Understanding G Protein Selectivity of Muscarinic Acetylcholine Receptors Using Computational Methods


Abstract

1. Introduction
2. Results and Discussion
2.1. Structural Modeling and Generation of Receptor–G Protein Conformational Ensembles
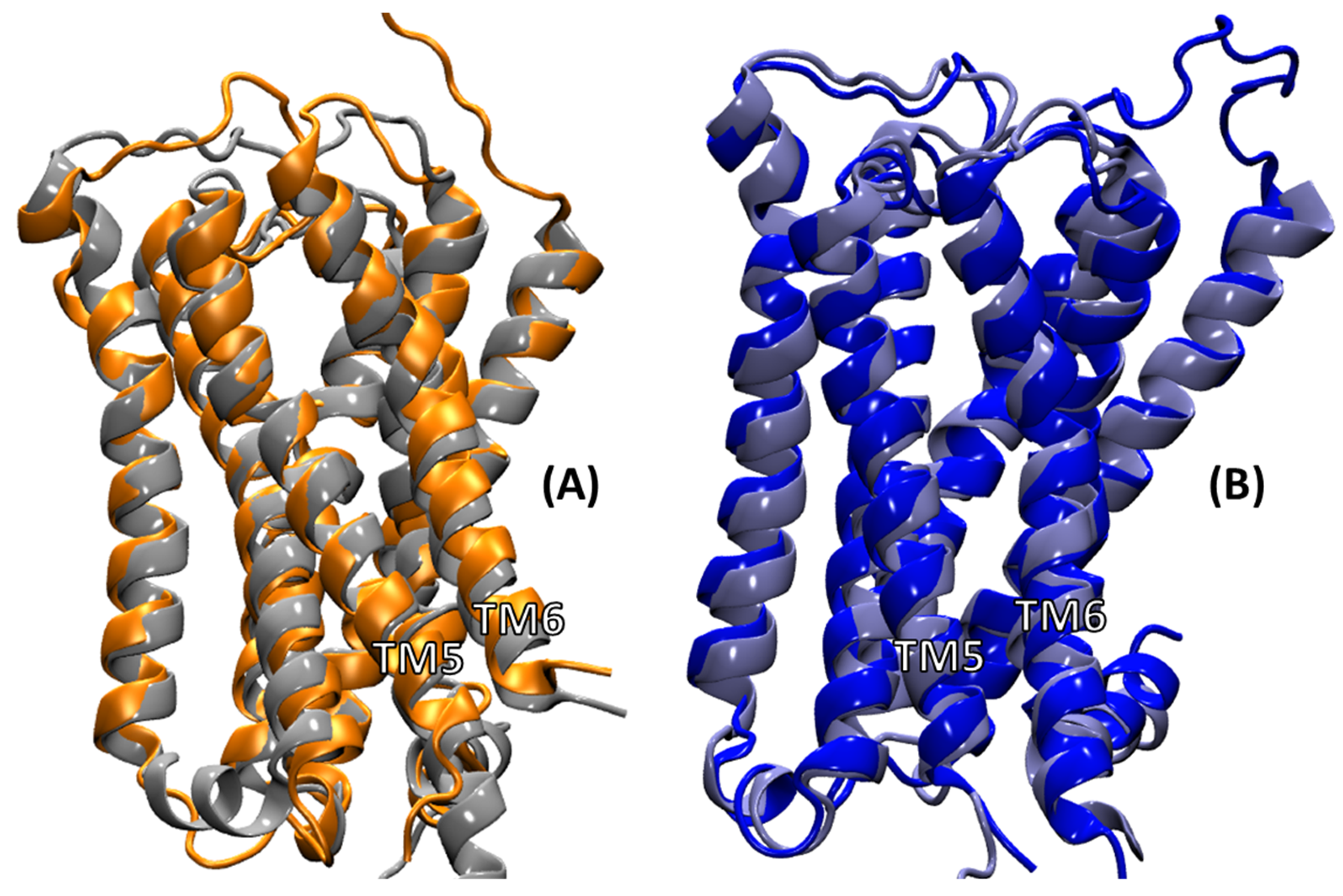
2.2. Comparison of MD Structures with cryo-EM Structures
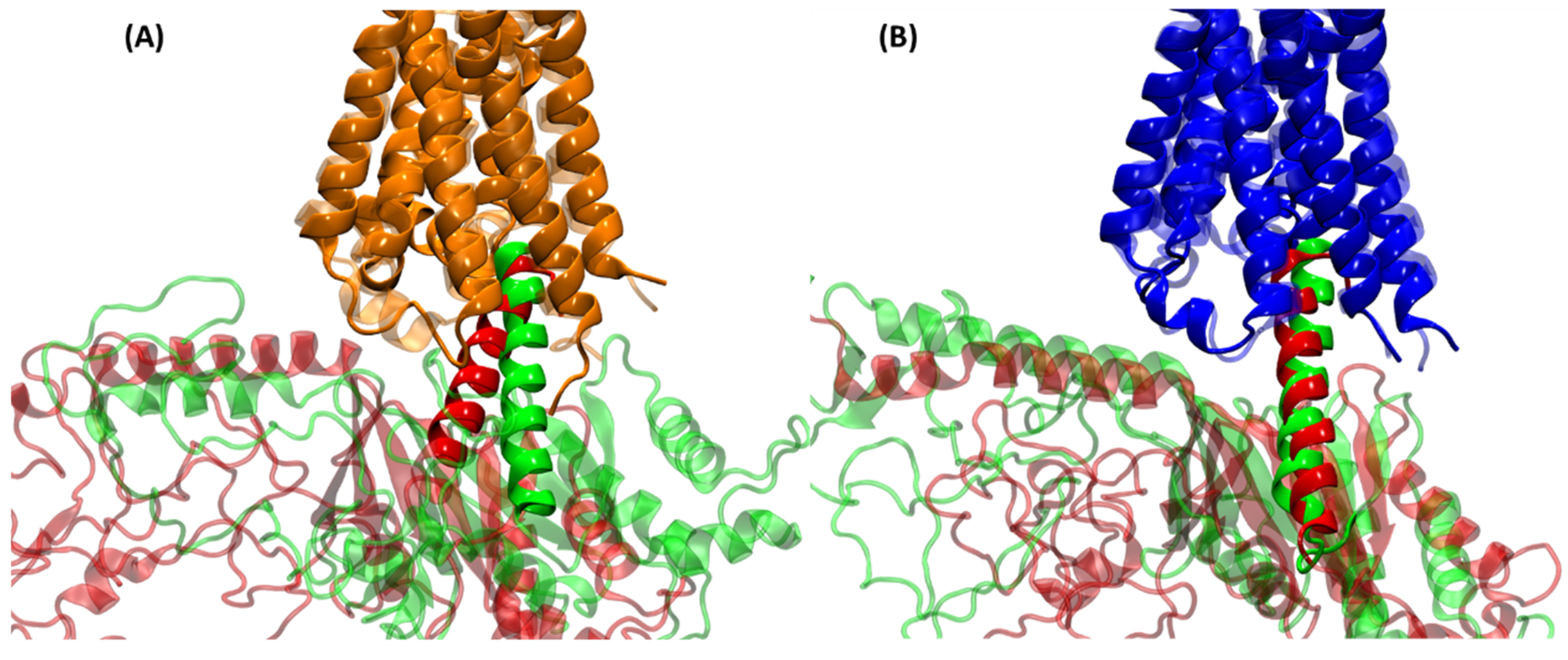
2.3. Structural Comparison of Cognate and Non-Cognate Complexes
2.4. Basis for the G Protein Selectivity of the M1 and M2 Receptors
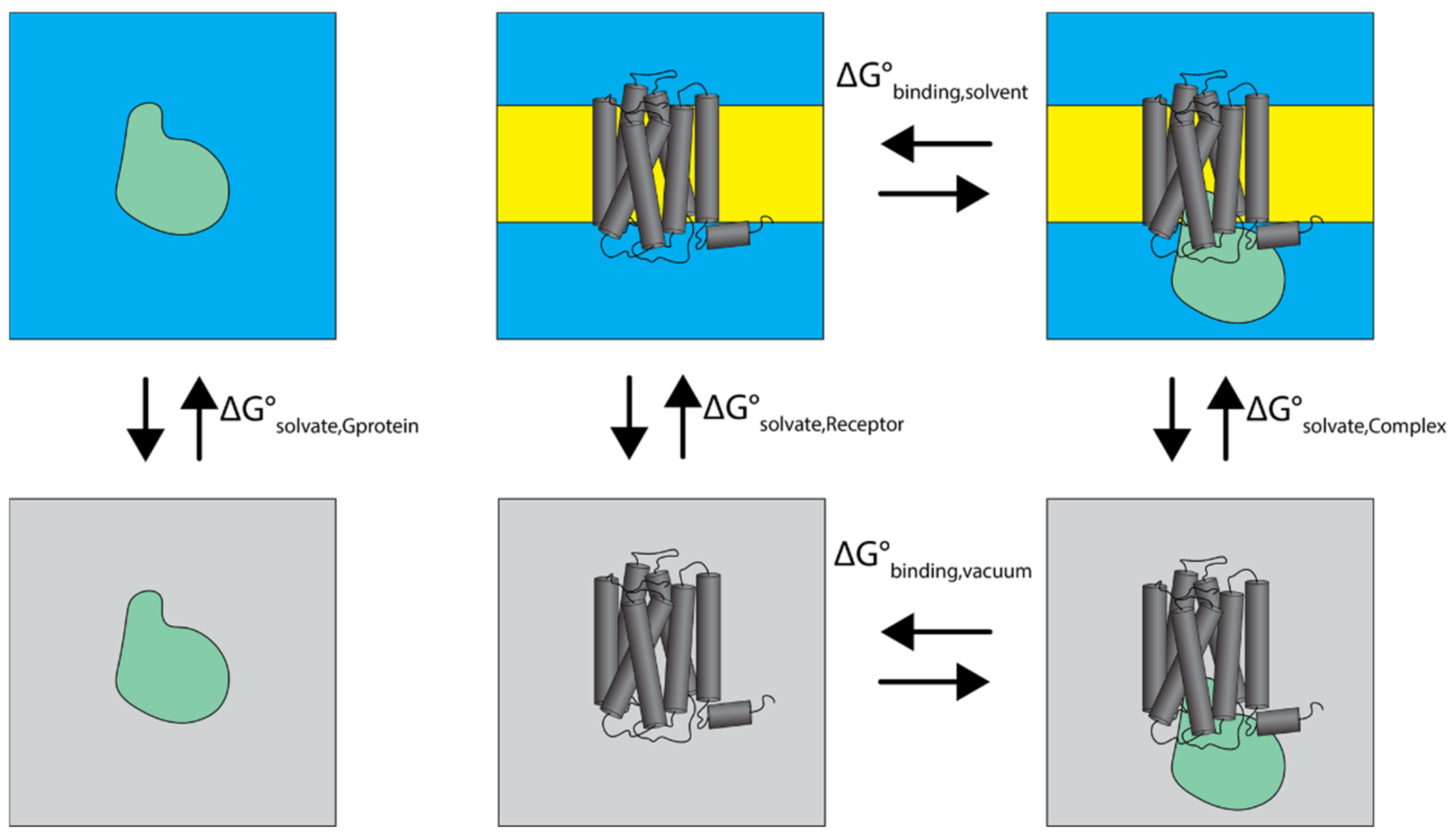
2.4.1. Thermodynamic Analysis of Cognate and Non-Cognate Complexes
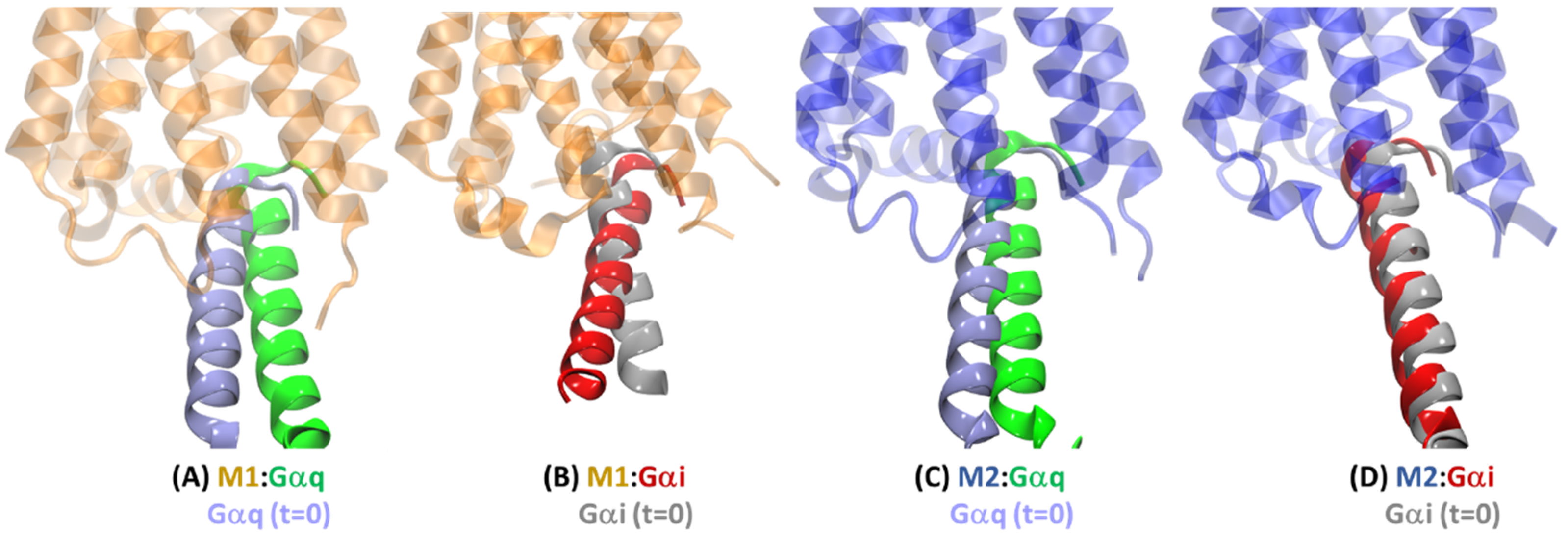
2.4.2. Receptor–G Protein Contacts in Cognate and Non-Cognate Complexes
2.4.3. Intra-Receptor Contacts in Cognate and Non-Cognate Complexes
3. Materials and Methods
3.1. Receptor–G Protein Complex Structure Preparation
3.2. Molecular Dynamics
- The lipids and waters were energy minimized for 2000 steps using a sequence of 1000 steepest descent steps and 1000 conjugate gradient steps while keeping the protein and ligand fixed. This removed any protein–solvent steric clashes and prepared the solvent for the next step.
- The lipids and waters were then relaxed using NPT equilibration MD for a simulation time of 250 ps while keeping the protein and ligand fixed. This allowed for any artificial air bubbles at the protein–solvent interface to be filled in by the solvent.
- The whole molecular system was then energy minimized for 2000 steps (like in Step 1) to allow the protein atoms to adjust to the relaxed solvent.
- The whole molecular system was then slowly heated from 0 K to 310 K at 1 atm pressure over a simulation time of 100 ps.
- The whole system was then relaxed using NPT equilibration at 310 K and 1 atm until the system density was stabilized, which typically occurred within 250 ps.
- Finally, the whole system was equilibrated for 0.5 µs. Simulation snapshots were saved every 10 ps for subsequent structural and thermodynamic analysis.
3.3. Trajectory Analysis
3.4. Binding Free Energy Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| GPCR | G-protein-coupled receptor |
| MD | Molecular dynamics |
| MMPBSA | Molecular mechanics/Poisson–Boltzmann surface area |
| VMD | Visual molecular dynamics |
| RMSD | Root-mean-squared deviation |
References
- Lefkowitz, R.J. Historical review: A brief history and personal retrospective of seven-transmembrane receptors. Trends Pharm. Sci. 2004, 25, 413–422. [Google Scholar] [CrossRef]
- Hill, S.J. G-protein-coupled receptors: Past, present and future. Br. J. Pharm. 2006, 147, S27–S37. [Google Scholar] [CrossRef]
- Galandrin, S.; Oligny-Longpre, G.; Bouvier, M. The evasive nature of drug efficacy: Implications for drug discovery. Trends Pharm. Sci. 2007, 28, 423–430. [Google Scholar] [CrossRef]
- Lundstrom, K. Latest development in drug discovery on G protein-coupled receptors. Curr. Protein Pept. Sci. 2006, 7, 465–470. [Google Scholar]
- Zalewska, M.; Siara, M.; Sajewicz, W. G protein-coupled receptors: Abnormalities in signal transmission, disease states and pharmacotherapy. Acta Pol. Pharm. 2014, 71, 229–243. [Google Scholar]
- Reiter, E.; Lefkowitz, R.J. GRKs and beta-arrestins: Roles in receptor silencing, trafficking and signaling. Trends Endocrinol. Metab. 2006, 17, 159–165. [Google Scholar] [CrossRef]
- Barki-Harrington, L.; Rockman, H.A. Beta-arrestins: Multifunctional cellular mediators. Physiology 2008, 23, 17–22. [Google Scholar] [CrossRef]
- Kenakin, T.; Miller, L.J. Seven Transmembrane Receptors as Shapeshifting Proteins: The Impact of Allosteric Modulation and Functional Selectivity on New Drug Discovery. Pharmacol. Rev. 2010, 62, 265–304. [Google Scholar] [CrossRef]
- Walters, R.W.; Shukla, A.K.; Kovacs, J.J.; Violin, J.D.; DeWire, S.M.; Lam, C.M.; Chen, J.R.; Muehlbauer, M.J.; Whalen, E.J.; Lefkowitz, R.J. beta-Arrestin1 mediates nicotinic acid-induced flushing, but not its antilipolytic effect, in mice. J. Clin. Investig. 2009, 119, 1312–1321. [Google Scholar] [CrossRef]
- Kenakin, T.P. Biased signalling and allosteric machines: New vistas and challenges for drug discovery. Br. J. Pharmacol. 2012, 165, 1659–1669. [Google Scholar] [CrossRef]
- Whalen, E.J.; Rajagopal, S.; Lefkowitz, R.J. Therapeutic potential of beta-arrestin- and G protein-biased agonists. Trends Mol. Med. 2011, 17, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T. Functional selectivity and biased receptor signaling. J. Pharm. Exp. 2011, 336, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Caulfield, M.P.; Birdsall, N.J. International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharm. Rev. 1998, 50, 279–290. [Google Scholar] [PubMed]
- Griffin, M.T.; Figueroa, K.W.; Liller, S.; Ehlert, F.J. Estimation of agonist activity at G protein-coupled receptors: Analysis of M2 muscarinic receptor signaling through Gi/o,Gs, and G15. J. Pharm. Exp. 2007, 321, 1193–1207. [Google Scholar] [CrossRef] [PubMed]
- Michal, P.; El-Fakahany, E.E.; Dolezal, V. Muscarinic M2 receptors directly activate Gq/11 and Gs G-proteins. J. Pharm. Exp. 2007, 320, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Horn, F.; van der Wenden, E.M.; Oliveira, L.; IJzerman, A.P.; Vriend, G. Receptors coupling to G proteins: Is there a signal behind the sequence? Proteins 2000, 41, 448–459. [Google Scholar] [CrossRef]
- Flock, T.; Hauser, A.S.; Lund, N.; Gloriam, D.E.; Balaji, S.; Babu, M.M. Selectivity determinants of GPCR-G-protein binding. Nature 2017, 545, 317–322. [Google Scholar] [CrossRef]
- Yabuki, Y.; Muramatsu, T.; Hirokawa, T.; Mukai, H.; Suwa, M. GRIFFIN: A system for predicting GPCR-G-protein coupling selectivity using a support vector machine and a hidden Markov model. Nucleic Acids Res. 2005, 33, W148–W153. [Google Scholar] [CrossRef]
- Vaidehi, N.; Kenakin, T. The role of conformational ensembles of seven transmembrane receptors in functional selectivity. Curr. Opin. Pharm. 2010, 10, 775–781. [Google Scholar] [CrossRef]
- Abrol, R.; Kim, S.K.; Bray, J.K.; Trzaskowski, B.; Goddard, W.A., 3rd. Conformational ensemble view of G protein-coupled receptors and the effect of mutations and ligand binding. Methods Enzym. 2013, 520, 31–48. [Google Scholar] [CrossRef]
- Manglik, A.; Kobilka, B. The role of protein dynamics in GPCR function: Insights from the beta2AR and rhodopsin. Curr. Opin. Cell Biol. 2014, 27, 136–143. [Google Scholar] [CrossRef]
- Abrol, R.; Trzaskowski, B.; Goddard, W.A.; Nesterov, A.; Olave, I.; Irons, C. Ligand- and mutation-induced conformational selection in the CCR5 chemokine G protein-coupled receptor. Proc. Natl. Acad. Sci. USA 2014, 111, 13040–13045. [Google Scholar] [CrossRef]
- Ahn, K.H.; Scott, C.E.; Abrol, R.; Goddard, W.A.; Kendall, D.A. Computationally-predicted CB1 cannabinoid receptor mutants show distinct patterns of salt-bridges that correlate with their level of constitutive activity reflected in G protein coupling levels, thermal stability, and ligand binding. Proteins 2013, 81, 1304–1317. [Google Scholar] [CrossRef]
- Scott, C.E.; Abrol, R.; Ahn, K.H.; Kendall, D.A.; Goddard, W.A. Molecular basis for dramatic changes in cannabinoid CB1 G protein-coupled receptor activation upon single and double point mutations. Protein Sci. 2013, 22, 101–113. [Google Scholar] [CrossRef]
- Rasmussen, S.G.; DeVree, B.T.; Zou, Y.; Kruse, A.C.; Chung, K.Y.; Kobilka, T.S.; Thian, F.S.; Chae, P.S.; Pardon, E.; Calinski, D.; et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 2011, 477, 549–555. [Google Scholar] [CrossRef]
- Chung, K.Y.; Rasmussen, S.G.; Liu, T.; Li, S.; DeVree, B.T.; Chae, P.S.; Calinski, D.; Kobilka, B.K.; Woods, V.L., Jr.; Sunahara, R.K. Conformational changes in the G protein Gs induced by the beta2 adrenergic receptor. Nature 2011, 477, 611–615. [Google Scholar] [CrossRef]
- Draper-Joyce, C.J.; Khoshouei, M.; Thal, D.M.; Liang, Y.L.; Nguyen, A.T.N.; Furness, S.G.B.; Venugopal, H.; Baltos, J.A.; Plitzko, J.M.; Danev, R.; et al. Structure of the adenosine-bound human adenosine A. Nature 2018, 558, 559–563. [Google Scholar] [CrossRef]
- Maeda, S.; Qu, Q.; Robertson, M.J.; Skiniotis, G.; Kobilka, B.K. Structures of the M1 and M2 muscarinic acetylcholine receptor/G-protein complexes. Science 2019, 364, 552–557. [Google Scholar] [CrossRef]
- Safdari, H.A.; Pandey, S.; Shukla, A.K.; Dutta, S. Illuminating GPCR Signaling by Cryo-EM. Trends Cell Biol. 2018, 28, 591–594. [Google Scholar] [CrossRef]
- Rosenbaum, D.M.; Rasmussen, S.G.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef]
- Hanson, M.A.; Stevens, R.C. Discovery of new GPCR biology: One receptor structure at a time. Structure 2009, 17, 8–14. [Google Scholar] [CrossRef]
- Heifetz, A.; Schertler, G.F.; Seifert, R.; Tate, C.G.; Sexton, P.M.; Gurevich, V.V.; Fourmy, D.; Cherezov, V.; Marshall, F.H.; Storer, R.I.; et al. GPCR structure, function, drug discovery and crystallography: Report from Academia-Industry International Conference (UK Royal Society) Chicheley Hall, 1–2 September 2014. Naunyn Schmiedebergs Arch. Pharm. 2015. [Google Scholar] [CrossRef]
- Garcia-Nafria, J.; Tate, C.G. Cryo-EM structures of GPCRs coupled to Gs, Gi and Go. Mol. Cell. Endocrinol. 2019, 488, 1–13. [Google Scholar] [CrossRef]
- Sandhu, M.; Touma, A.M.; Dysthe, M.; Sadler, F.; Sivaramakrishnan, S.; Vaidehi, N. Conformational plasticity of the intracellular cavity of GPCR-G-protein complexes leads to G-protein promiscuity and selectivity. Proc. Natl. Acad. Sci. USA 2019, 116, 11956–11965. [Google Scholar] [CrossRef]
- Ilyaskina, O.S.; Lemoine, H.; Bunemann, M. Lifetime of muscarinic receptor-G-protein complexes determines coupling efficiency and G-protein subtype selectivity. Proc. Natl. Acad. Sci. USA 2018, 115, 5016–5021. [Google Scholar] [CrossRef]
- Greene, D.; Botello-Smith, W.M.; Follmer, A.; Xiao, L.; Lambros, E.; Luo, R. Modeling Membrane Protein-Ligand Binding Interactions: The Human Purinergic Platelet Receptor. J. Phys. Chem. B 2016, 120, 12293–12304. [Google Scholar] [CrossRef]
- Botello-Smith, W.M.; Luo, R. Applications of MMPBSA to Membrane Proteins I: Efficient Numerical Solutions of Periodic Poisson-Boltzmann Equation. J. Chem. Inf. Model. 2015, 55, 2187–2199. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of threedimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Methods in Neurosciences; Sealfon, S.C., Ed.; Academic: New York, NY, USA, 1995; Volume 25, pp. 366–428. [Google Scholar]
- Flock, T.; Ravarani, C.N.J.; Sun, D.; Venkatakrishnan, A.J.; Kayikci, M.; Tate, C.G.; Veprintsev, D.B.; Babu, M.M. Universal allosteric mechanism for Galpha activation by GPCRs. Nature 2015, 524, 173–179. [Google Scholar] [CrossRef]
- Kostenis, E.; Conklin, B.R.; Wess, J. Molecular basis of receptor/G protein coupling selectivity studied by coexpression of wild type and mutant m2 muscarinic receptors with mutant G alpha(q) subunits. Biochemistry 1997, 36, 1487–1495. [Google Scholar] [CrossRef]
- Liu, J.; Conklin, B.R.; Blin, N.; Yun, J.; Wess, J. Identification of a receptor/G-protein contact site critical for signaling specificity and G-protein activation. Proc. Natl. Acad. Sci. USA 1995, 92, 11642–11646. [Google Scholar] [CrossRef]
- Bluml, K.; Mutschler, E.; Wess, J. Identification of an intracellular tyrosine residue critical for muscarinic receptor-mediated stimulation of phosphatidylinositol hydrolysis. J. Biol. Chem. 1994, 269, 402–405. [Google Scholar]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Krivov, G.G.; Shapovalov, M.V.; Dunbrack, R.L., Jr. Improved prediction of protein side-chain conformations with SCWRL4. Proteins 2009, 77, 778–795. [Google Scholar] [CrossRef]
- Jo, S.; Cheng, X.; Lee, J.; Kim, S.; Park, S.J.; Patel, D.S.; Beaven, A.H.; Lee, K.I.; Rui, H.; Park, S.; et al. CHARMM-GUI 10 years for biomolecular modeling and simulation. J. Comput. Chem. 2017, 38, 1114–1124. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef]
- Price, D.J.; Brooks, C.L., 3rd. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef]
- Kim, S.; Lee, J.; Jo, S.; Brooks, C.L., 3rd; Lee, H.S.; Im, W. CHARMM-GUI ligand reader and modeler for CHARMM force field generation of small molecules. J. Comput. Chem. 2017, 38, 1879–1886. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., 3rd; Darden, T.; Gohlke, h.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, c.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., 3rd. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R:G Binding Free Energy (kcal/mol) | M1:Gαi | M1:Gαq | M2:Gαi | M2:Gαq |
|---|---|---|---|---|
| MD1 | −318 | −233 | −259 | −272 |
| MD2 | −252 | −353 | −285 | −184 |
| MD3 | −221 | −327 | −325 | −281 |
| MD4 | −228 | −363 | −253 | −253 |
| Average | −270 | −319 | −280 | −248 |
| StdDev | 42 | 59 | 33 | 44 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santiago, L.J.; Abrol, R. Understanding G Protein Selectivity of Muscarinic Acetylcholine Receptors Using Computational Methods. Int. J. Mol. Sci. 2019, 20, 5290. https://doi.org/10.3390/ijms20215290
Santiago LJ, Abrol R. Understanding G Protein Selectivity of Muscarinic Acetylcholine Receptors Using Computational Methods. International Journal of Molecular Sciences. 2019; 20(21):5290. https://doi.org/10.3390/ijms20215290
Chicago/Turabian StyleSantiago, Luis Jaimes, and Ravinder Abrol. 2019. "Understanding G Protein Selectivity of Muscarinic Acetylcholine Receptors Using Computational Methods" International Journal of Molecular Sciences 20, no. 21: 5290. https://doi.org/10.3390/ijms20215290
APA StyleSantiago, L. J., & Abrol, R. (2019). Understanding G Protein Selectivity of Muscarinic Acetylcholine Receptors Using Computational Methods. International Journal of Molecular Sciences, 20(21), 5290. https://doi.org/10.3390/ijms20215290