Using PAR4 Inhibition as an Anti-Thrombotic Approach: Why, How, and When?


Abstract
:1. Introduction
2. What is the Function of PAR4 on Platelets?
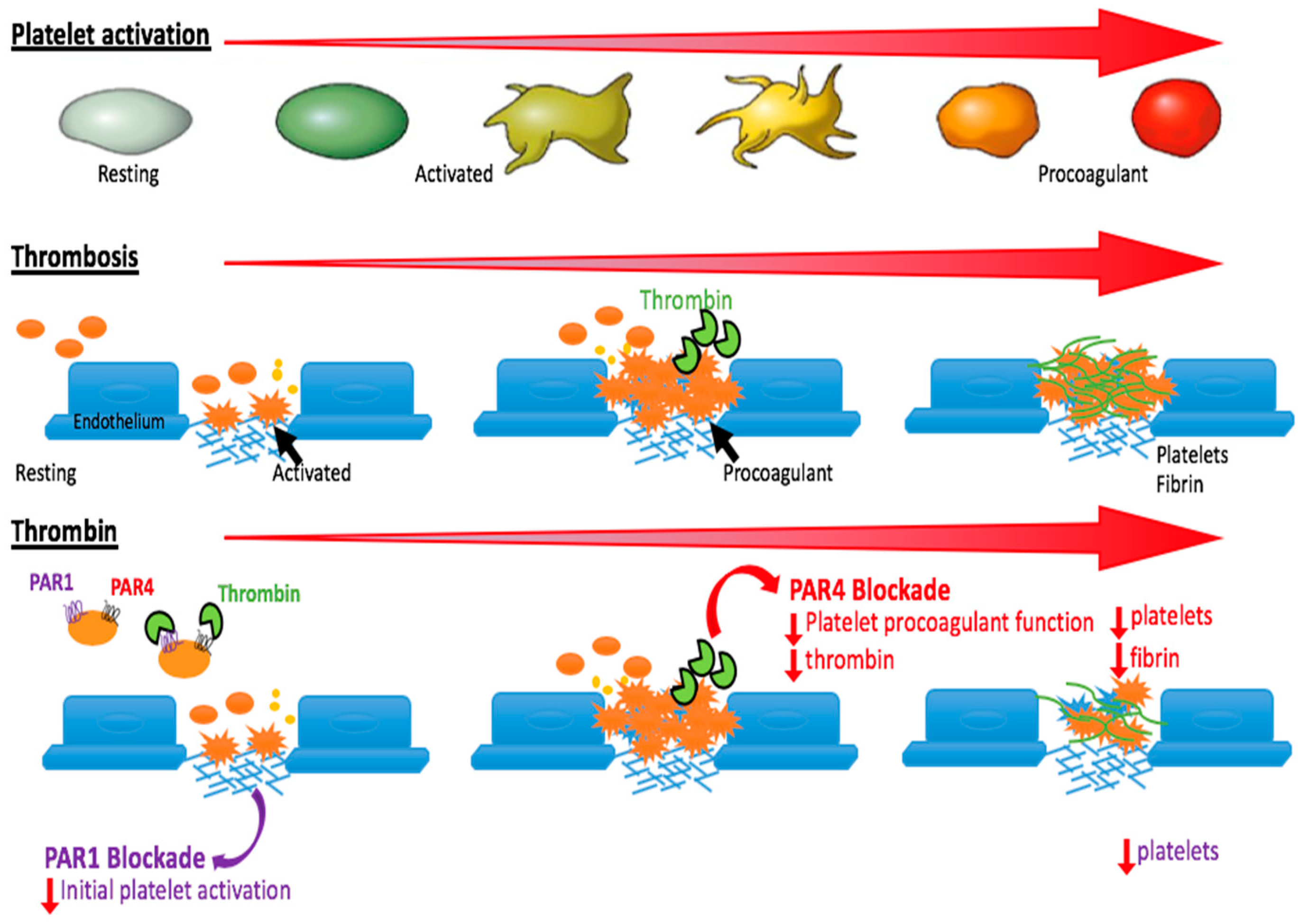
3. Why Target PAR4 for Anti-Thrombotic Therapy?
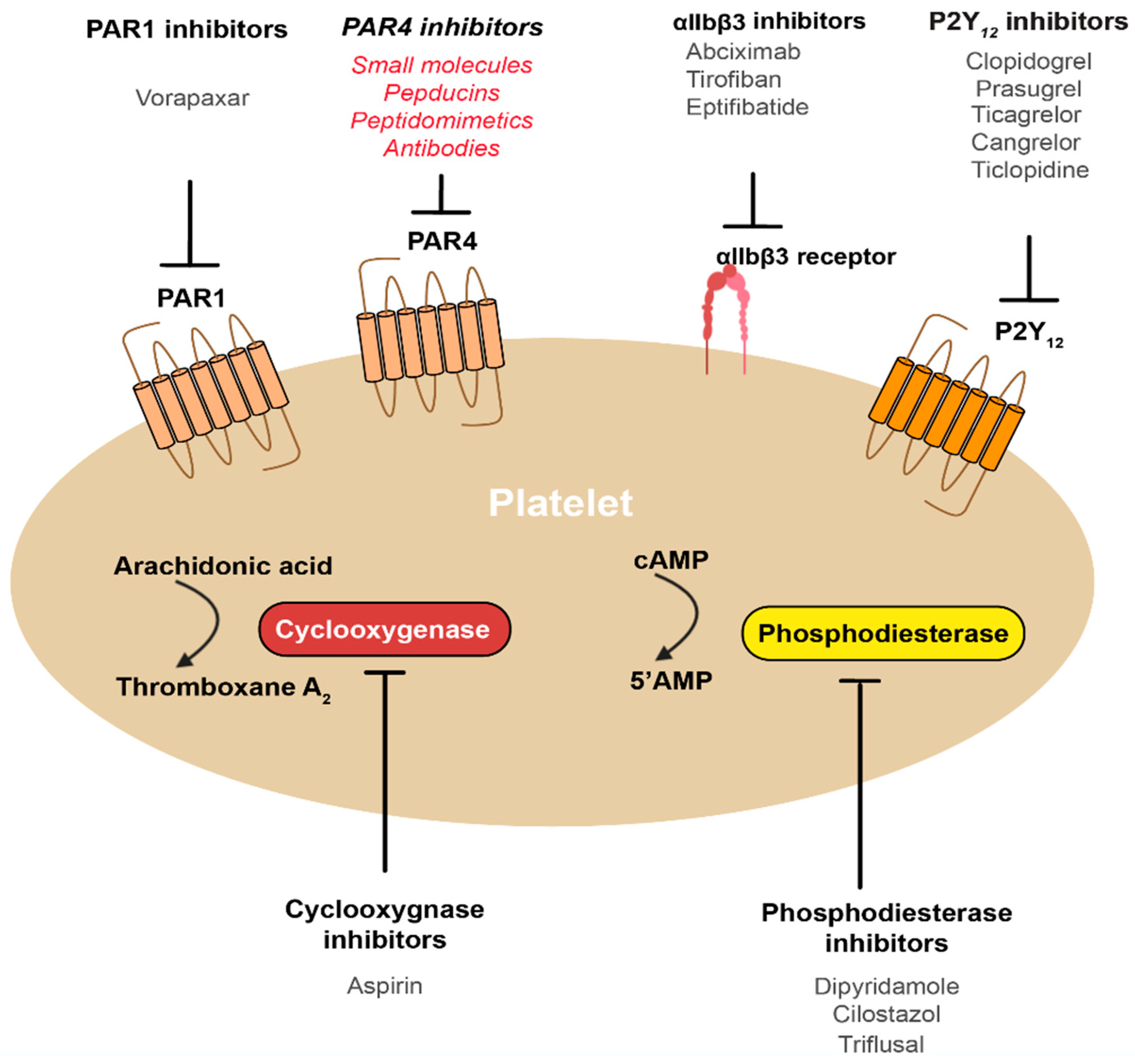
4. How Can We Best Target PAR4?
5. When Can We Best Target PAR4?
6. What Are the Major Issues to Consider for PAR4 Inhibitors?
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). Lancet 1996, 348, 1329–1339. [Google Scholar] [CrossRef]
- Lette, J.; Tatum, J.L.; Fraser, S.; Miller, D.D.; Waters, D.D.; Heller, G.; Stanton, E.B.; Bom, H.S.; Leppo, J.; Nattel, S. Safety of dipyridamole testing in 73,806 patients: The Multicenter Dipyridamole Safety Study. J. Nucl. Cardiol. 1995, 2, 3–17. [Google Scholar] [CrossRef]
- van den Merkhof, L.F.; Zijlstra, F.; Olsson, H.; Grip, L.; Veen, G.; Bar, F.W.; van den Brand, M.J.; Simoons, M.L.; Verheugt, F.W. Abciximab in the treatment of acute myocardial infarction eligible for primary percutaneous transluminal coronary angioplasty. Results of the Glycoprotein Receptor Antagonist Patency Evaluation (GRAPE) pilot study. J. Am. Coll Cardiol. 1999, 33, 1528–1532. [Google Scholar] [CrossRef]
- PURSUIT Investigators. Inhibition of platelet glycoprotein IIb/IIIa with eptifibatide in patients with acute coronary syndromes. N. Engl. J. Med. 1998, 339, 436–443. [Google Scholar] [CrossRef] [PubMed]
- PRISM-PLUS Investigators. Inhibition of the platelet glycoprotein IIb/IIIa receptor with tirofiban in unstable angina and non-Q-wave myocardial infarction. N. Engl. J. Med. 1998, 338, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- Bliden, K.; Chaudhary, R.; Kuliopulos, A.; Tran, H.; Taheri, H.; Tehrani, B.; Rosenblatt, A.; Navarese, E.; Tantry, U.S.; Gurbel, P. Effects of Vorapaxar on Clot Characteristics, Coagulation, Inflammation, and Platelet and Endothelial Function in Patients Treated with Mono-and Dual-Antiplatelet Therapy. J. Thromb. Haemost. 2019. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, S.; Becker, R.C. PAR-1 inhibitors: A novel class of antiplatelet agents for the treatment of patients with atherothrombosis. Handb. Exp. Pharmacol. 2012, 210, 239–260. [Google Scholar]
- Tricoci, P.; Huang, Z.; Held, C.; Moliterno, D.J.; Armstrong, P.W.; Van de Werf, F.; White, H.D.; Aylward, P.E.; Wallentin, L.; Chen, E.; et al. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N. Engl. J. Med. 2012, 366, 20–33. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, P.J.; Molino, M.; Kahn, M.; Brass, L.F. Protease activated receptors: Theme and variations. Oncogene 2001, 20, 1570–1581. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.K.; Wheaton, V.I.; Hung, D.T.; Charo, I.; Coughlin, S.R. Domains specifying thrombin-receptor interaction. Nature 1991, 353, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Andrade-Gordon, P.; Derian, C.K.; Maryanoff, B.E.; Zhang, H.C.; Addo, M.F.; Cheung, W.; Damiano, B.P.; D’Andrea, M.R.; Darrow, A.L.; de Garavilla, L.; et al. Administration of a potent antagonist of protease-activated receptor-1 (PAR-1) attenuates vascular restenosis following balloon angioplasty in rats. J. Pharmacol. Exp. Ther. 2001, 298, 34–42. [Google Scholar] [PubMed]
- Kahn, M.L.; Zheng, Y.W.; Huang, W.; Bigornia, V.; Zeng, D.; Moff, S.; Farese, R.V., Jr.; Tam, C.; Coughlin, S.R. A dual thrombin receptor system for platelet activation. Nature 1998, 394, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Antoniak, S.; Cardenas, J.C.; Buczek, L.J.; Church, F.C.; Mackman, N.; Pawlinski, R. Protease-Activated Receptor 1 Contributes to Angiotensin II-Induced Cardiovascular Remodeling and Inflammation. Cardiology 2017, 136, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.C.; Seiffert, D.; Bird, J.E.; Watson, C.A.; Bostwick, J.S.; Giancarli, M.; Allegretto, N.; Hua, J.; Harden, D.; Guay, J.; et al. Blockade of protease-activated receptor-4 (PAR4) provides robust antithrombotic activity with low bleeding. Sci. Transl. Med. 2017, 9, 371. [Google Scholar] [CrossRef] [PubMed]
- Seeley, S.; Covic, L.; Jacques, S.L.; Sudmeier, J.; Baleja, J.D.; Kuliopulos, A. Structural basis for thrombin activation of a protease-activated receptor: Inhibition of intramolecular liganding. Chem. Biol. 2003, 10, 1033–1041. [Google Scholar] [CrossRef] [PubMed]
- Jacques, S.L.; Kuliopulos, A. Protease-activated receptor-4 uses dual prolines and an anionic retention motif for thrombin recognition and cleavage. Biochem. J. 2003, 376, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Covic, L.; Gresser, A.L.; Kuliopulos, A. Biphasic kinetics of activation and signaling for PAR1 and PAR4 thrombin receptors in platelets. Biochemistry 2000, 39, 5458–5467. [Google Scholar] [CrossRef] [PubMed]
- French, S.L.; Arthur, J.F.; Lee, H.; Nesbitt, W.S.; Andrews, R.K.; Gardiner, E.E.; Hamilton, J.R. Inhibition of protease-activated receptor 4 impairs platelet procoagulant activity during thrombus formation in human blood. J. Thromb. Haemost. 2016, 14, 1642–1654. [Google Scholar] [CrossRef] [PubMed]
- Covic, L.; Gresser, A.L.; Talavera, J.; Swift, S.; Kuliopulos, A. Activation and inhibition of G protein-coupled receptors by cell-penetrating membrane-tethered peptides. Proc. Natl. Acad. Sci. USA 2002, 99, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Duvernay, M.; Young, S.; Gailani, D.; Schoenecker, J.; Hamm, H.E. Protease-activated receptor (PAR) 1 and PAR4 differentially regulate factor V expression from human platelets. Mol. Pharmacol. 2013, 83, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Resendiz, J.C.; Kroll, M.H.; Lassila, R. Protease-activated receptor-induced Akt activation--regulation and possible function. J. Thromb. Haemost. 2007, 5, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- French, S.L.; Thalmann, C.; Bray, P.F.; Macdonald, L.E.; Murphy, A.J.; Sleeman, M.W.; Hamilton, J.R. A function-blocking PAR4 antibody is markedly antithrombotic in the face of a hyperreactive PAR4 variant. Blood Adv. 2018, 2, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Andersen, H.; Greenberg, D.L.; Fujikawa, K.; Xu, W.; Chung, D.W.; Davie, E.W. Protease-activated receptor 1 is the primary mediator of thrombin-stimulated platelet procoagulant activity. Proc. Natl. Acad. Sci. USA 1999, 96, 11189–11193. [Google Scholar] [CrossRef] [PubMed]
- Keuren, J.F.; Wielders, S.J.; Ulrichts, H.; Hackeng, T.; Heemskerk, J.W.; Deckmyn, H.; Bevers, E.M.; Lindhout, T. Synergistic effect of thrombin on collagen-induced platelet procoagulant activity is mediated through protease-activated receptor-1. Arterioscler Thromb. Vasc. Biol. 2005, 25, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- French, S.L.; Arthur, J.F.; Tran, H.A.; Hamilton, J.R. Approval of the first protease-activated receptor antagonist: Rationale, development, significance, and considerations of a novel anti-platelet agent. Blood Rev. 2015, 29, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Srinivasan, Y.; Arlow, D.H.; Fung, J.J.; Palmer, D.; Zheng, Y.; Green, H.F.; Pandey, A.; Dror, R.O.; Shaw, D.E.; et al. High-resolution crystal structure of human protease-activated receptor 1. Nature 2012, 492, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Olie, R.H.; van der Meijden, P.E.J.; Spronk, H.M.H.; van Oerle, R.; Barvik, S.; Bonarjee, V.V.S.; Ten Cate, H.; Nilsen, D.W.T. Effects of the PAR-1 Antagonist Vorapaxar on Platelet Activation and Coagulation Biomarkers in Patients with Stable Coronary Artery Disease. TH Open 2019, 3, e259–e262. [Google Scholar] [CrossRef] [PubMed]
- Abdulsattar, Y.; Ternas, T.; Garcia, D. Vorapaxar: Targeting a novel antiplatelet pathway. Pharmacol. Therap. 2011, 36, 564–568. [Google Scholar]
- Morrow, D.A.; Braunwald, E.; Bonaca, M.P.; Ameriso, S.F.; Dalby, A.J.; Fish, M.P.; Fox, K.A.; Lipka, L.J.; Liu, X.; Nicolau, J.C.; et al. Vorapaxar in the secondary prevention of atherothrombotic events. N. Engl. J. Med. 2012, 366, 1404–1413. [Google Scholar] [CrossRef] [PubMed]
- Vranckx, P.; White, H.D.; Huang, Z.; Mahaffey, K.W.; Armstrong, P.W.; Van de Werf, F.; Moliterno, D.J.; Wallentin, L.; Held, C.; Aylward, P.E.; et al. Validation of BARC Bleeding Criteria in Patients With Acute Coronary Syndromes: The TRACER Trial. J. Am. Coll. Cardiol. 2016, 67, 2135–2144. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.M.; Andrews, R.K. Illustrated State-of-the-Art Capsules of the ISTH 2019 Congress in Melbourne, Australia. Res. Pract. Thromb. Haemost. 2019, 3, 431–497. [Google Scholar] [CrossRef] [PubMed]
- Hollenberg, M.D.; Saifeddine, M.; Sandhu, S.; Houle, S.; Vergnolle, N. Proteinase-activated receptor-4: Evaluation of tethered ligand-derived peptides as probes for receptor function and as inflammatory agonists in vivo. Br. J. Pharmacol. 2004, 143, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Leger, A.J.; Jacques, S.L.; Kaneider, N.C.; Derian, C.K.; Andrade-Gordon, P.; Covic, L.; Kuliopulos, A. Blocking the protease-activated receptor 1–4 heterodimer in platelet-mediated thrombosis. Circulation 2006, 113, 1244–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Covic, L.; Misra, M.; Badar, J.; Singh, C.; Kuliopulos, A. Pepducin-based intervention of thrombin-receptor signaling and systemic platelet activation. Nat. Med. 2002, 8, 1161–1165. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Hwang, T.L.; Liao, C.H.; Kuo, S.C.; Lee, F.Y.; Lee, C.Y.; Teng, C.M. Selective inhibition of protease-activated receptor 3-dependent platelet activation by YD-3. Thromb. Haemost. 2002, 87, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.J.; Ismat, F.A.; Wang, Z.; Cerra, M.; Narayan, H.; Raftis, J.; Gray, T.J.; Connell, S.; Garonzik, S.; Ma, X.; et al. PAR4 (Protease-Activated Receptor 4) Antagonism With BMS-986120 Inhibits Human Ex Vivo Thrombus Formation. Arterioscler. Thromb. Vasc Biol. 2018, 38, 448–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, P.C.; Waston, C.A.; Bostwick, J.; Banville, J.; Wexler, R.R.; Priestley, E.S.; Marinier, A.; Bouvier, M.; Gordon, D.; Schumacher, W.; et al. An orally-active small-molecule antagonist of the platelet protease-activated receptor-4, BMS-986141, prevents aeterial thrombosis with low bleeding liability in cynomolgus monkeys. Circulation 2017, 136, A13794. [Google Scholar]
- Mumaw, M.M.; de la Fuente, M.; Noble, D.N.; Nieman, M.T. Targeting the anionic region of human protease-activated receptor 4 inhibits platelet aggregation and thrombosis without interfering with hemostasis. J. Thromb. Haemost. 2014, 12, 1331–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, R.; Noorbakhsh, F.; Defea, K.; Hollenberg, M.D. Targeting proteinase-activated receptors: Therapeutic potential and challenges. Nat. Rev. Drug Discov. 2012, 11, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Moschonas, I.C.; Kellici, T.F.; Mavromoustakos, T.; Stathopoulos, P.; Tsikaris, V.; Magafa, V.; Tzakos, A.G.; Tselepis, A.D. Molecular requirements involving the human platelet protease-activated receptor-4 mechanism of activation by peptide analogues of its tethered-ligand. Platelets 2017, 28, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.R.; Trejo, J. Challenges and Opportunities in Protease-Activated Receptor Drug Development. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 349–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stampfuss, J.J.; Schror, K.; Weber, A.A. Inhibition of platelet thromboxane receptor function by a thrombin receptor-targeted pepducin. Nat. Med. 2003, 9, 1447. [Google Scholar] [CrossRef] [PubMed]
- Rwibasira Rudinga, G.; Khan, G.J.; Kong, Y. Protease-Activated Receptor 4 (PAR4): A Promising Target for Antiplatelet Therapy. Int. J. Mol. Sci. 2018, 19, 573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, S.L.; Hamilton, J.R. Drugs targeting protease-activated receptor-4 improve the anti-thrombotic therapeutic window. Ann. Transl. Med. 2017, 5, 464. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, M.; McIntosh, K.; Bushell, T.; Sloan, G.; Plevin, R. Proteinase-activated receptors (PARs) as targets for antiplatelet therapy. Biochem. Soc. Trans. 2016, 44, 606–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, C.P.; Steg, G.; Bhatt, D.L. The management of antiplatelet therapy in acute coronary syndrome patients with thrombocytopenia: A clinical conundrum. Eur. Heart J. 2017, 38, 3488–3492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jneid, H.; Bhatt, D.L.; Corti, R.; Badimon, J.J.; Fuster, V.; Francis, G.S. Aspirin and clopidogrel in acute coronary syndromes: Therapeutic insights from the CURE study. Arch. Intern. Med. 2003, 163, 1145–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasalic, L.; Wing-Lun, E.; Lau, J.K.; Campbell, H.; Pennings, G.J.; Lau, E.; Connor, D.; Liang, H.P.; Muller, D.; Kritharides, L.; et al. Novel assay demonstrates that coronary artery disease patients have heightened procoagulant platelet response. J. Thromb. Haemost. 2018, 16, 1198–1210. [Google Scholar] [CrossRef] [PubMed]
- Bathina, S.; Srinivas, N.; Das, U.N. Streptozotocin produces oxidative stress, inflammation and decreases BDNF concentrations to induce apoptosis of RIN5F cells and type 2 diabetes mellitus in Wistar rats. Biochem. Biophys. Res. Commun. 2017, 486, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, H.; Nakayama, M.; Morimoto, T.; Uemura, S.; Kanauchi, M.; Doi, N.; Jinnouchi, H.; Sugiyama, S.; Saito, Y.; Japanese Primary Prevention of Atherosclerosis With Aspirin for Diabetes Trial Investigators. Low-dose aspirin for primary prevention of atherosclerotic events in patients with type 2 diabetes: A randomized controlled trial. JAMA 2008, 300, 2134–2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, P.M.; Peterson, E.D.; Wang, L.; Magid, D.J.; Fihn, S.D.; Larsen, G.C.; Jesse, R.A.; Rumsfeld, J.S. Incidence of death and acute myocardial infarction associated with stopping clopidogrel after acute coronary syndrome. JAMA 2008, 299, 532–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehilli, J.; Kastrati, A.; Schuhlen, H.; Dibra, A.; Dotzer, F.; von Beckerath, N.; Bollwein, H.; Pache, J.; Dirschinger, J.; Berger, P.P.; et al. Antithrombotic Regimen: Is Abciximab a Superior Way to Eliminate Elevated Thrombotic Risk in Diabetics Study, I. Randomized clinical trial of abciximab in diabetic patients undergoing elective percutaneous coronary interventions after treatment with a high loading dose of clopidogrel. Circulation 2004, 110, 3627–3635. [Google Scholar] [PubMed] [Green Version]
- Pavic, G.; Grandoch, M.; Dangwal, S.; Jobi, K.; Rauch, B.H.; Doller, A.; Oberhuber, A.; Akhyari, P.; Schror, K.; Fischer, J.W.; et al. Thrombin receptor protease-activated receptor 4 is a key regulator of exaggerated intimal thickening in diabetes mellitus. Circulation 2014, 130, 1700–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scridon, A.; Marginean, A.; Hutanu, A.; Chinezu, L.; Gheban, D.; Perian, M.; Vantu, A.; Ghertescu, D.; Fisca, P.C.; Serban, R.C.; et al. Vascular protease-activated receptor 4 upregulation, increased platelet aggregation, and coronary lipid deposits induced by long-term dabigatran administration - results from a diabetes animal model. J. Thromb. Haemost. 2019, 17, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Gryka, R.J.; Buckley, L.F.; Anderson, S.M. Vorapaxar: The Current Role and Future Directions of a Novel Protease-Activated Receptor Antagonist for Risk Reduction in Atherosclerotic Disease. Drugs R D 2017, 17, 65–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitrugno, A.; Tassi Yunga, S.; Sylman, J.L.; Zilberman-Rudenko, J.; Shirai, T.; Hebert, J.F.; Kayton, R.; Zhang, Y.; Nan, X.; Shatzel, J.J.; et al. The role of coagulation and platelets in colon cancer-associated thrombosis. Am. J. Physiol. Cell Physiol. 2019, 316, C264–C273. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Jiang, P.; Xiang, Y.; Zhang, Y.; Zhu, Z.; Zhang, C.; Lee, S.; Lee, W.; Zhang, Y. Increased expression of protease-activated receptor 4 and Trefoil factor 2 in human colorectal cancer. PLoS ONE 2015, 10, e0122678. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Zhang, M.; Tuma, R.F.; Kunapuli, S.P. Deficiency of PAR4 attenuates cerebral ischemia/reperfusion injury in mice. J. Cereb. Blood Flow Metab. 2010, 30, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.F.; Andersen, H.; Whitmore, T.E.; Presnell, S.R.; Yee, D.P.; Ching, A.; Gilbert, T.; Davie, E.W.; Foster, D.C. Cloning and characterization of human protease-activated receptor 4. Proc. Natl. Acad Sci. USA 1998, 95, 6642–6646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asfaha, S.; Cenac, N.; Houle, S.; Altier, C.; Papez, M.D.; Nguyen, C.; Steinhoff, M.; Chapman, K.; Zamponi, G.W.; Vergnolle, N. Protease-activated receptor-4: A novel mechanism of inflammatory pain modulation. Br. J. Pharmacol. 2007, 150, 176–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, S.L.; Hamilton, J.R. Perinatal lethality of Par4(-/-) mice delivered by primiparous dams reveals spontaneous bleeding in mice without platelet thrombin receptor function. Platelets 2018, 29, 196–198. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, K.; Schmedes, C.M.; Houston, E.R.; Butler, E.; Mackman, N.; Antoniak, S. Protease-activated receptor 4 protects mice from Coxsackievirus B3 and H1N1 influenza A virus infection. Cell Immunol. 2019, 344, 103949. [Google Scholar] [CrossRef] [PubMed]
- de Stoppelaar, S.F.; Van’t Veer, C.; van den Boogaard, F.E.; Nieuwland, R.; Hoogendijk, A.J.; de Boer, O.J.; Roelofs, J.J.; van der Poll, T. Protease activated receptor 4 limits bacterial growth and lung pathology during late stage Streptococcus pneumoniae induced pneumonia in mice. Thromb. Haemost. 2013, 110, 582–592. [Google Scholar] [PubMed] [Green Version]
- Suo, Z.; Wu, M.; Citron, B.A.; Gao, C.; Festoff, B.W. Persistent protease-activated receptor 4 signaling mediates thrombin-induced microglial activation. J. Biol. Chem. 2003, 278, 31177–31183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asokananthan, N.; Graham, P.T.; Fink, J.; Knight, D.A.; Bakker, A.J.; McWilliam, A.S.; Thompson, P.J.; Stewart, G.A. Activation of protease-activated receptor (PAR)-1, PAR-2, and PAR-4 stimulates IL-6, IL-8, and prostaglandin E2 release from human respiratory epithelial cells. J. Immunol. 2002, 168, 3577–3585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergnolle, N.; Derian, C.K.; D’Andrea, M.R.; Steinhoff, M.; Andrade-Gordon, P. Characterization of thrombin-induced leukocyte rolling and adherence: A potential proinflammatory role for proteinase-activated receptor-4. J. Immunol. 2002, 169, 1467–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houle, S.; Papez, M.D.; Ferazzini, M.; Hollenberg, M.D.; Vergnolle, N. Neutrophils and the kallikrein-kinin system in proteinase-activated receptor 4-mediated inflammation in rodents. Br. J. Pharmacol. 2005, 146, 670–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busso, N.; Chobaz-Peclat, V.; Hamilton, J.; Spee, P.; Wagtmann, N.; So, A. Essential role of platelet activation via protease activated receptor 4 in tissue factor-initiated inflammation. Arthritis. Res. Ther. 2008, 10, R42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Andrea, M.R.; Saban, M.R.; Nguyen, N.B.; Andrade-Gordon, P.; Saban, R. Expression of protease-activated receptor-1, -2, -3, and -4 in control and experimentally inflamed mouse bladder. Am. J. Pathol. 2003, 162, 907–923. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, J.R.; Frauman, A.G.; Cocks, T.M. Increased expression of protease-activated receptor-2 (PAR2) and PAR4 in human coronary artery by inflammatory stimuli unveils endothelium-dependent relaxations to PAR2 and PAR4 agonists. Circ. Res. 2001, 89, 92–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Wang, Z.; Zhang, Z.; Zhang, R.; Yu, L. Capsaicin up-regulates protease-activated receptor-4 mRNA and protein in primary cultured dorsal root ganglion neurons. Cell Mol. Neurobiol. 2013, 33, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Edelstein, L.C.; Simon, L.M.; Lindsay, C.R.; Kong, X.; Teruel-Montoya, R.; Tourdot, B.E.; Chen, E.S.; Ma, L.; Coughlin, S.; Nieman, M.; et al. Common variants in the human platelet PAR4 thrombin receptor alter platelet function and differ by race. Blood 2014, 124, 3450–3458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morikawa, Y.; Kato, H.; Kashiwagi, H.; Nishiura, N.; Akuta, K.; Honda, S.; Kanakura, Y.; Tomiyama, Y. Protease-activated receptor-4 (PAR4) variant influences on platelet reactivity induced by PAR4-activating peptide through altered Ca(2+) mobilization and ERK phosphorylation in healthy Japanese subjects. Thromb. Res. 2018, 162, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Heenkenda, M.K.; Lindahl, T.L.; Osman, A. Frequency of PAR4 Ala120Thr variant associated with platelet reactivity significantly varies across sub-Saharan African populations. Blood 2018, 132, 2103–2106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tricoci, P.; Neely, M.; Whitley, M.J.; Edelstein, L.C.; Simon, L.M.; Shaw, C.; Fortina, P.; Moliterno, D.J.; Armstrong, P.W.; Aylward, P.; et al. Effects of genetic variation in protease activated receptor 4 after an acute coronary syndrome: Analysis from the TRACER trial. Blood Cells Mol. Dis. 2018, 72, 37–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitley, M.J.; Henke, D.M.; Ghazi, A.; Nieman, M.; Stoller, M.; Simon, L.M.; Chen, E.; Vesci, J.; Holinstat, M.; McKenzie, S.E.; et al. The protease-activated receptor 4 Ala120Thr variant alters platelet responsiveness to low-dose thrombin and protease-activated receptor 4 desensitization, and is blocked by non-competitive P2Y12 inhibition. J. Thromb. Haemost. 2018, 16, 2501–2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tourdot, B.E.; Stoveken, H.; Trumbo, D.; Yeung, J.; Kanthi, Y.; Edelstein, L.C.; Bray, P.F.; Tall, G.G.; Holinstat, M. Genetic Variant in Human PAR (Protease-Activated Receptor) 4 Enhances Thrombus Formation Resulting in Resistance to Antiplatelet Therapeutics. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1632–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Drug Class | Antagonist | Target Site | IC50 | Stage of Development | Key Reference |
|---|---|---|---|---|---|
| Peptidomimetic | tc-YPGKF-NH2 | Ligand binding site * | 100 µM | Tool compound | Hollenberg et al., 2004 [32] |
| Pepducin | P4pal-i1 | First intracellular loop | ≈ 5 µM | Preclinical | Leger et al., 200 [33] |
| P4pal-10 | Third intracellular loop | ≈ 1 µM | Preclinical | Covic et al., 2002 [34] | |
| Small molecule | YD-3 | Ligand binding site * | 28 µM | Tool compound | Wu et al., 2002 [35] |
| BMS-986120 | Ligand binding site * | 8 nM | Phase I | Wong et al., 2016 [36] | |
| BMS-986141 | Ligand binding site * | 6 nM | Phase II | Wong et al., 2017 [37] | |
| Antibody | CAN12 (rabbit polyclonal) | N-terminal anionic cluster | 10 ng/mL | Tool compound | Mumaw et al., 2014 [38] |
| RC3 (human monoclonal) | Thrombin cleavage site | 5 µg/mL | Preclinical | French et al., 2018 [22] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Tarlac, V.; Hamilton, J.R. Using PAR4 Inhibition as an Anti-Thrombotic Approach: Why, How, and When? Int. J. Mol. Sci. 2019, 20, 5629. https://doi.org/10.3390/ijms20225629
Li S, Tarlac V, Hamilton JR. Using PAR4 Inhibition as an Anti-Thrombotic Approach: Why, How, and When? International Journal of Molecular Sciences. 2019; 20(22):5629. https://doi.org/10.3390/ijms20225629
Chicago/Turabian StyleLi, Simeng, Volga Tarlac, and Justin R. Hamilton. 2019. "Using PAR4 Inhibition as an Anti-Thrombotic Approach: Why, How, and When?" International Journal of Molecular Sciences 20, no. 22: 5629. https://doi.org/10.3390/ijms20225629
APA StyleLi, S., Tarlac, V., & Hamilton, J. R. (2019). Using PAR4 Inhibition as an Anti-Thrombotic Approach: Why, How, and When? International Journal of Molecular Sciences, 20(22), 5629. https://doi.org/10.3390/ijms20225629