholar] [CrossRef]
- Marx, G. Mechanism of fibrin coagulation based on selective, cation-driven, protofibril association. Biopolymers 1988, 27, 763–774. [Google Scholar] [CrossRef]
- Marx, G.; Eldor, A. The procoagulant effect of zinc on fibrin clot formation. Am. J. Hematol. 1985, 19, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Marx, G. Modulation of thrombin activity by zinc. Ann. N. Y. Acad. Sci. 1986, 485, 421–424. [Google Scholar] [CrossRef]
- Marx, G.; Hopmeier, P. Zinc inhibits FPA release and increases fibrin turbidity. Am. J. Hematol. 1986, 22, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Scully, M.F.; Kakkar, V.V. Structural features of fibrinogen associated with binding to chelated zinc. Biochim. Biophys. Acta. 1982, 700, 130–135. [Google Scholar] [CrossRef]
- Marx, G. Divalent cations induce protofibril gelation. Am. J. Hematol. 1988, 27, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Fatah, K.; Hessel, B. Effect of zinc ions on fibrin network structure. Blood Coagul. Fibrinolysis 1998, 9, 629–635. [Google Scholar] [CrossRef]
- Marx, G.; Hopmeier, P.; Gurfel, D. Zinc alters fibrin ultrastructure. Thromb. Haemost. 1987, 57, 73–76. [Google Scholar] [CrossRef]
- Marx, G.; Harari, N. Albumin indirectly modulates fibrin and protofibrin ultrastructure. Biochemistry 1989, 28, 8242–8248. [Google Scholar] [CrossRef]
- Müller, F.; Mutch, N.J.; Schenk, W.A.; Smith, S.A.; Esterl, L.; Spronk, H.M.; Schmidbauer, S.; Gahl, W.A.; Morrissey, J.H.; Renné, T. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell 2009, 139, 1143–1156. [Google Scholar] [CrossRef]
- Wu, Y. Contact pathway of coagulation and inflammation. Thromb. J. 2015, 13, 17. [Google Scholar] [CrossRef]
- Schmaier, A.H. The contact activation and kallikrein/kinin systems: Pathophysiologic and physiologic activities. J. Thromb. Haemost. 2016, 14, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Ehrenfeld, P.; Millan, C.; Matus, C.E.; Figueroa, J.E.; Burgos, R.A.; Nualart, F.; Bhoola, K.D.; Figueroa, C.D. Activation of kinin B1 receptors induces chemotaxis of human neutrophils. J. Leukoc. Biol. 2006, 80, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Tubek, S.; Grzanka, P.; Tubek, I. Role of zinc in hemostasis: A review. Biol. Trace Elem. Res. 2008, 121, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sobieszczańska, M.; Tubek, S. Zinc in Hemostasis. In Encyclopedia of Metalloproteins; Kretsinger, R.H., Uversky, V.N., Permyakov, E.A., Eds.; Springer: New York, NY, USA, 2013. [Google Scholar]
- Schousboe, I. Contact activation in human plasma is triggered by zinc ion modulation of factor XII (Hageman factor). Blood Coagul. Fibrinolysis 1993, 4, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, M.M.; Day, D.E.; Halvorson, H.R.; Olson, S.T.; Shore, J.D. Surface-independent acceleration of factor XII activation by zinc ions. II. Direct binding and fluorescence studies. J. Biol. Chem. 1993, 268, 12477–12483. [Google Scholar]
- Baglia, F.A.; Badellino, K.O.; Li, C.Q.; Lopez, J.A.; Walsh, P.N. Factor XI binding to the platelet glycoprotein Ib-IX-V complex promotes factor XI activation by thrombin. J. Biol. Chem. 2002, 277, 1662–1668. [Google Scholar] [CrossRef]
- Sobczak, A.I.S.; Pitt, S.J.; Stewart, A.J. Influence of zinc on glycosaminoglycan neutralisation during coagulation. Metallomics 2018, 10, 1180–1190. [Google Scholar] [CrossRef]
- Joseph, K.; Ghebrehiwet, B.; Peerschke, E.I.; Reid, K.B.; Kaplan, A.P. Identification of the zinc-dependent endothelial cell binding protein for high molecular weight kininogen and factor XII: Identity with the receptor that binds to the globular “heads” of C1q (gC1q-R). Proc. Natl. Acad. Sci. USA 1996, 93, 8552–8557. [Google Scholar] [CrossRef]
- Poon, I.K.; Patel, K.K.; Davis, D.S.; Parish, C.R.; Hulett, M.D. Histidine-rich glycoprotein: The Swiss Army knife of mammalian plasma. Blood 2011, 117, 2093–2101. [Google Scholar] [CrossRef]
- Shigekiyo, T.; Ohshima, T.; Oka, H.; Tomonari, A.; Azuma, H.; Saito, S. Congenital histidine-rich glycoprotein deficiency. Thromb. Haemost. 1993, 70, 263–265. [Google Scholar] [CrossRef]
- Khan, S.; Dickerman, J.D. Hereditary thrombophilia. Thromb. J. 2006, 4, 15. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stewart, A.J.; Blindauer, C.A.; Sadler, P.J. Plasma fatty acid levels may regulate the Zn2+-dependent activities of histidine-rich glycoprotein. Biochimie 2009, 91, 1518–1522. [Google Scholar] [CrossRef] [PubMed]
- Coverdale, J.P.C.; Khazaipoul, S.; Arya, S.; Stewart, A.J.; Blindauer, C.A. Crosstalk between zinc and free fatty acids in plasma. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, R.; Wang, Z.; Jing, Z.; Wang, S.; Ma, J. S100A8/A9 in Inflammation. Front. Immunol. 2018, 9, 1298. [Google Scholar] [CrossRef]
- Sampson, B.; Fagerhol, M.K.; Sunderkötter, C.; Golden, B.E.; Richmond, P.; Klein, N.; Kovar, I.Z.; Beattie, J.H.; Wolska-Kuśnierz, B.; Saito, Y.; et al. Hyperzincaemia and hypercalprotectinaemia: A new disorder of zinc metabolism. Lancet 2002, 360, 1742–1745. [Google Scholar] [CrossRef]
- Larsen, S.B.; Grove, E.L.; Pareek, M.; Kristensen, S.D.; Hvas, A.M. Calprotectin and platelet aggregation in patients with stable coronary artery disease. PLoS ONE 2015, 10, e0125992. [Google Scholar] [CrossRef]
- Larsen, S.B.; Grove, E.L.; Wurtz, M.; Neergaard-Petersen, S.; Hvas, A.M.; Kristensen, S.D. The influence of low-grade inflammation on platelets in patients with stable coronary artery disease. Thromb. Haemost. 2015, 114, 519–529. [Google Scholar] [CrossRef]
- Lood, C.; Tydén, H.; Gullstrand, B.; Jönsen, A.; Källberg, E.; Mörgelin, M.; Kahn, R.; Gunnarsson, I.; Leanderson, T.; Ivars, F.; et al. Platelet-Derived S100A8/A9 and Cardiovascular Disease in Systemic Lupus Erythematosus. Arthr. Rheumatol. 2016, 68, 1970–1980. [Google Scholar] [CrossRef]
- Prakash, A.; Bharti, K.; Majeed, A.B. Zinc: Indications in brain disorders. Fundam. Clin. Pharmacol. 2015, 29, 131–149. [Google Scholar] [CrossRef]
- Koh, J.Y. Zinc and disease of the brain. Mol. Neurobiol. 2001, 24, 99–106. [Google Scholar] [CrossRef]
- Lee, J.M.; Zipfel, G.J.; Park, K.H.; He, Y.Y.; Hsu, C.Y.; Choi, D.W. Zinc translocation accelerates infarction after mild transient focal ischemia. Neuroscience 2002, 115, 871–878. [Google Scholar] [CrossRef]
- Diener, H.C.; Schneider, D.; Lampl, Y.; Bornstein, N.M.; Kozak, A.; Rosenberg, G. DP-b99, a membrane-activated metal ion chelator, as neuroprotective therapy in ischemic stroke. Stroke 2008, 39, 1774–1778. [Google Scholar] [CrossRef] [PubMed]
- Angel, I.B.A.; Horovitz, T.; Taler, G.; Krakovsky, M.; Resnitsky, D.; Rosenberg, G.; Striem, S.; Friedman, J.E.; Kozak, A. Metal ion chelation in neurodegenerative disorders. Drug Dev. Res. 2002, 56, 300–309. [Google Scholar] [CrossRef]
- Krakovsky, M.P.M.; Angel, I.; Kozak, A. A novel membrane targeted compound active against global and focal ischemia. In Physiological Imaging of the Brain with PET; Gjedde, A.S.H., Knudsen, G.M., Paulson, O.B., Eds.; Academic Press: Cambridge, MA, USA, 2001; pp. 347–352. [Google Scholar]
- Fujimura, M.; Morita-Fujimura, Y.; Noshita, N.; Sugawara, T.; Kawase, M.; Chan, P.H. The cytosolic antioxidant copper/zinc-superoxide dismutase prevents the early release of mitochondrial cytochrome c in ischemic brain after transient focal cerebral ischemia in mice. J. Neurosci. 2000, 20, 2817–2824. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Kondo, T.; Epstein, C.J.; Chan, P.H. Overexpression of CuZn-superoxide dismutase reduces hippocampal injury after global ischemia in transgenic mice. Stroke 1997, 28, 1797–1804. [Google Scholar] [CrossRef] [PubMed]
- Helal, G.K. Systemic administration of Zn2+ during the reperfusion phase of transient cerebral ischaemia protects rat hippocampus against iron-catalysed postischaemic injury. Clin. Exp. Pharmacol. Physiol. 2008, 35, 775–781. [Google Scholar] [CrossRef]
- Aquilani, R.; Baiardi, P.; Scocchi, M.; Iadarola, P.; Verri, M.; Sessarego, P.; Boschi, F.; Pasini, E.; Pastoris, O.; Viglio, S. Normalization of zinc intake enhances neurological retrieval of patients suffering from ischemic strokes. Nutr. Neurosci. 2009, 12, 219–225. [Google Scholar] [CrossRef]
- Frederickson, C.J.; Suh, S.W.; Silva, D.; Frederickson, C.J.; Thompson, R.B. Importance of zinc in the central nervous system: The zinc-containing neuron. J. Nutr. 2000, 130 (Suppl. S5), 1471S–1483S. [Google Scholar] [CrossRef]
- Choi, D.W.; Koh, J.Y. Zinc and brain injury. Annu. Rev. Neurosci. 1998, 21, 347–375. [Google Scholar] [CrossRef]
- Sensi, S.L.; Paoletti, P.; Bush, A.I.; Sekler, I. Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 2009, 10, 780–791. [Google Scholar] [CrossRef]
- Noh, K.M.; Yokota, H.; Mashiko, T.; Castillo, P.E.; Zukin, R.S.; Bennett, M.V. Blockade of calcium-permeable AMPA receptors protects hippocampal neurons against global ischemia-induced death. Proc. Natl. Acad. Sci. USA 2005, 102, 12230–12235. [Google Scholar] [CrossRef] [PubMed]
- Bollimuntha, S.; Pani, B.; Singh, B.B. Neurological and Motor Disorders: Neuronal Store-Operated Ca2+ Signaling: An Overview and Its Function. Adv. Exp. Med. Biol. 2017, 993, 535–556. [Google Scholar] [PubMed]
- Secondo, A.; Bagetta, G.; Amantea, D. On the Role of Store-Operated Calcium Entry in Acute and Chronic Neurodegenerative Diseases. Front. Mol. Neurosci. 2018, 11, 87. [Google Scholar] [CrossRef] [PubMed]
- Berna-Erro, A.; Braun, A.; Kraft, R.; Kleinschnitz, C.; Schuhmann, M.K.; Stegner, D.; Wultsch, T.; Eilers, J.; Meuth, S.G.; Stoll, G.; et al. STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci. Signal. 2009, 2, ra67. [Google Scholar] [CrossRef] [PubMed]
- Kiedrowski, L. Cytosolic zinc release and clearance in hippocampal neurons exposed to glutamate—The role of pH and sodium. J. Neurochem. 2011, 117, 231–243. [Google Scholar] [CrossRef]
- Chubanov, V.; Schafer, S.; Ferioli, S.; Gudermann, T. Natural and Synthetic Modulators of the TRPM7 Channel. Cells 2014, 3, 1089–1101. [Google Scholar] [CrossRef]
- Lin, J.; Xiong, Z.G. TRPM7 is a unique target for therapeutic intervention of stroke. Int. J. Physiol. Pathophysiol. Pharmacol. 2017, 9, 211–216. [Google Scholar]
- Gotru, S.K.; Chen, W.; Kraft, P.; Becker, I.C.; Wolf, K.; Stritt, S.; Zierler, S.; Hermanns, H.M.; Rao, D.; Perraud, A.-L.; et al. TRPM7 Kinase Controls Calcium Responses in Arterial Thrombosis and Stroke in Mice. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 344–352. [Google Scholar] [CrossRef]
- Stritt, S.; Nurden, P.; Favier, R.; Favier, M.; Ferioli, S.; Gotru, S.K.; van Eeuwijk, J.M.M.; Schulze, H.; Nurden, A.T.; Lambert, M.P.; et al. Defects in TRPM7 channel function deregulate thrombopoiesis through altered cellular Mg2+ homeostasis and cytoskeletal architecture. Nat. Commun. 2016, 7, 11097. [Google Scholar] [CrossRef]
- Tsuda, M.; Imaizumi, K.; Katayama, T.; Kitagawa, K.; Wanaka, A.; Tohyama, M.; Takagi, T. Expression of zinc transporter gene, ZnT-1, is induced after transient forebrain ischemia in the gerbil. J. Neurosci. 1997, 17, 6678–6684. [Google Scholar] [CrossRef]
- Malairaman, U.; Dandapani, K.; Katyal, A. Effect of Ca2EDTA on zinc mediated inflammation and neuronal apoptosis in hippocampus of an in vivo mouse model of hypobaric hypoxia. PLoS ONE 2014, 9, e110253. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.M. Recombinant tissue plasminogen activator for the treatment of acute ischemic stroke. Bayl. Univ. Med Cent. Proc. 2011, 24, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.K. Inherited platelet function disorders: Overview and disorders of granules, secretion, and signal transduction. Hematol. Oncol. Clin. North Am. 2013, 27, 585–611. [Google Scholar] [CrossRef] [PubMed]
- Sandrock, K.; Zieger, B. Current Strategies in Diagnosis of Inherited Storage Pool Defects. Transfus. Med. Hemother. 2010, 37, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Albers, C.A.; Cvejic, A.; Favier, R.; Bouwmans, E.E.; Alessi, M.C.; Bertone, P.; Jordan, G.; Kettleborough, R.N.; Kiddle, G.; Kostadima, M.; et al. Exome sequencing identifies NBEAL2 as the causative gene for gray platelet syndrome. Nat. Genet. 2011, 43, 735–737. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, J.A.; Bennett, C.; van der Weyden, L.; McKinney, H.; Chin, M.; Nurden, P.; McIntyre, Z.; Cambridge, E.L.; Estabel, J.; Wardle-Jones, H.; et al. Gray platelet syndrome: Proinflammatory megakaryocytes and α-granule loss cause myelofibrosis and confer metastasis resistance in mice. Blood 2014, 124, 3624–3635. [Google Scholar] [CrossRef]
- Deppermann, C.; Cherpokova, D.; Nurden, P.; Schulz, J.-N.; Thielmann, I.; Kraft, P.; Vogtle, T.; Kleinschnitz, C.; Dütting, S.; Krohne, G.; et al. Gray platelet syndrome and defective thrombo-inflammation in Nbeal2-deficient mice. J. Clin. Investig. 2013, 123, 3331–3342. [Google Scholar] [CrossRef]
- Gunay-Aygun, M.; Falik-Zaccai, T.C.; Vilboux, T.; Zivony-Elboum, Y.; Gumruk, F.; Cetin, M.; Khayat, M.; Boerkoel, C.F.; Kfir, N.; Huang, Y.; et al. NBEAL2 is mutated in gray platelet syndrome and is required for biogenesis of platelet α-granules. Nat. Genet. 2011, 43, 732–734. [Google Scholar] [CrossRef]
- Rendu, F.; Brohard-Bohn, B. The platelet release reaction: Granules’ constituents, secretion and functions. Platelets 2001, 12, 261–273. [Google Scholar] [CrossRef]
- Lo, R.W.; Li, L.; Leung, R.; Pluthero, F.G.; Kahr, W.H.A. NBEAL2 (Neurobeachin-Like 2) Is Required for Retention of Cargo Proteins by α-Granules During Their Production by Megakaryocytes. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2435–2447. [Google Scholar] [CrossRef]
- Harper, M.T.; van den Bosch, M.T.; Hers, I.; Poole, A.W. Platelet dense granule secretion defects may obscure alpha-granule secretion mechanisms: Evidence from Munc13-4-deficient platelets. Blood 2015, 125, 3034–3036. [Google Scholar] [CrossRef] [PubMed][Green Version]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).