Quantification of Mitochondrial Oxidative Phosphorylation in Metabolic Disease: Application to Type 2 Diabetes

 ,
,
Abstract
:1. Introduction
Definitions
2. Mitochondrial Oxidative Phosphorylation (MOP)
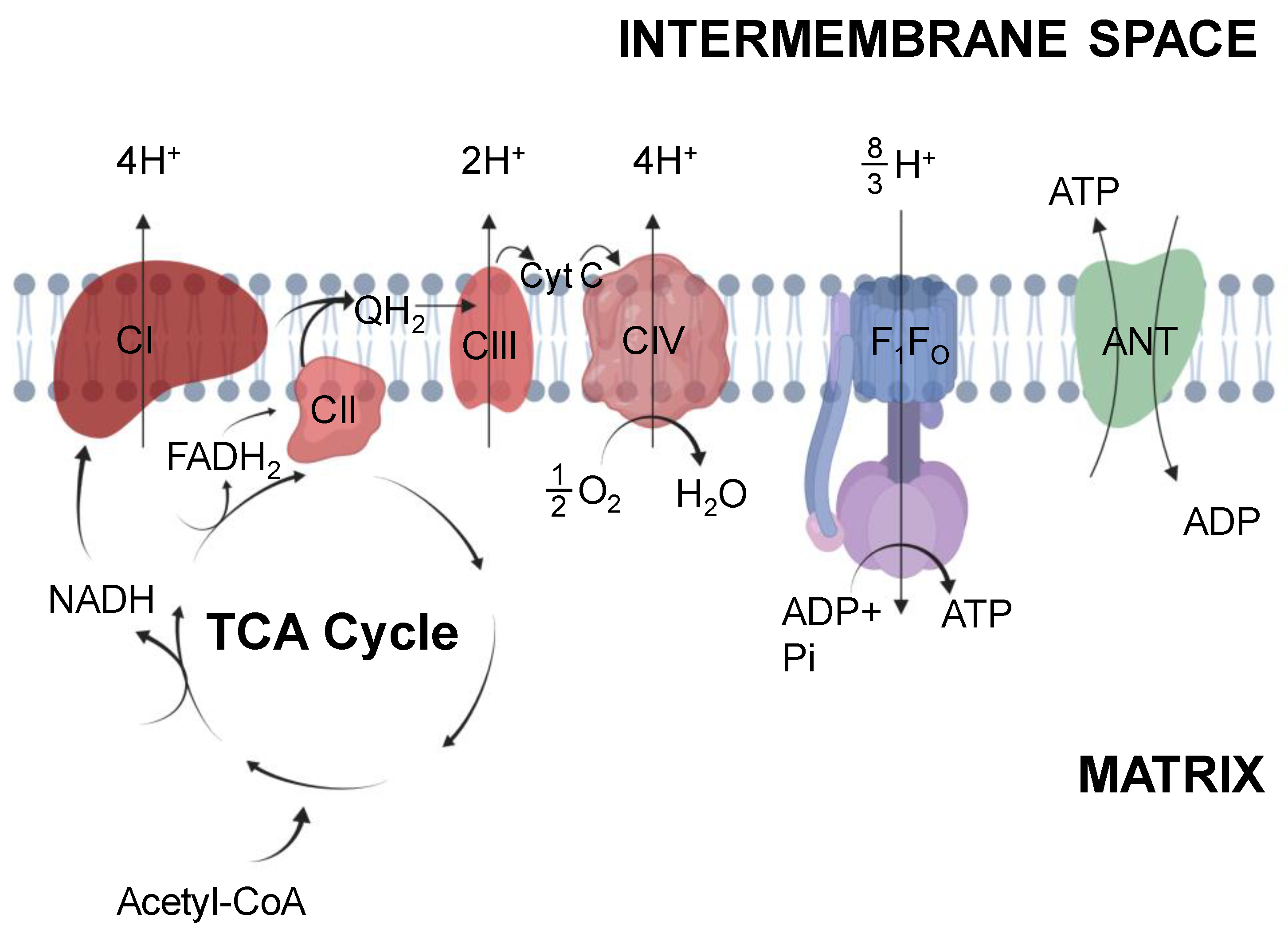
2.1. Tricarboxylic Acid Cycle
2.2. Electron Transport System
3. Measurement of Mitochondrial Function
3.1. Mitochondrial Function Quantified In Vitro
3.2. Mitochondrial Function Quantified In Vivo
4. Obesity, Physical Inactivity, and Mop Capacity
4.1. Obesity Effect on MOP Capacity
4.2. Physical Activity and MOP Capacity
4.3. Summary
5. Human Type 2 Diabetes Mitochondrial Function
5.1. Human Type 2 Diabetes Mitochondrial Function In Vitro
5.2. Human Type 2 Diabetes Mitochondrial Function In Vivo
5.3. What Is Missing?
6. Rat Models of Type 2 Diabetes
6.1. Zucker Diabetic Fatty (ZDF) Rat
6.1.1. Mitochondrial Function in Vitro
6.1.2. Mitochondrial Function in Vivo
6.2. Goto-Kakizaki (GK) Rat
6.2.1. Mitochondrial Function in Vitro
6.2.2. Mitochondrial Function in Vivo
7. Summary and Future Directions
Funding
Conflicts of Interest
References
- Wiseman, R.W.; Jeneson, J.A.L. Noninvasive Assessment of Mitochondrial Function Using Nuclear Magnetic Resonance Spectroscopy. In Drug-Induced Mitochondrial Dysfunction; Wiley-Blackwell: Hoboken, NJ, USA, 2008; pp. 555–574. ISBN 978-0-470-37253-1. [Google Scholar]
- Larsen, S.; Nielsen, J.; Hansen, C.N.; Nielsen, L.B.; Wibrand, F.; Stride, N.; Schroder, H.D.; Boushel, R.; Helge, J.W.; Dela, F.; et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 2012, 590, 3349–3360. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.A.; Wiseman Robert, W. Chapter 13: The Metabolic Systems: Control of ATP Synthesis in Skeletal Muscle. In ACSM’s Advanced Exercise Physiology; Lippincott Williams & Wilkins: Baltimore, MD, USA, 2011. [Google Scholar]
- Frisbee, J.C.; Lewis, M.T.; Wiseman, R.W. Skeletal muscle performance in metabolic disease: Microvascular or mitochondrial limitation or both? Microcirculation 2018, 26, e12517. [Google Scholar] [CrossRef] [PubMed]
- CDC National Diabetes Statistics Report|Data & Statistics|Diabetes|CDC. Available online: https://www.cdc.gov/diabetes/data/statistics/statistics-report.html (accessed on 22 January 2019).
- Rowley, W.R.; Bezold, C.; Arikan, Y.; Byrne, E.; Krohe, S. Diabetes 2030: Insights from Yesterday, Today, and Future Trends. Popul. Health Manag. 2017, 20, 6–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Diabetes Association Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 2011, 34, S62–S69. [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Laakso, M. Cardiovascular Disease in Type 2 Diabetes from Population to Man to Mechanisms: The Kelly West Award Lecture 2008. Diabetes Care 2010, 33, 442–449. [Google Scholar] [CrossRef]
- Fang, Z.Y.; Sharman, J.; Prins, J.B.; Marwick, T.H. Determinants of Exercise Capacity in Patients with Type 2 Diabetes. Diabetes Care 2005, 28, 1643–1648. [Google Scholar] [CrossRef]
- Regensteiner, J.G.; Sippel, J.; McFarling, E.T.; Wolfel, E.E.; Hiatt, W.R. Effects of non-insulin-dependent diabetes on oxygen consumption during treadmill exercise. Med. Sci. Sports Exerc. 1995, 27, 875–881. [Google Scholar] [CrossRef]
- Reusch, J.E.B.; Bridenstine, M.; Regensteiner, J.G. Type 2 diabetes mellitus and exercise impairment. Rev. Endocr. Metab. Disord. 2013, 14, 77–86. [Google Scholar] [CrossRef]
- Wahl, M.P.; Scalzo, R.L.; Regensteiner, J.G.; Reusch, J.E.B. Mechanisms of Aerobic Exercise Impairment in Diabetes: A Narrative Review. Front. Endocrinol. (Lausanne) 2018, 9, 181. [Google Scholar] [CrossRef]
- Holloszy, J.O.; Schultz, J.; Kusnierkiewicz, J.; Hagberg, J.M.; Ehsani, A.A. Effects of Exercise on Glucose Tolerance and Insulin Resistance. Acta Med. Scand. 1986, 220, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.T.; Lujan, H.L.; Tonson, A.; Wiseman, R.W.; DiCarlo, S.E. Obesity and inactivity, not hyperglycemia, cause exercise intolerance in individuals with type 2 diabetes: Solving the obesity and inactivity versus hyperglycemia causality dilemma. Med. Hypotheses 2019, 123, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Poitras, V.J.; Bentley, R.F.; Hopkins-Rosseel, D.H.; LaHaye, S.A.; Tschakovsky, M.E. Lack of independent effect of type 2 diabetes beyond characteristic comorbidities and medications on small muscle mass exercising muscle blood flow and exercise tolerance. Physiol. Rep. 2015, 3, e12487. [Google Scholar] [CrossRef] [PubMed]
- Slade, J.M.; Towse, T.F.; Gossain, V.V.; Meyer, R.A. Peripheral microvascular response to muscle contraction is unaltered by early diabetes but decreases with age. J. Appl. Physiol. 2011, 111, 1361–1371. [Google Scholar] [CrossRef] [Green Version]
- DeFronzo, R.A.; Tripathy, D. Skeletal Muscle Insulin Resistance Is the Primary Defect in Type 2 Diabetes. Diabetes Care 2009, 32, S157–S163. [Google Scholar] [CrossRef] [Green Version]
- Wahren, J.; Felig, P.; Hagenfeldt, L. Physical exercise and fuel homeostasis in diabetes mellitus. Diabetologia 1978, 14, 213–222. [Google Scholar] [CrossRef] [Green Version]
- Sahlin, K. Muscle glucose metabolism during exercise. Ann. Med. 1990, 22, 85–89. [Google Scholar]
- Richter, E.A.; Hargreaves, M. Exercise, GLUT4, and Skeletal Muscle Glucose Uptake. Physiol. Rev. 2013, 93, 993–1017. [Google Scholar] [CrossRef] [Green Version]
- Wasserman, D.H. Four grams of glucose. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E11–E21. [Google Scholar] [CrossRef]
- Heath, G.W.; Gavin, J.R.; Hinderliter, J.M.; Hagberg, J.M.; Bloomfield, S.A.; Holloszy, J.O. Effects of exercise and lack of exercise on glucose tolerance and insulin sensitivity. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1983, 55, 512–517. [Google Scholar] [CrossRef]
- Holloszy, J.O. Regulation of Mitochondrial Biogenesis and GLUT4 Expression by Exercise. In Comprehensive Physiology; American Cancer Society: Atlanta, GA, USA, 2011; pp. 921–940. ISBN 978-0-470-65071-4. [Google Scholar]
- Knowler, W.C.; Barrett-Connor, E.; Fowler, S.E.; Hamman, R.F.; Lachin, J.M.; Walker, E.A.; Nathan, D.M.; Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N. Engl. J. Med. 2002, 346, 393–403. [Google Scholar] [PubMed]
- Lemaster, K.A.; Frisbee, S.J.; Dubois, L.; Tzemos, N.; Wu, F.; Lewis, M.T.; Wiseman, R.W.; Frisbee, J.C. Chronic atorvastatin and exercise can partially reverse established skeletal muscle microvasculopathy in metabolic syndrome. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H855–H870. [Google Scholar] [CrossRef] [PubMed]
- Mikus, C.R.; Oberlin, D.J.; Libla, J.; Boyle, L.J.; Thyfault, J.P. Glycaemic control is improved by 7 days of aerobic exercise training in patients with type 2 diabetes. Diabetologia 2012, 55, 1417–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanford, K.I.; Goodyear, L.J. Exercise and type 2 diabetes: Molecular mechanisms regulating glucose uptake in skeletal muscle. Adv. Physiol. Educ. 2014, 38, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.A.; Foley, J.M. Cellular Processes Integrating the Metabolic Response to Exercise. In Comprehensive Physiology; American Cancer Society: Atlanta, GA, USA, 1996; pp. 841–869. ISBN 978-0-470-65071-4. [Google Scholar]
- Fitts, R.H.; Booth, F.W.; Winder, W.W.; Holloszy, J.O. Skeletal muscle respiratory capacity, endurance, and glycogen utilization. Am. J. Physiol. 1975, 228, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Holloszy, J.O. Biochemical Adaptations in Muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J. Biol. Chem. 1967, 242, 2278–2282. [Google Scholar]
- Meex, R.C.R.; Schrauwen-Hinderling, V.B.; Moonen-Kornips, E.; Schaart, G.; Mensink, M.; Phielix, E.; van de Weijer, T.; Sels, J.-P.; Schrauwen, P.; Hesselink, M.K.C. Restoration of muscle mitochondrial function and metabolic flexibility in type 2 diabetes by exercise training is paralleled by increased myocellular fat storage and improved insulin sensitivity. Diabetes 2010, 59, 572–579. [Google Scholar] [CrossRef]
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of Mitochondria in Human Skeletal Muscle in Type 2 Diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar] [CrossRef] [Green Version]
- Lowell, B.B.; Shulman, G.I. Mitochondrial dysfunction and type 2 diabetes. Science 2005, 307, 384–387. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.-S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Ferguson, S.J. Bioenergetics, 4th ed.; Academic Press: Amsterdam, The Netherlands, 2013; ISBN 978-0-12-388431-2. [Google Scholar]
- Koebmann, B.J.; Westerhoff, H.V.; Snoep, J.L.; Nilsson, D.; Jensen, P.R. The Glycolytic Flux in Escherichia coli Is Controlled by the Demand for ATP. J. Bacteriol. 2002, 184, 3909–3916. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Jeneson, J.A.; Wiseman, R.W.; Westerhoff, H.V.; Kushmerick, M.J. The signal transduction function for oxidative phosphorylation is at least second order in ADP. J. Biol. Chem. 1996, 271, 27995–27998. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.; Westerhoff, H.V. Sigmoidal relation between mitochondrial respiration and log ([ATP]/[ADP])out under conditions of extramitochondrial ATP utilization. Implications for the control and thermodynamics of oxidative phosphorylation. Biochemistry 1988, 27, 7832–7840. [Google Scholar] [CrossRef]
- Hart, C.R.; Lanza, I.R. Mitochondrial ADP Sensitivity and Transport: New Insights into Diet-Induced Mitochondrial Impairments. Diabetes 2018, 67, 2152–2153. [Google Scholar] [CrossRef]
- Lerner, E.; Shug, A.L.; Elson, C.; Shrago, E. Reversible Inhibition of Adenine Nucleotide Translocation by Long Chain Fatty Acyl Coenzyme A Esters in Liver Mitochondria of Diabetic and Hibernating Animals. J. Biol. Chem. 1972, 247, 1513–1519. [Google Scholar]
- Ludzki, A.; Paglialunga, S.; Smith, B.K.; Herbst, E.A.F.; Allison, M.K.; Heigenhauser, G.J.; Neufer, P.D.; Holloway, G.P. Rapid Repression of ADP Transport by Palmitoyl-CoA Is Attenuated by Exercise Training in Humans: A Potential Mechanism to Decrease Oxidative Stress and Improve Skeletal Muscle Insulin Signaling. Diabetes 2015, 64, 2769–2779. [Google Scholar] [CrossRef] [Green Version]
- Miotto, P.M.; LeBlanc, P.J.; Holloway, G.P. High-Fat Diet Causes Mitochondrial Dysfunction as a Result of Impaired ADP Sensitivity. Diabetes 2018, 67, 2199–2205. [Google Scholar] [CrossRef] [Green Version]
- Watt, I.N.; Montgomery, M.G.; Runswick, M.J.; Leslie, A.G.W.; Walker, J.E. Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria. Proc. Natl. Acad. Sci. USA 2010, 107, 16823–16827. [Google Scholar] [CrossRef] [Green Version]
- Kasper, J.D.; Meyer, R.A.; Beard, D.A.; Wiseman, R.W. Effects of altered Pyruvate Dehydrogenase Activity on contracting skeletal muscle bioenergetics. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 316, R76–R86. [Google Scholar] [CrossRef]
- Timmons, J.A.; Poucher, S.M.; Constantin-Teodosiu, D.; Macdonald, I.A.; Greenhaff, P.L. Metabolic responses from rest to steady state determine contractile function in ischemic skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 1997, 273, E233–E238. [Google Scholar] [CrossRef] [PubMed]
- Vinnakota, K.C.; Singhal, A.; Van den Bergh, F.; Bagher-Oskouei, M.; Wiseman, R.W.; Beard, D.A. Open-Loop Control of Oxidative Phosphorylation in Skeletal and Cardiac Muscle Mitochondria by Ca(2.). Biophys. J. 2016, 110, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Wollenman, L.C.; Vander Ploeg, M.R.; Miller, M.L.; Zhang, Y.; Bazil, J.N. The effect of respiration buffer composition on mitochondrial metabolism and function. PLoS ONE 2017, 12, e0187523. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, C.G.R.; Kane, D.A.; Lanza, I.R.; Neufer, P.D. Methods for Assessing Mitochondrial Function in Diabetes. Diabetes 2013, 62, 1041–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jørgensen, W.; Jelnes, P.; Rud, K.A.; Hansen, L.L.; Grunnet, N.; Quistorff, B. Progression of type 2 diabetes in GK rats affects muscle and liver mitochondria differently: Pronounced reduction of complex II flux is observed in liver only. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E515–E523. [Google Scholar] [CrossRef]
- Lewis, M.T.; Kasper, J.D.; Bazil, J.N.; Frisbee, J.C.; Wiseman, R.W. Skeletal muscle energetics are compromised only during high-intensity contractions in the Goto-Kakizaki rat model of type 2 diabetes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 317, R356–R368. [Google Scholar] [CrossRef]
- Fisher-Wellman, K.H.; Davidson, M.T.; Narowski, T.M.; Lin, C.-T.; Koves, T.R.; Muoio, D.M. Mitochondrial Diagnostics: A Multiplexed Assay Platform for Comprehensive Assessment of Mitochondrial Energy Fluxes. Cell Rep. 2018, 24, 3593–3606.e10. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.; Nelson, M.D.; Hamaoka, T.; Jacobs, R.A.; Pearson, J.; Subudhi, A.W.; Jenkins, N.T.; Bartlett, M.F.; Fitzgerald, L.F.; Miehm, J.D.; et al. Commentaries on Viewpoint: Principles, insights, and potential pitfalls of the noninvasive determination of muscle oxidative capacity by near-infrared spectroscopy. J. Appl. Physiol. 2018, 124, 249–255. [Google Scholar]
- Hamaoka, T.; McCully, K.K.; Quaresima, V.; Yamamoto, K.; Chance, B. Near-infrared spectroscopy/imaging for monitoring muscle oxygenation and oxidative metabolism in healthy and diseased humans. JBO 2007, 12, 062105. [Google Scholar] [CrossRef]
- Koga, S.; Rossiter, H.B.; Heinonen, I.; Musch, T.I.; Poole, D.C. Dynamic Heterogeneity of Exercising Muscle Blood Flow and O2 Utilization. Med. Sci. Sports Exerc. 2014, 46, 860–876. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.E.; Southern, W.M.; Reynolds, M.A.; McCully, K.K. A cross-validation of near-infrared spectroscopy measurements of skeletal muscle oxidative capacity with phosphorus magnetic resonance spectroscopy. J. Appl. Physiol. 2013, 115, 1757–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, C.R.; Layec, G.; Trinity, J.D.; Le Fur, Y.; Gifford, J.R.; Clifton, H.L.; Richardson, R.S. Oxygen availability and skeletal muscle oxidative capacity in patients with peripheral arterial disease: Implications from in vivo and in vitro assessments. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H897–H909. [Google Scholar] [CrossRef] [PubMed]
- Jeneson, J.A.; Wiseman, R.W.; Kushmerick, M.J. Non-invasive quantitative 31P MRS assay of mitochondrial function in skeletal muscle in situ. Mol. Cell. Biochem. 1997, 174, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Kemp, G.J. Quantifying Skeletal Muscle Mitochondrial Function In Vivo by 31P Magnetic Resonance Spectroscopy. In Mitochondrial Dysfunction Caused by Drugs and Environmental Toxicants; Will, Y., Dykens, J.A., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2018; pp. 443–456. ISBN 978-1-119-32972-5. [Google Scholar]
- Meyer, R.A.; Foley, J.M. Testing models of respiratory control in skeletal muscle. Med. Sci. Sports Exerc. 1994, 26, 52–57. [Google Scholar] [CrossRef]
- Rossiter, H.B.; Ward, S.A.; Kowalchuk, J.M.; Howe, F.A.; Griffiths, J.R.; Whipp, B.J. Dynamic asymmetry of phosphocreatine concentration and O2 uptake between the on- and off-transients of moderate- and high-intensity exercise in humans. J. Physiol. 2002, 541, 991–1002. [Google Scholar] [CrossRef]
- Adams, G.R.; Foley, J.M.; Meyer, R.A. Muscle buffer capacity estimated from pH changes during rest-to-work transitions. J. Appl. Physiol. 1990, 69, 968–972. [Google Scholar] [CrossRef] [Green Version]
- Meyer, R.A.; Sweeney, H.L.; Kushmerick, M.J. A simple analysis of the “phosphocreatine shuttle. Am. J. Physiol. 1984, 246, C365–C377. [Google Scholar] [CrossRef]
- Lawson, J.W.; Veech, R.L. Effects of pH and free Mg2+ on the Keq of the creatine kinase reaction and other phosphate hydrolyses and phosphate transfer reactions. J. Biol. Chem. 1979, 254, 6528–6537. [Google Scholar]
- Sundberg, C.W.; Prost, R.W.; Fitts, R.H.; Hunter, S.K. Bioenergetic basis for the increased fatigability with ageing. J. Physiol. 2019, 597, 4943–4957. [Google Scholar] [CrossRef]
- Sundberg, C.W.; Fitts, R.H. Bioenergetic basis of skeletal muscle fatigue. Curr. Opin. Physiol. 2019, 10, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, F.; Wang, H.; Wu, W.; Zhang, X.; Tian, C.; Yu, H.; Liu, R.; Zhu, B.; Zhang, B.; et al. Non-invasive assessment of phosphate metabolism and oxidative capacity in working skeletal muscle in healthy young Chinese volunteers using (31)P Magnetic Resonance Spectroscopy. PeerJ 2016, 4, e2259. [Google Scholar] [CrossRef] [PubMed]
- Paganini, A.T.; Foley, J.M.; Meyer, R.A. Linear dependence of muscle phosphocreatine kinetics on oxidative capacity. Am. J. Physiol. 1997, 272, C501–C510. [Google Scholar] [CrossRef] [PubMed]
- McCully, K.K.; Fielding, R.A.; Evans, W.J.; Leigh, J.S.; Posner, J.D. Relationships between in vivo and in vitro measurements of metabolism in young and old human calf muscles. J. Appl. Physiol. 1993, 75, 813–819. [Google Scholar] [CrossRef]
- van den Broek, N.M.A.; De Feyter, H.M.M.L.; de Graaf, L.; Nicolay, K.; Prompers, J.J. Intersubject differences in the effect of acidosis on phosphocreatine recovery kinetics in muscle after exercise are due to differences in proton efflux rates. Am. J. Physiol. Cell Physiol. 2007, 293, C228–C237. [Google Scholar] [CrossRef] [Green Version]
- Forbes, S.C.; Paganini, A.T.; Slade, J.M.; Towse, T.F.; Meyer, R.A. Phosphocreatine recovery kinetics following low- and high-intensity exercise in human triceps surae and rat posterior hindlimb muscles. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R161–R170. [Google Scholar] [CrossRef] [Green Version]
- Harkema, S.J.; Meyer, R.A. Effect of acidosis on control of respiration in skeletal muscle. Am. J. Physiol. 1997, 272, C491–C500. [Google Scholar] [CrossRef]
- Jubrias, S.A.; Crowther, G.J.; Shankland, E.G.; Gronka, R.K.; Conley, K.E. Acidosis inhibits oxidative phosphorylation in contracting human skeletal muscle in vivo. J. Physiol. 2003, 553, 589–599. [Google Scholar] [CrossRef]
- Layec, G.; Malucelli, E.; Le Fur, Y.; Manners, D.; Yashiro, K.; Testa, C.; Cozzone, P.J.; Iotti, S.; Bendahan, D. Effects of exercise-induced intracellular acidosis on the phosphocreatine recovery kinetics: A 31P MRS study in three muscle groups in humans. NMR Biomed. 2013, 26, 1403–1411. [Google Scholar] [CrossRef]
- McCully, K.K.; Smith, S.; Rajaei, S.; Leigh, J.S.; Natelson, B.H. Muscle metabolism with blood flow restriction in chronic fatigue syndrome. J. Appl. Physiol. 2004, 96, 871–878. [Google Scholar] [CrossRef]
- Walsh, B.; Tiivel, T.; Tonkonogi, M.; Sahlin, K. Increased concentrations of P(i) and lactic acid reduce creatine-stimulated respiration in muscle fibers. J. Appl. Physiol. 2002, 92, 2273–2276. [Google Scholar] [CrossRef] [PubMed]
- Walter, G.; Vandenborne, K.; McCully, K.K.; Leigh, J.S. Noninvasive measurement of phosphocreatine recovery kinetics in single human muscles. Am. J. Physiol. 1997, 272, C525–C534. [Google Scholar] [CrossRef] [PubMed]
- Ouriel, K. Peripheral arterial disease. Lancet 2001, 358, 1257–1264. [Google Scholar] [CrossRef]
- Goodwin, M.L.; Harris, J.E.; Hernández, A.; Gladden, L.B. Blood Lactate Measurements and Analysis during Exercise: A Guide for Clinicians. J. Diabetes Sci. Technol. 2007, 1, 558–569. [Google Scholar] [CrossRef] [Green Version]
- Cree-Green, M.; Scalzo, R.L.; Harrall, K.; Newcomer, B.R.; Schauer, I.E.; Huebschmann, A.G.; McMillin, S.; Brown, M.S.; Orlicky, D.; Knaub, L.; et al. Supplemental Oxygen Improves In Vivo Mitochondrial Oxidative Phosphorylation Flux in Sedentary Obese Adults With Type 2 Diabetes. Diabetes 2018, 67, 1369–1379. [Google Scholar] [CrossRef] [Green Version]
- Frisbee, J.C. Impaired skeletal muscle perfusion in obese Zucker rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R1124–R1134. [Google Scholar] [CrossRef] [Green Version]
- Haseler, L.J.; Lin, A.; Hoff, J.; Richardson, R.S. Oxygen availability and PCr recovery rate in untrained human calf muscle: Evidence of metabolic limitation in normoxia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R2046–R2051. [Google Scholar] [CrossRef]
- Larsen, S.; Ara, I.; Rabøl, R.; Andersen, J.L.; Boushel, R.; Dela, F.; Helge, J.W. Are substrate use during exercise and mitochondrial respiratory capacity decreased in arm and leg muscle in type 2 diabetes? Diabetologia 2009, 52, 1400–1408. [Google Scholar] [CrossRef] [Green Version]
- Wilkerson, D.P.; Poole, D.C.; Jones, A.M.; Fulford, J.; Mawson, D.M.; Ball, C.I.; Shore, A.C. Older type 2 diabetic males do not exhibit abnormal pulmonary oxygen uptake and muscle oxygen utilization dynamics during submaximal cycling exercise. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R685–R692. [Google Scholar] [CrossRef]
- Booth, F.W.; Roberts, C.K.; Thyfault, J.P.; Ruegsegger, G.N.; Toedebusch, R.G. Role of Inactivity in Chronic Diseases: Evolutionary Insight and Pathophysiological Mechanisms. Physiol. Rev. 2017, 97, 1351–1402. [Google Scholar] [CrossRef]
- Heo, J.-W.; No, M.-H.; Park, D.-H.; Kang, J.-H.; Seo, D.Y.; Han, J.; Neufer, P.D.; Kwak, H.-B. Effects of exercise on obesity-induced mitochondrial dysfunction in skeletal muscle. Korean J. Physiol. Pharm. 2017, 21, 567–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.; Bruce, C.R.; Beale, S.M.; Hoehn, K.L.; So, T.; Rolph, M.S.; Cooney, G.J. Excess Lipid Availability Increases Mitochondrial Fatty Acid Oxidative Capacity in Muscle. Diabetes 2007, 56, 2085–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamman, R.F.; Wing, R.R.; Edelstein, S.L.; Lachin, J.M.; Bray, G.A.; Delahanty, L.; Hoskin, M.; Kriska, A.M.; Mayer-Davis, E.J.; Pi-Sunyer, X.; et al. Effect of Weight Loss With Lifestyle Intervention on Risk of Diabetes. Diabetes Care 2006, 29, 2102–2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnard, C.; Durand, A.; Peyrol, S.; Chanseaume, E.; Chauvin, M.-A.; Morio, B.; Vidal, H.; Rieusset, J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J. Clin. Investig. 2008, 118, 789–800. [Google Scholar] [CrossRef]
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.-T.; Price, J.W.; Kang, L.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investig. 2009, 119, 573–581. [Google Scholar] [CrossRef]
- Konopka, A.R.; Asante, A.; Lanza, I.R.; Robinson, M.M.; Johnson, M.L.; Man, C.D.; Cobelli, C.; Amols, M.H.; Irving, B.A.; Nair, K.S. Defects in Mitochondrial Efficiency and H2O2 Emissions in Obese Women Are Restored to a Lean Phenotype With Aerobic Exercise Training. Diabetes 2015, 64, 2104–2115. [Google Scholar] [CrossRef] [Green Version]
- Coyle, E.F.; Martin, W.H.; Sinacore, D.R.; Joyner, M.J.; Hagberg, J.M.; Holloszy, J.O. Time course of loss of adaptations after stopping prolonged intense endurance training. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1984, 57, 1857–1864. [Google Scholar] [CrossRef]
- Holloszy, J.O.; Coyle, E.F. Adaptations of skeletal muscle to endurance exercise and their metabolic consequences. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1984, 56, 831–838. [Google Scholar] [CrossRef]
- Holloszy, J.O.; Kohrt, W.M. Regulation of carbohydrate and fat metabolism during and after exercise. Annu. Rev. Nutr. 1996, 16, 121–138. [Google Scholar] [CrossRef]
- Holloszy, J.O.; Oscai, L.B.; Don, I.J.; Molé, P.A. Mitochondrial citric acid cycle and related enzymes: Adaptive response to exercise. Biochem. Biophys. Res. Commun. 1970, 40, 1368–1373. [Google Scholar] [CrossRef]
- Hurley, B.; Goldberg, A.P.; Hagberg, J.M.; John, O. Holloszy: An Enduring Legacy in Exercise Physiology, Aging, and Muscle Research. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 588–589. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Leigh, J.S.; Clark, B.J.; Maris, J.; Kent, J.; Nioka, S.; Smith, D. Control of oxidative metabolism and oxygen delivery in human skeletal muscle: A steady-state analysis of the work/energy cost transfer function. Proc Natl Acad Sci USA 1985, 82, 8384–8388. [Google Scholar] [CrossRef] [PubMed]
- Dudley, G.A.; Tullson, P.C.; Terjung, R.L. Influence of mitochondrial content on the sensitivity of respiratory control. J. Biol. Chem. 1987, 262, 9109–9114. [Google Scholar] [PubMed]
- Meyer, R.A. A linear model of muscle respiration explains monoexponential phosphocreatine changes. Am. J. Physiol. 1988, 254, C548–C553. [Google Scholar] [CrossRef]
- Meyer, R.A. Linear dependence of muscle phosphocreatine kinetics on total creatine content. Am. J. Physiol. Cell Physiol. 1989, 257, C1149–C1157. [Google Scholar] [CrossRef]
- Henriksson, J.; Reitman, J.S. Time course of changes in human skeletal muscle succinate dehydrogenase and cytochrome oxidase activities and maximal oxygen uptake with physical activity and inactivity. Acta Physiol. Scand. 1977, 99, 91–97. [Google Scholar] [CrossRef]
- Katzmarzyk, P.T. Physical Activity, Sedentary Behavior, and Health: Paradigm Paralysis or Paradigm Shift? Diabetes 2010, 59, 2717–2725. [Google Scholar] [CrossRef] [Green Version]
- Katzmarzyk, P.T.; Church, T.S.; Craig, C.L.; Bouchard, C. Sitting time and mortality from all causes, cardiovascular disease, and cancer. Med. Sci. Sports Exerc. 2009, 41, 998–1005. [Google Scholar] [CrossRef]
- Credeur, D.P.; Reynolds, L.J.; Holwerda, S.W.; Vranish, J.R.; Young, B.E.; Wang, J.; Thyfault, J.P.; Fadel, P.J. Influence of physical inactivity on arterial compliance during a glucose challenge. Exp. Physiol. 2018, 103, 483–494. [Google Scholar] [CrossRef]
- Reynolds, L.J.; Credeur, D.P.; Holwerda, S.W.; Leidy, H.J.; Fadel, P.J.; Thyfault, J.P. Acute inactivity impairs glycemic control but not blood flow to glucose ingestion. Med. Sci. Sports Exerc. 2015, 47, 1087–1094. [Google Scholar] [CrossRef]
- Vranish, J.R.; Young, B.E.; Stephens, B.Y.; Kaur, J.; Padilla, J.; Fadel, P.J. Brief periods of inactivity reduce leg microvascular, but not macrovascular, function in healthy young men. Exp. Physiol. 2018, 103, 1425–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, P.R.; Montgomery, J.A.; Wong, T.S.; Booth, F.W. Cytochrome c protein-synthesis rates and mRNA contents during atrophy and recovery in skeletal muscle. Biochem. J. 1987, 241, 257–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, H.E.; Dudley, G.A.; Hather, B.; Tesch, P.A. Work capacity and metabolic and morphologic characteristics of the human quadriceps muscle in response to unloading. Clin. Physiol. 1993, 13, 337–347. [Google Scholar] [CrossRef]
- Hikida, R.S.; Gollnick, P.D.; Dudley, G.A.; Convertino, V.A.; Buchanan, P. Structural and metabolic characteristics of human skeletal muscle following 30 days of simulated microgravity. Aviat. Space Environ. Med. 1989, 60, 664–670. [Google Scholar]
- Kenny, H.C.; Rudwill, F.; Breen, L.; Salanova, M.; Blottner, D.; Heise, T.; Heer, M.; Blanc, S.; O’Gorman, D.J. Bed rest and resistive vibration exercise unveil novel links between skeletal muscle mitochondrial function and insulin resistance. Diabetologia 2017, 60, 1491–1501. [Google Scholar] [CrossRef]
- Booth, F.W.; Roberts, C.K.; Laye, M.J. Lack of exercise is a major cause of chronic diseases. Compr. Physiol. 2012, 2, 1143–1211. [Google Scholar]
- Lanza, I.R.; Nair, K.S. Muscle mitochondrial changes with aging and exercise. Am. J. Clin. Nutr. 2009, 89, 467S–471S. [Google Scholar] [CrossRef]
- van Tienen, F.H.J.; Praet, S.F.E.; de Feyter, H.M.; van den Broek, N.M.; Lindsey, P.J.; Schoonderwoerd, K.G.C.; de Coo, I.F.M.; Nicolay, K.; Prompers, J.J.; Smeets, H.J.M.; et al. Physical Activity Is the Key Determinant of Skeletal Muscle Mitochondrial Function in Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2012, 97, 3261–3269. [Google Scholar] [CrossRef]
- Casaburi, R.; Patessio, A.; Ioli, F.; Zanaboni, S.; Donner, C.F.; Wasserman, K. Reductions in Exercise Lactic Acidosis and Ventilation as a Result of Exercise Training in Patients with Obstructive Lung Disease. Am. Rev. Respir. Dis. 1991, 143, 9–18. [Google Scholar] [CrossRef]
- Mlinac, M.E.; Feng, M.C. Assessment of Activities of Daily Living, Self-Care, and Independence. Arch. Clin. Neuropsychol. 2016, 31, 506–516. [Google Scholar] [CrossRef] [Green Version]
- Petersen, K.F.; Befroy, D.; Dufour, S.; Dziura, J.; Ariyan, C.; Rothman, D.L.; Di Pietro, L.; Cline, G.W.; Shulman, G.I. Mitochondrial Dysfunction in the Elderly: Possible Role in Insulin Resistance. Science 2003, 300, 1140–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fealy, C.E.; Mulya, A.; Axelrod, C.L.; Kirwan, J.P. Mitochondrial dynamics in skeletal muscle insulin resistance and type 2 diabetes. Transl. Res. 2018, 202, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Franquesa, A.; Patti, M.-E. Insulin Resistance and Mitochondrial Dysfunction. Adv. Exp. Med. Biol. 2017, 982, 465–520. [Google Scholar] [PubMed]
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 2014, 4, R1–R15. [Google Scholar] [CrossRef]
- Mogensen, M.; Sahlin, K.; Fernström, M.; Glintborg, D.; Vind, B.F.; Beck-Nielsen, H.; Højlund, K. Mitochondrial Respiration Is Decreased in Skeletal Muscle of Patients With Type 2 Diabetes. Diabetes 2007, 56, 1592–1599. [Google Scholar] [CrossRef] [Green Version]
- Phielix, E.; Schrauwen-Hinderling, V.B.; Mensink, M.; Lenaers, E.; Meex, R.; Hoeks, J.; Kooi, M.E.; Moonen-Kornips, E.; Sels, J.-P.; Hesselink, M.K.C.; et al. Lower intrinsic ADP-stimulated mitochondrial respiration underlies in vivo mitochondrial dysfunction in muscle of male type 2 diabetic patients. Diabetes 2008, 57, 2943–2949. [Google Scholar] [CrossRef]
- Ritov, V.B.; Menshikova, E.V.; He, J.; Ferrell, R.E.; Goodpaster, B.H.; Kelley, D.E. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes 2005, 54, 8–14. [Google Scholar] [CrossRef]
- Abdul-Ghani, M.A.; Jani, R.; Chavez, A.; Molina-Carrion, M.; Tripathy, D.; Defronzo, R.A. Mitochondrial reactive oxygen species generation in obese non-diabetic and type 2 diabetic participants. Diabetologia 2009, 52, 574–582. [Google Scholar] [CrossRef] [Green Version]
- Boushel, R.; Gnaiger, E.; Schjerling, P.; Skovbro, M.; Kraunsøe, R.; Dela, F. Patients with type 2 diabetes have normal mitochondrial function in skeletal muscle. Diabetologia 2007, 50, 790–796. [Google Scholar] [CrossRef] [Green Version]
- Hey-Mogensen, M.; Højlund, K.; Vind, B.F.; Wang, L.; Dela, F.; Beck-Nielsen, H.; Fernström, M.; Sahlin, K. Effect of physical training on mitochondrial respiration and reactive oxygen species release in skeletal muscle in patients with obesity and type 2 diabetes. Diabetologia 2010, 53, 1976–1985. [Google Scholar] [CrossRef] [Green Version]
- Scheuermann-Freestone, M.; Madsen, P.L.; Manners, D.; Blamire, A.M.; Buckingham, R.E.; Styles, P.; Radda, G.K.; Neubauer, S.; Clarke, K. Abnormal Cardiac and Skeletal Muscle Energy Metabolism in Patients With Type 2 Diabetes. Circulation 2003, 107, 3040–3046. [Google Scholar] [CrossRef] [PubMed]
- Schrauwen-Hinderling, V.B.; Kooi, M.E.; Hesselink, M.K.C.; Jeneson, J.A.L.; Backes, W.H.; van Echteld, C.J.A.; van Engelshoven, J.M.A.; Mensink, M.; Schrauwen, P. Impaired in vivo mitochondrial function but similar intramyocellular lipid content in patients with type 2 diabetes mellitus and BMI-matched control subjects. Diabetologia 2007, 50, 113–120. [Google Scholar] [CrossRef] [PubMed]
- De Feyter, H.M.; Lenaers, E.; Houten, S.M.; Schrauwen, P.; Hesselink, M.K.; Wanders, R.J.A.; Nicolay, K.; Prompers, J.J. Increased intramyocellular lipid content but normal skeletal muscle mitochondrial oxidative capacity throughout the pathogenesis of type 2 diabetes. FASEB J. 2008, 22, 3947–3955. [Google Scholar] [CrossRef]
- Praet, S.F.E.; De Feyter, H.M.M.; Jonkers, R.A.M.; Nicolay, K.; van Pul, C.; Kuipers, H.; van Loon, L.J.C.; Prompers, J.J. 31P MR spectroscopy and in vitro markers of oxidative capacity in type 2 diabetes patients. MAGMA 2006, 19, 321–331. [Google Scholar] [CrossRef]
- Bratic, I.; Trifunovic, A. Mitochondrial energy metabolism and ageing. Biochim. Biophys. Acta (BBA) Bioenerg. 2010, 1797, 961–967. [Google Scholar] [CrossRef] [Green Version]
- Vanderwall, C.; Randall Clark, R.; Eickhoff, J.; Carrel, A.L. BMI is a poor predictor of adiposity in young overweight and obese children. BMC Pediatr. 2017, 17, 135. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H.; Liu, J.; Ma, J. [The association between body mass index, waist circumference with body fat percent, and abdominal fat rate in overweight and obese pupils]. Zhonghua Yu Fang Yi Xue Za Zhi 2013, 47, 603–607. [Google Scholar]
- Widhalm, K.; Schönegger, K. BMI: Does it really reflect body fat mass? J. Pediatr. 1999, 134, 522–523. [Google Scholar] [CrossRef]
- De Feyter, H.M.; van den Broek, N.M.A.; Praet, S.F.E.; Nicolay, K.; van Loon, L.J.C.; Prompers, J.J. Early or advanced stage type 2 diabetes is not accompanied by in vivo skeletal muscle mitochondrial dysfunction. Eur. J. Endocrinol. 2008, 158, 643–653. [Google Scholar] [CrossRef] [Green Version]
- Challiss, R.A.; Vranic, M.; Radda, G.K. Bioenergetic changes during contraction and recovery in diabetic rat skeletal muscle. Am. J. Physiol. 1989, 256, E129–E137. [Google Scholar] [CrossRef]
- Cieslar, J.H.; Dobson, G.P. Free [ADP] and Aerobic Muscle Work Follow at Least Second Order Kinetics in Rat Gastrocnemius in Vivo. J. Biol. Chem. 2000, 275, 6129–6134. [Google Scholar] [CrossRef] [PubMed]
- Giannesini, B.; Izquierdo, M.; Le Fur, Y.; Cozzone, P.J.; Fingerle, J.; Himber, J.; Künnecke, B.; Von Kienlin, M.; Bendahan, D. New experimental setup for studying strictly noninvasively skeletal muscle function in rat using 1H-magnetic resonance (MR) imaging and 31P-MR spectroscopy. Magn. Reson. Med. 2005, 54, 1058–1064. [Google Scholar] [CrossRef] [PubMed]
- Lenaers, E.; De Feyter, H.M.; Hoeks, J.; Schrauwen, P.; Schaart, G.; Nabben, M.; Nicolay, K.; Prompers, J.J.; Hesselink, M.K.C. Adaptations in mitochondrial function parallel, but fail to rescue, the transition to severe hyperglycemia and hyperinsulinemia: A study in Zucker diabetic fatty rats. Obesity (Silver Spring) 2010, 18, 1100–1107. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C. Fiber Types in Mammalian Skeletal Muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, J.A.; Taylor, A. Fatigue of maintained voluntary muscle contraction in man. J. Physiol. 1972, 220, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ameredes, B.T.; Brechue, W.F.; Stainsby, W.N. Mechanical and metabolic determination of VO2 and fatigue during repetitive isometric contractions in situ. J. Appl. Physiol. 1998, 84, 1909–1916. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, K.; Ramarao, P. Animal models in type 2 diabetes research: An overview. Indian J. Med. Res. 2007, 125, 451. [Google Scholar]
- Peterson, R.G.; Shaw, W.N.; Neel, M.-A.; Little, L.A.; Eichberg, J. Zucker Diabetic Fatty Rat as a Model for Non-insulin-dependent Diabetes Mellitus. ILAR J. 1990, 32, 16–19. [Google Scholar] [CrossRef] [Green Version]
- Shiota, M.; Printz, R.L. Diabetes in Zucker Diabetic Fatty Rat. In Animal Models in Diabetes Research; Joost, H.-G., Al-Hasani, H., Schürmann, A., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 103–123. ISBN 978-1-62703-068-7. [Google Scholar]
- Frisbee, J.C.; Delp, M.D. Vascular function in the metabolic syndrome and the effects on skeletal muscle perfusion: Lessons from the obese Zucker rat. Essays Biochem. 2006, 42, 145–161. [Google Scholar] [CrossRef]
- Beilby, J. Definition of Metabolic Syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association Conference on Scientific Issues Related to Definition. Clin. Biochem. Rev. 2004, 25, 195–198. [Google Scholar]
- Holloway, G.P.; Gurd, B.J.; Snook, L.A.; Lally, J.; Bonen, A. Compensatory Increases in Nuclear PGC1α Protein Are Primarily Associated With Subsarcolemmal Mitochondrial Adaptations in ZDF Rats. Diabetes 2010, 59, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Wessels, B.; Ciapaite, J.; van den Broek, N.M.A.; Houten, S.M.; Nicolay, K.; Prompers, J.J. Pioglitazone treatment restores in vivo muscle oxidative capacity in a rat model of diabetes. Diabetes Obes. Metab. 2015, 17, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Wessels, B.; Ciapaite, J.; van den Broek, N.M.A.; Nicolay, K.; Prompers, J.J. Metformin Impairs Mitochondrial Function in Skeletal Muscle of Both Lean and Diabetic Rats in a Dose-Dependent Manner. PLoS ONE 2014, 9, e100525. [Google Scholar] [CrossRef] [PubMed]
- Bonen, A.; Holloway, G.P.; Tandon, N.N.; Han, X.-X.; McFarlan, J.; Glatz, J.F.C.; Luiken, J.J.F.P. Cardiac and skeletal muscle fatty acid transport and transporters and triacylglycerol and fatty acid oxidation in lean and Zucker diabetic fatty rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1202–R1212. [Google Scholar] [CrossRef] [Green Version]
- van Bree, B.W.J.; Lenaers, E.; Nabben, M.; Briedé, J.J.; Jörgensen, J.A.; Schaart, G.; Schrauwen, P.; Hoeks, J.; Hesselink, M.K.C. A genistein-enriched diet neither improves skeletal muscle oxidative capacity nor prevents the transition towards advanced insulin resistance in ZDF rats. Sci Rep 2016, 6, 22854. [Google Scholar] [CrossRef]
- Sparks, L.M.; Gemmink, A.; Phielix, E.; Bosma, M.; Schaart, G.; Moonen-Kornips, E.; Jörgensen, J.A.; Nascimento, E.B.M.; Hesselink, M.K.C.; Schrauwen, P.; et al. ANT1-mediated fatty acid-induced uncoupling as a target for improving myocellular insulin sensitivity. Diabetologia 2016, 59, 1030–1039. [Google Scholar] [CrossRef] [Green Version]
- Hood, D.A.; Gorski, J.; Terjung, R.L. Oxygen cost of twitch and tetanic isometric contractions of rat skeletal muscle. Am. J. Physiol. 1986, 250, E449–E456. [Google Scholar] [CrossRef]
- Jeneson, J.A.; Westerhoff, H.V.; Brown, T.R.; Van Echteld, C.J.; Berger, R. Quasi-linear relationship between Gibbs free energy of ATP hydrolysis and power output in human forearm muscle. Am. J. Physiol. 1995, 268, C1474–C1484. [Google Scholar] [CrossRef] [Green Version]
- Pörtner, H.O.; Heisler, N.; Grieshaber, M.K. Anaerobiosis and acid-base status in marine invertebrates: A theoretical analysis of proton generation by anaerobic metabolism. J. Comp. Physiol. B 1984, 155, 1–12. [Google Scholar] [CrossRef]
- Roos, A.; Boron, W.F. Intracellular pH. Physiol. Rev. 1981, 61, 296–434. [Google Scholar] [CrossRef]
- Wiseman, R.W.; Ellington, W.R.; Rosanske, R.C. Effects of extracellular pH and D-lactate efflux on regulation of intracellular pH during isotonic contractions in a molluscan muscle: A 31p-nuclear magnetic resonance study. J. Exp. Zool. 1989, 252, 228–236. [Google Scholar] [CrossRef]
- Akash, M.S.; Rehman, K.; Chen, S. Goto-Kakizaki rats: Its suitability as non-obese diabetic animal model for spontaneous type 2 diabetes mellitus. Curr. Diabetes Rev. 2013, 9, 387–396. [Google Scholar] [CrossRef]
- Goto, Y.; Kakizaki, M.; Masaki, N. Spontaneous Diabetes Produced by Selective Breeding of Normal Wistar Rats. Proc. Jpn. Acad. 1975, 51, 80–85. [Google Scholar] [CrossRef] [Green Version]
- Miralles, F.; Portha, B. Early development of beta-cells is impaired in the GK rat model of type 2 diabetes. Diabetes 2001, 50 (Suppl. S1), S84–S88. [Google Scholar] [CrossRef]
- Nie, J.; Xue, B.; Sukumaran, S.; Jusko, W.J.; DuBois, D.C.; Almon, R.R. Differential Muscle Gene Expression as a Function of Disease Progression in Goto-Kakizaki Diabetic Rats. Mol. Cell. Endocrinol. 2011, 338, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Sena, C.M.; Barosa, C.; Nunes, E.; Seiça, R.; Jones, J.G. Sources of endogenous glucose production in the Goto-Kakizaki diabetic rat. Diabetes Metab. 2007, 33, 296–302. [Google Scholar] [CrossRef]
- Portha, B.; Giroix, M.-H.; Tourrel-Cuzin, C.; Le-Stunff, H.; Movassat, J. The GK Rat: A Prototype for the Study of Non-overweight Type 2 Diabetes. In Animal Models in Diabetes Research; Joost, H.-G., Al-Hasani, H., Schürmann, A., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 125–159. ISBN 978-1-62703-067-0. [Google Scholar]
- Alameddine, A.; Fajloun, Z.; Bourreau, J.; Gauquelin-Koch, G.; Yuan, M.; Gauguier, D.; Derbre, S.; Ayer, A.; Custaud, M.A.; Navasiolava, N. The Cardiovascular Effects of Salidroside in the Goto-Kakizaki Diabetic Rat Model. Journal of Phydiology and Pharmacology 2015, 2, 249–257. [Google Scholar]
- Lai, N.; Kummitha, C.; Hoppel, C. Defects in skeletal muscle subsarcolemmal mitochondria in a non-obese model of type 2 diabetes mellitus. PLoS ONE 2017, 12, e0183978. [Google Scholar] [CrossRef]
- Liu, Y.; Mei, X.; Li, J.; Lai, N.; Yu, X. Mitochondrial function assessed by 31P MRS and BOLD MRI in non-obese type 2 diabetic rats. Physiol. Rep. 2016, 4, e12890. [Google Scholar] [CrossRef]
- Macia, M.; Pecchi, E.; Vilmen, C.; Desrois, M.; Lan, C.; Portha, B.; Bernard, M.; Bendahan, D.; Giannesini, B. Insulin Resistance Is Not Associated with an Impaired Mitochondrial Function in Contracting Gastrocnemius Muscle of Goto-Kakizaki Diabetic Rats In Vivo. PLoS ONE 2015, 10, e0129579. [Google Scholar] [CrossRef]
- Hölscher, M.E.; Bode, C.; Bugger, H. Diabetic Cardiomyopathy: Does the Type of Diabetes Matter? Int. J. Mol. Sci. 2016, 17, 2136. [Google Scholar] [CrossRef] [PubMed]
- Laakso, M. Hyperglycemia and cardiovascular disease in type 2 diabetes. Diabetes 1999, 48, 937–942. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.T.; Lujan, H.L.; Wiseman, R.W.; DiCarlo, S.E. The Hypertension Advantage and Natural Selection: Since Type 2 Diabetes Associates with Co-morbidities and Premature Death, Why have the Genetic Variants Remained in the Human Genome? Med. Hypotheses 2019, 129, 109237. [Google Scholar] [CrossRef]
- Roussel, R.; Steg, P.G.; Mohammedi, K.; Marre, M.; Potier, L. Prevention of cardiovascular disease through reduction of glycaemic exposure in type 2 diabetes: A perspective on glucose-lowering interventions. Diabetes Obes. Metab. 2018, 20, 238–244. [Google Scholar] [CrossRef]
- Frisbee, J.C.; Lewis, M.T.; Kasper, J.D.; Chantler, P.D.; Wiseman, R.W. Type 2 diabetes mellitus in the Goto-Kakizaki rat impairs microvascular function and contributes to premature skeletal muscle fatigue. J. Appl. Physiol. 2018, 126, 626–637. [Google Scholar] [CrossRef]
- Rhee, S.Y.; Kim, Y.S. Peripheral Arterial Disease in Patients with Type 2 Diabetes Mellitus. Diabetes Metab. J. 2015, 39, 283–290. [Google Scholar] [CrossRef]
- Bhati, P.; Shenoy, S.; Hussain, M.E. Exercise training and cardiac autonomic function in type 2 diabetes mellitus: A systematic review. Diabetes Metab. Syndr. Clin. Res. Rev. 2018, 12, 69–78. [Google Scholar] [CrossRef]
- Parasoglou, P.; Rao, S.; Slade, J.M. Declining Skeletal Muscle Function in Diabetic Peripheral Neuropathy. Clin. Ther. 2017, 39, 1085–1103. [Google Scholar] [CrossRef]
- Connett, R.J.; Honig, C.R.; Gayeski, T.E.; Brooks, G.A. Defining hypoxia: A systems view of VO2, glycolysis, energetics, and intracellular PO2. J. Appl. Physiol. 1990, 68, 833–842. [Google Scholar] [CrossRef]
- Lanza, I.R.; Tevald, M.A.; Befroy, D.E.; Kent-Braun, J.A. Intracellular energetics and critical PO2 in resting ischemic human skeletal muscle in vivo. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R1415–R1422. [Google Scholar] [CrossRef]
- Marcinek, D.J.; Ciesielski, W.A.; Conley, K.E.; Schenkman, K.A. Oxygen regulation and limitation to cellular respiration in mouse skeletal muscle in vivo. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1900–H1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowell, L.B.; Saltin, B.; Kiens, B.; Christensen, N.J. Is peak quadriceps blood flow in humans even higher during exercise with hypoxemia? Am. J. Physiol. 1986, 251, H1038–H1044. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.M.; Bunch, F.T.; Cavros, N.G.; Dippel, E.J. Multidisciplinary approach to the diagnosis and management of patients with peripheral arterial disease. Clin. Interv. Aging 2015, 10, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Soguel Schenkel, N.; Burdet, L.; de Muralt, B.; Fitting, J.W. Oxygen saturation during daily activities in chronic obstructive pulmonary disease. Eur. Respir. J. 1996, 9, 2584–2589. [Google Scholar] [CrossRef] [PubMed]
- Stainsby, W.N.; Otis, A.B. Blood flow, blood oxygen tension, oxygen uptake, and oxygen transport in skeletal muscle. Am. J. Physiol. 1964, 206, 858–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertinini, I.; Luchinat, C. NMR Paramagnetic Molecules in Biological Systems 1986.
- Richardson, R.S.; Noyszewski, E.A.; Kendrick, K.F.; Leigh, J.S.; Wagner, P.D. Myoglobin O2 desaturation during exercise. Evidence of limited O2 transport. J. Clin. Investig. 1995, 96, 1916–1926. [Google Scholar] [CrossRef]
- Cree-Green, M.; Gupta, A.; Coe, G.V.; Baumgartner, A.D.; Pyle, L.; Reusch, J.E.B.; Brown, M.S.; Newcomer, B.R.; Nadeau, K.J. Insulin resistance in type 2 diabetes youth relates to serum free fatty acids and muscle mitochondrial dysfunction. J. Diabetes Complicat. 2017, 31, 141–148. [Google Scholar] [CrossRef]
- Sunoo, S.; Asano, K.; Mitsumori, F. 31P nuclear magnetic resonance study on changes in phosphocreatine and the intracellular pH in rat skeletal muscle during exercise at various inspired oxygen contents. Eur. J. Appl. Physiol. Occup. Physiol. 1996, 74, 305–310. [Google Scholar] [CrossRef]
- Haseler, L.J.; Hogan, M.C.; Richardson, R.S. Skeletal muscle phosphocreatine recovery in exercise-trained humans is dependent on O2availability. J. Appl. Physiol. 1999, 86, 2013–2018. [Google Scholar] [CrossRef]
- Haseler, L.J.; Lin, A.P.; Richardson, R.S. Skeletal muscle oxidative metabolism in sedentary humans: 31P-MRS assessment of O2 supply and demand limitations. J. Appl. Physiol. 2004, 97, 1077–1081. [Google Scholar] [CrossRef] [PubMed]
- Haseler, L.J.; Richardson, R.S.; Videen, J.S.; Hogan, M.C. Phosphocreatine hydrolysis during submaximal exercise: The effect of FIO2. J. Appl. Physiol. 1998, 85, 1457–1463. [Google Scholar] [CrossRef] [PubMed]
- Hogan, M.C.; Richardson, R.S.; Haseler, L.J. Human muscle performance and PCr hydrolysis with varied inspired oxygen fractions: A 31P-MRS study. J. Appl. Physiol. 1999, 86, 1367–1373. [Google Scholar] [CrossRef]
- Poole, D.C.; Copp, S.W.; Hirai, D.M.; Musch, T.I. Dynamics of muscle microcirculatory and blood-myocyte O2 flux during contractions: Dynamics of muscle microcirculatory O2 flux. Acta Physiol. 2011, 202, 293–310. [Google Scholar] [CrossRef]
- Poole, D.C.; Jones, A.M. Oxygen Uptake Kinetics. In Comprehensive Physiology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; ISBN 978-0-470-65071-4. [Google Scholar]
- Hirai, D.M.; Colburn, T.D.; Craig, J.C.; Hotta, K.; Kano, Y.; Musch, T.I.; Poole, D.C. Skeletal muscle interstitial O2 pressures: Bridging the gap between the capillary and myocyte. Microcirculation 2019, 26, e12497. [Google Scholar] [CrossRef]
- Misra, S.; Shishehbor, M.H.; Takahashi, E.A.; Aronow, H.D.; Brewster, L.P.; Bunte, M.C.; Kim, E.S.H.; Lindner, J.R.; Rich, K.; American Heart Association Council on Peripheral Vascular Disease; et al. Perfusion Assessment in Critical Limb Ischemia: Principles for Understanding and the Development of Evidence and Evaluation of Devices: A Scientific Statement From the American Heart Association. Circulation 2019, 140, e657–e672. [Google Scholar] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assumptions | |
|---|---|
| 1) | Equilibrium of the cytoplasmic creatine kinase reaction |
| 2) | Oxygen and substrate supply are not limiting during recovery |
| 3) | Glycolytic ATP production is negligible during recovery |
| 4) | Mitochondrial ATP:O and basal muscle oxygen consumption (VO2) are each constant |
| 5) | PCr resynthesis accounts for all but a negligible fraction of ATP consumed during recovery |
| 6) | Similar mitochondrial fractional volumes within the measured tissues |
| Model | Summary | |
|---|---|---|
| Genetically developed diabetic animals: | ||
| Obese: | ob/ob mouse | Pros: Develop resembling severe insulin resistance and type 2 diabetes in humans, homogeneous genetic background reduces variability. Cons: Highly genetic-determined diagnosis unlike mostly lifestyle disease in humans, limited and expensive, substantial maintenance often because of disease severity |
| db/db mouse | ||
| Obese Zucker rat | ||
| ZDF rat | ||
| OLETF rat | ||
| Obese rhesus monkey | ||
| Nonobese: | Cohen diabetic rat | |
| GK rat | ||
| Akita mouse | ||
| Diet-induced diabetic animals: | Pros: Diabetes progression similar to diabetes in obese populations overnutrition, toxicity from chemical induction avoided Cons: Generally, take a long time for development, frank hyperglycemia only apparent in certain animal models and therefore confounds development factors | |
| Obese: | Sand rat | |
| C57/BL 6J mouse | ||
| spiny mouse | ||
| Chemical, surgical-induced diabetic animals: | Pros: Controlled onset of diabetic progression, residual insulin secretion better maintains health, inexpensive, recapitulates human with reduced beta cell mass. Cons: Hyperglycemia results mostly because of cytotoxic effect on beta cells versus initial insulin resistance, difficult for long term experiments because of beta cell regeneration, toxic actions on other organ systems | |
| Non-obese: | HFD, low dose STZ rat or mouse | |
| Neonatal STZ rat | ||
| Partial pancreatectomized animals | ||
| Myriad transgenic murine models targeting insulin receptors, glucose transporters, etc. | Pros: Effect of single gene, better understanding of genetic contribution to type 2 diabetes. Cons: Highly expensive, translation to human population unclear | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewis, M.T.; Kasper, J.D.; Bazil, J.N.; Frisbee, J.C.; Wiseman, R.W. Quantification of Mitochondrial Oxidative Phosphorylation in Metabolic Disease: Application to Type 2 Diabetes. Int. J. Mol. Sci. 2019, 20, 5271. https://doi.org/10.3390/ijms20215271
Lewis MT, Kasper JD, Bazil JN, Frisbee JC, Wiseman RW. Quantification of Mitochondrial Oxidative Phosphorylation in Metabolic Disease: Application to Type 2 Diabetes. International Journal of Molecular Sciences. 2019; 20(21):5271. https://doi.org/10.3390/ijms20215271
Chicago/Turabian StyleLewis, Matthew T., Jonathan D. Kasper, Jason N. Bazil, Jefferson C. Frisbee, and Robert W. Wiseman. 2019. "Quantification of Mitochondrial Oxidative Phosphorylation in Metabolic Disease: Application to Type 2 Diabetes" International Journal of Molecular Sciences 20, no. 21: 5271. https://doi.org/10.3390/ijms20215271