Identification of MicroRNAs That Respond to Soybean Cyst Nematode Infection in Early Stages in Resistant and Susceptible Soybean Cultivars

Abstract
1. Introduction
2. Results
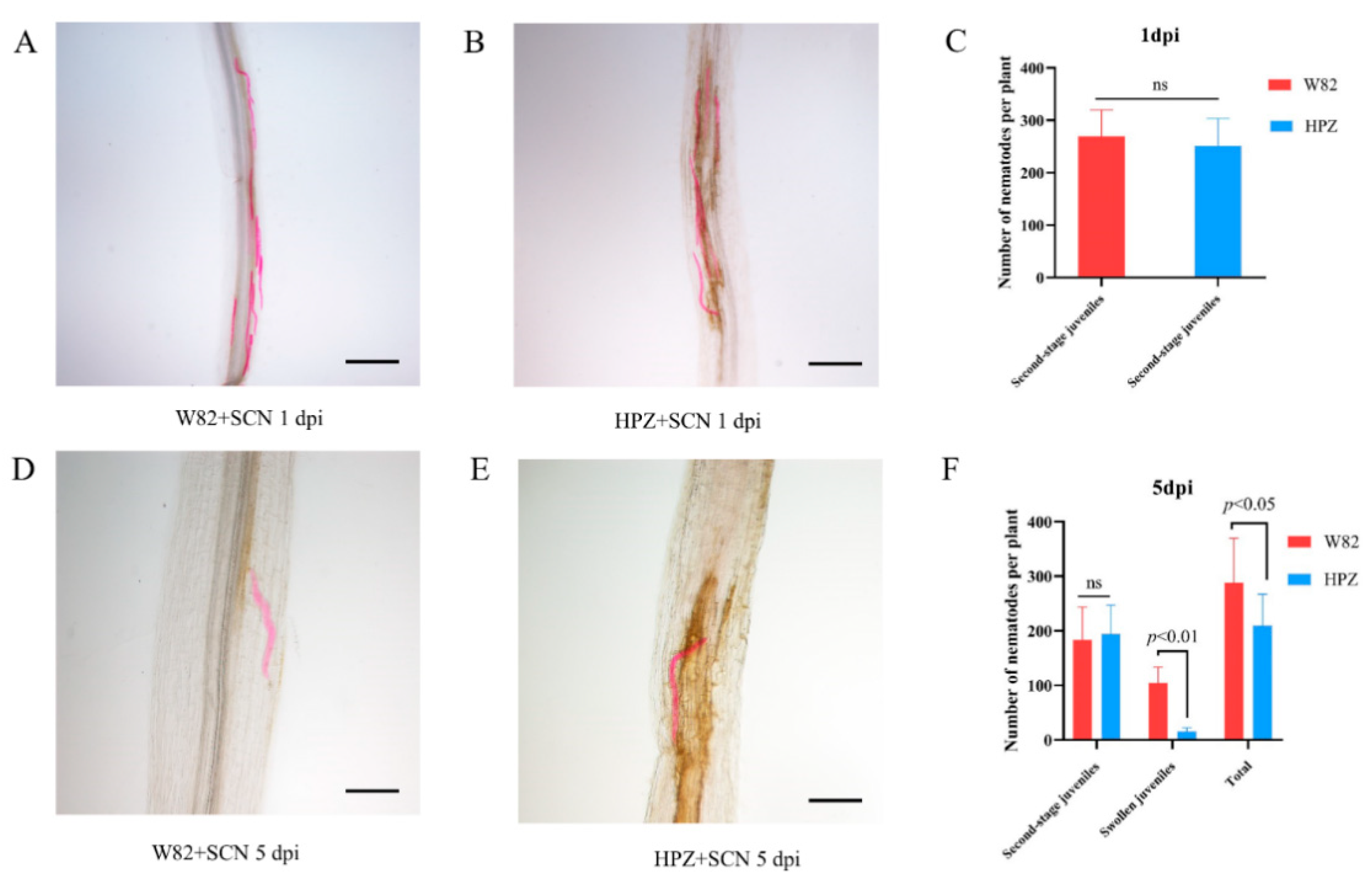
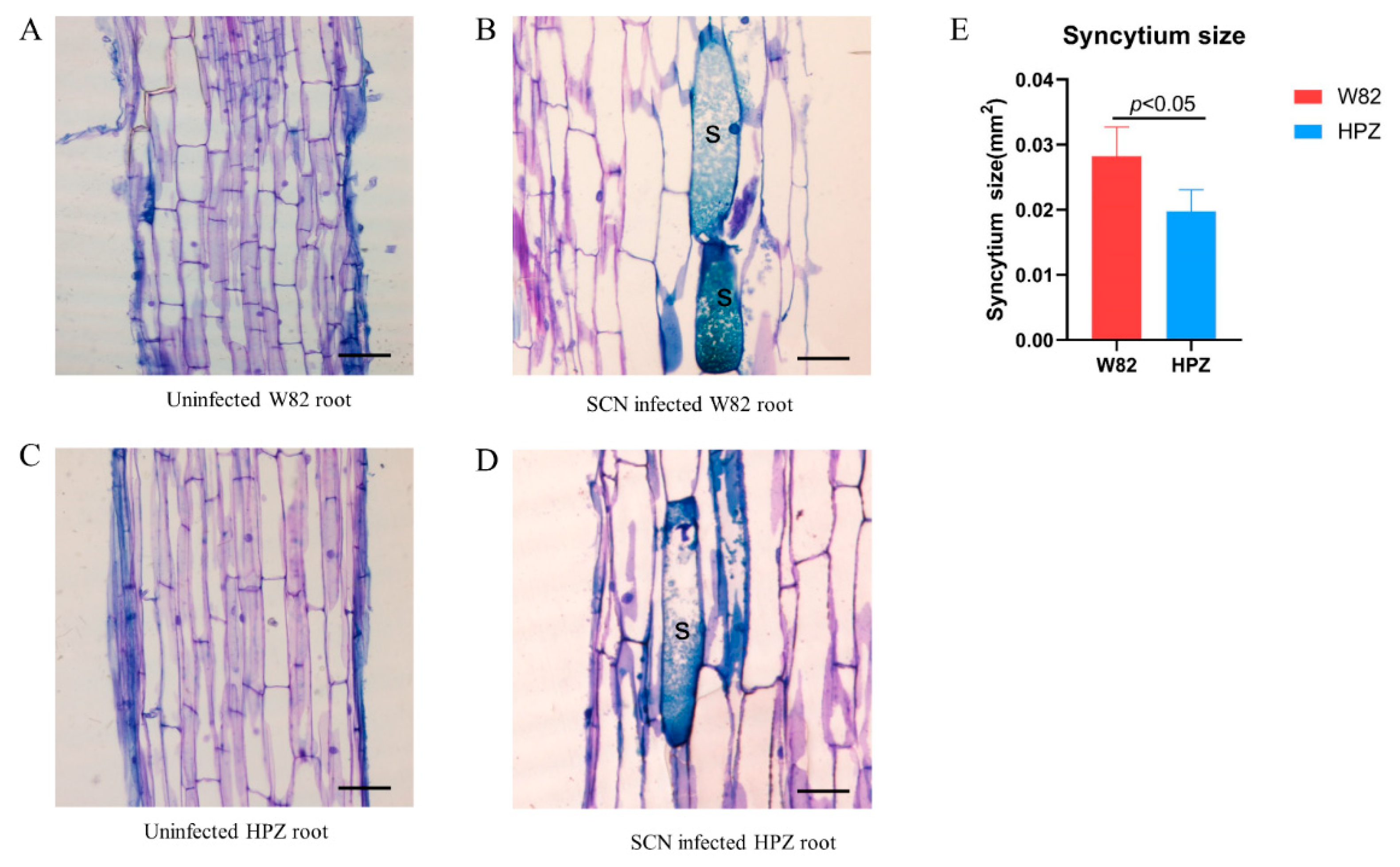
2.1. HPZ Was Highly Resistant to SCN During the Early Stages of Infection/HPZ Represses the Development of SCN and Syncytium
2.2. Small RNA Sequenced from the 24 Libraries Constructed from HPZ and W82 Roots
2.3. Identification of Known and Novel miRNAs
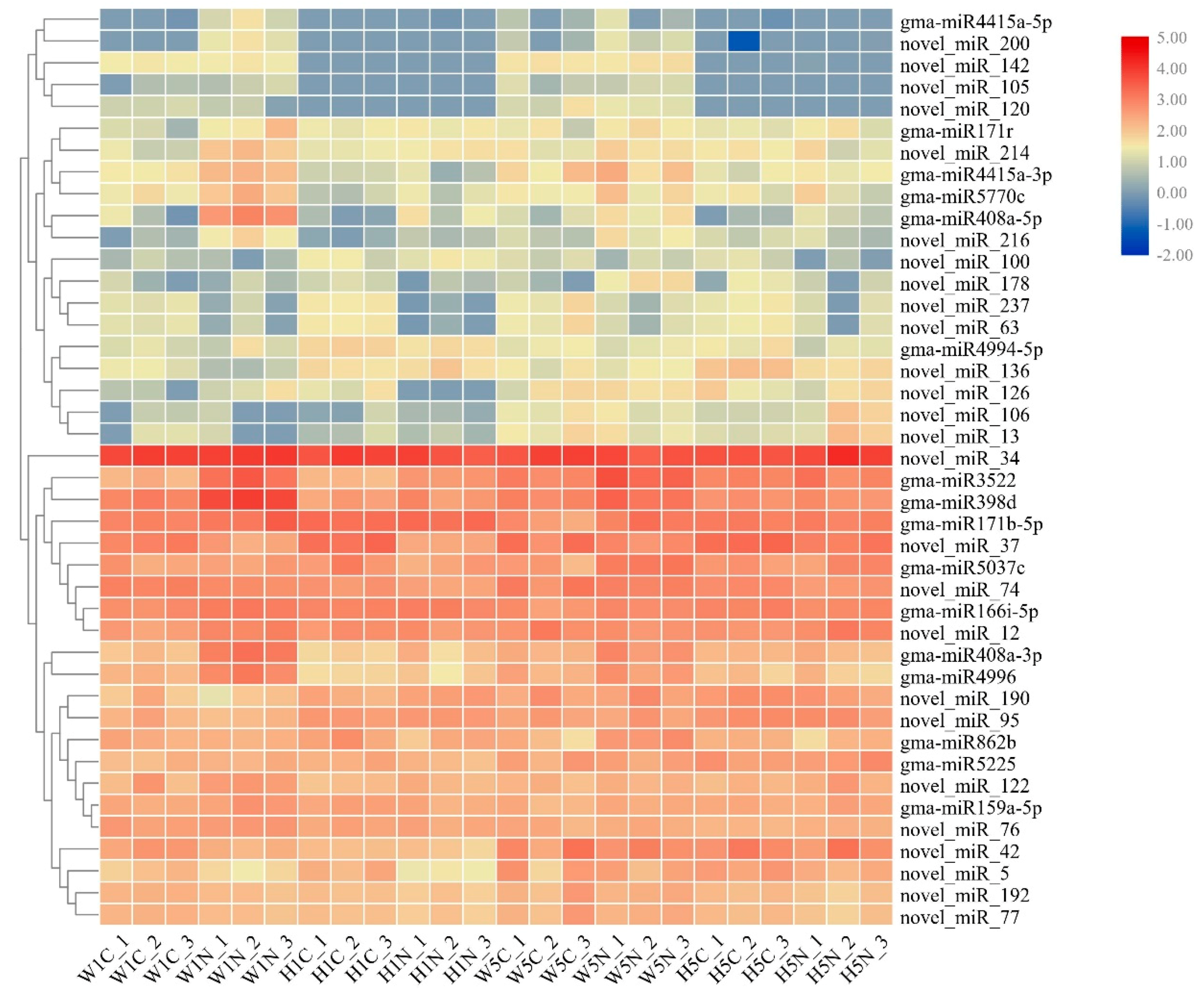
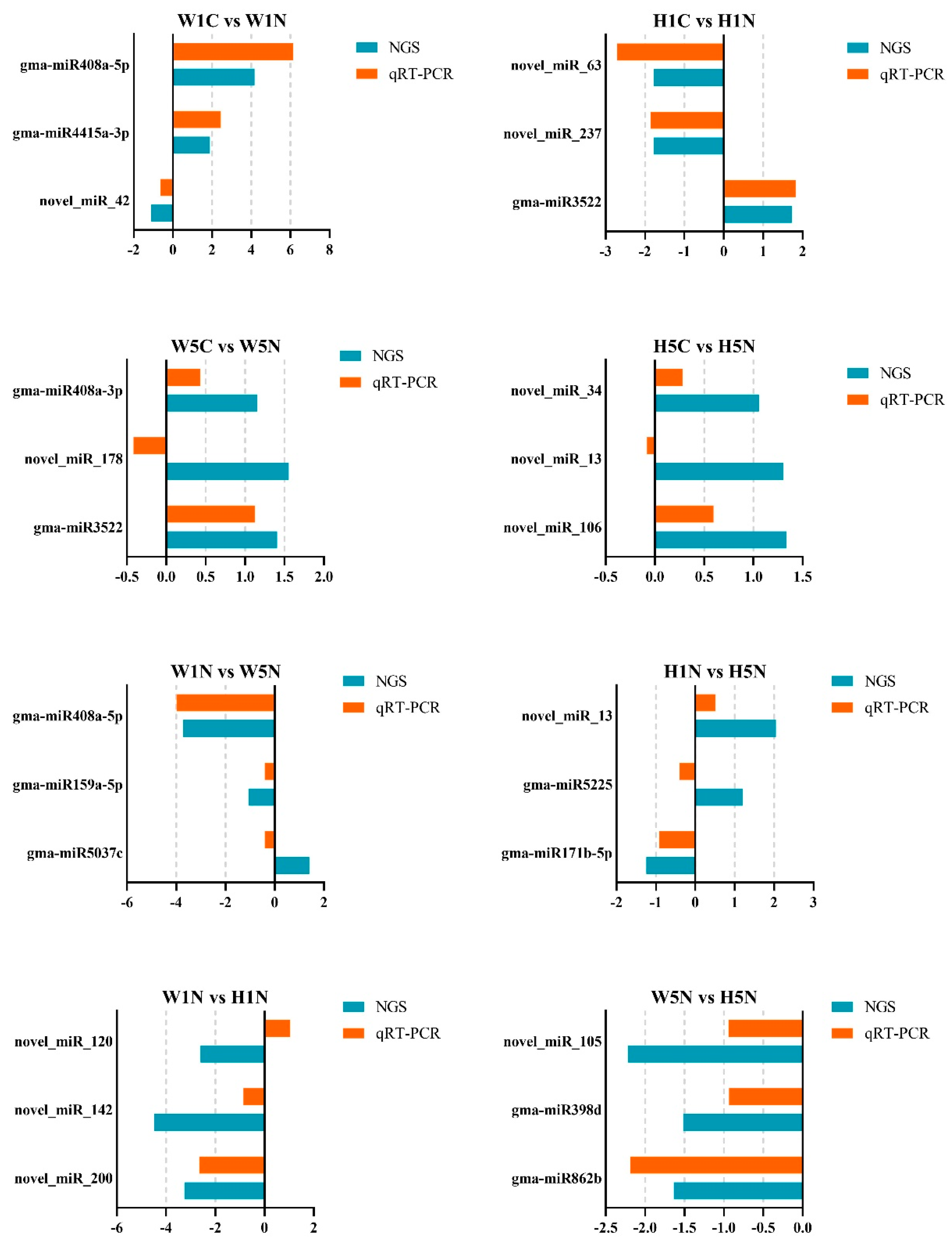
2.4. Analysis of Differences in miRNA Expression Between HPZ and W82
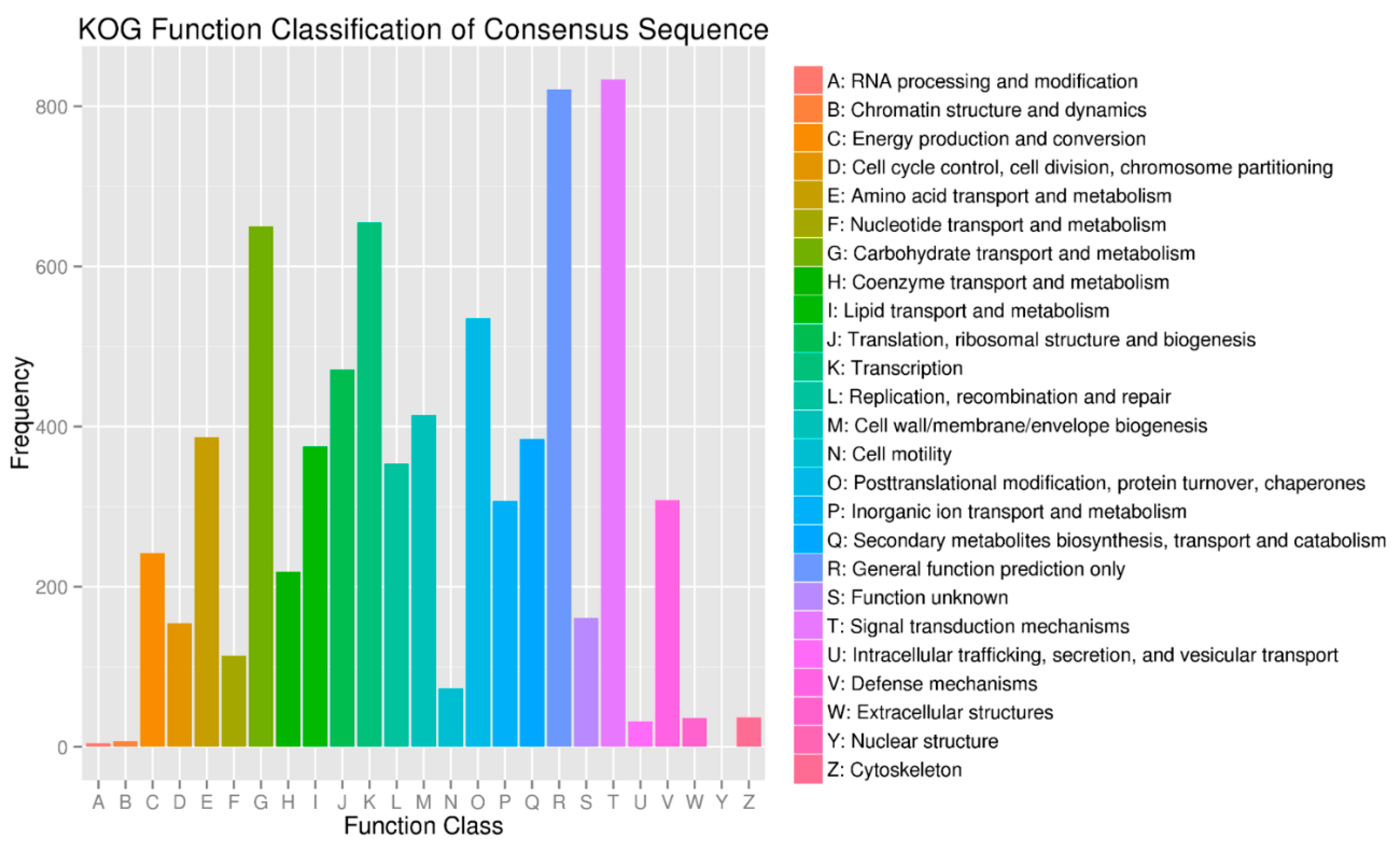
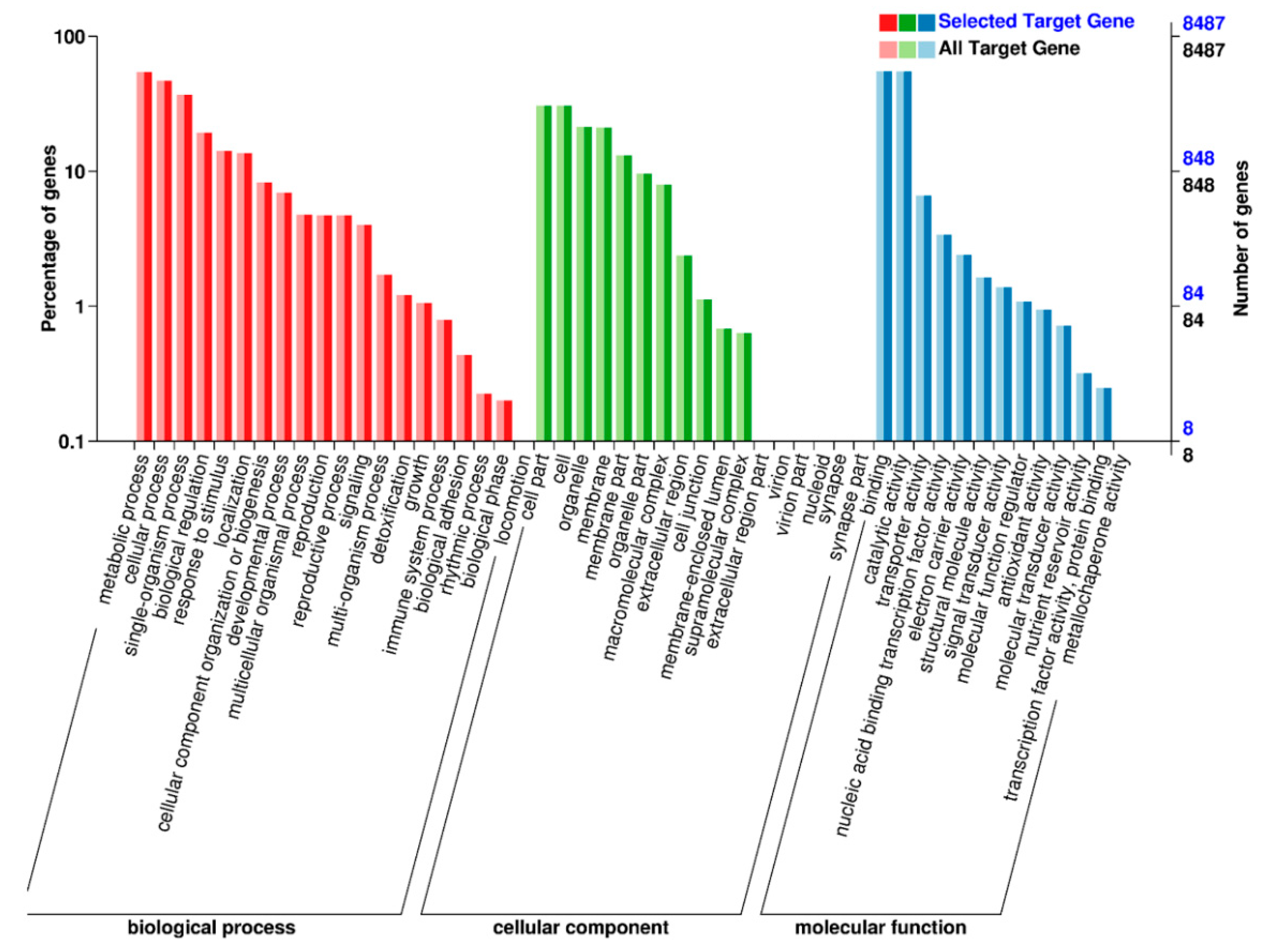
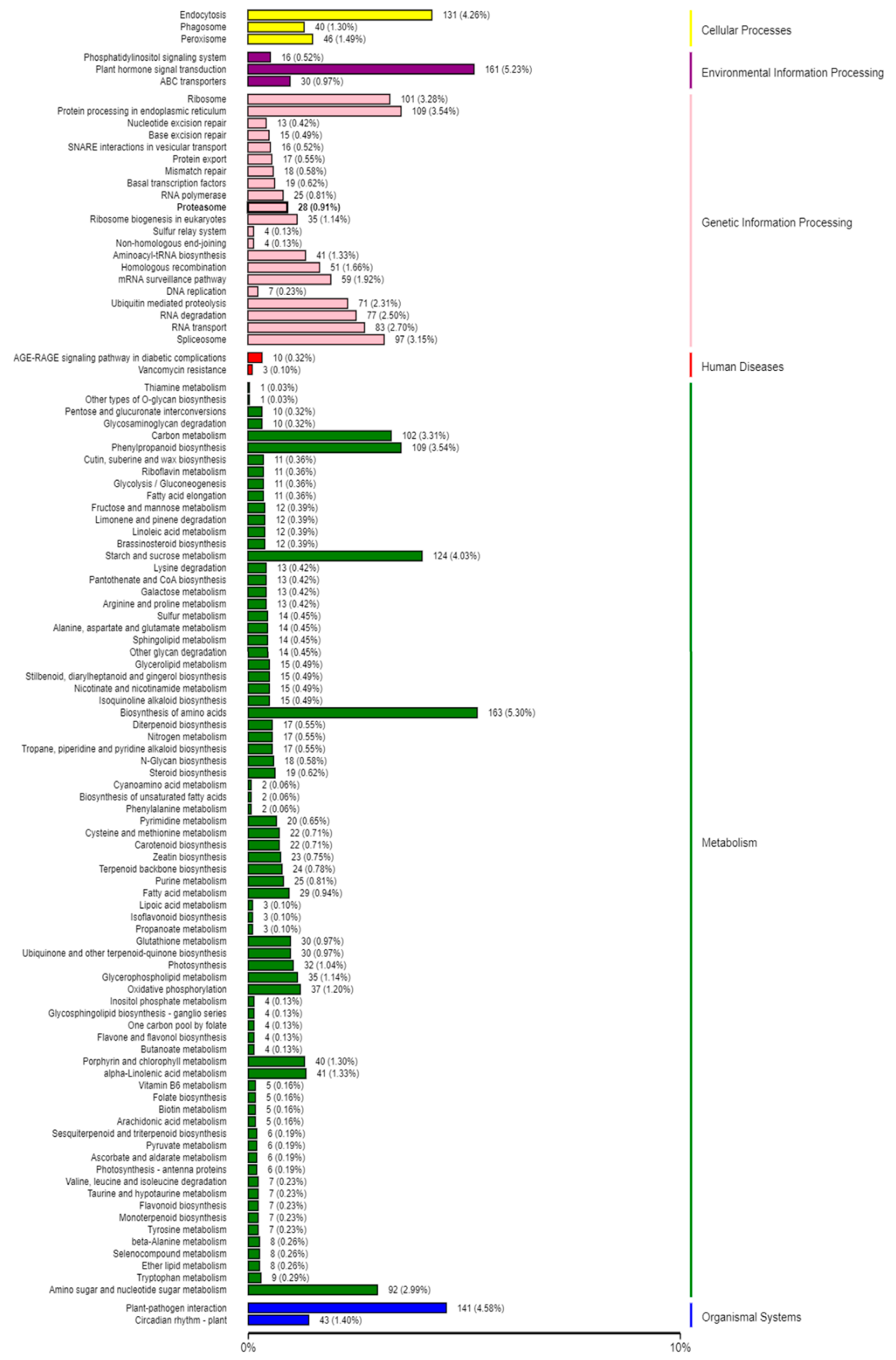
2.5. Target Prediction and Function Annotation
3. Discussion
4. Materials and Methods
4.1. Soybean, Nematode Population Culture
4.2. Nematode Inoculation, Penetration, Development Evaluation, and Histological Observation
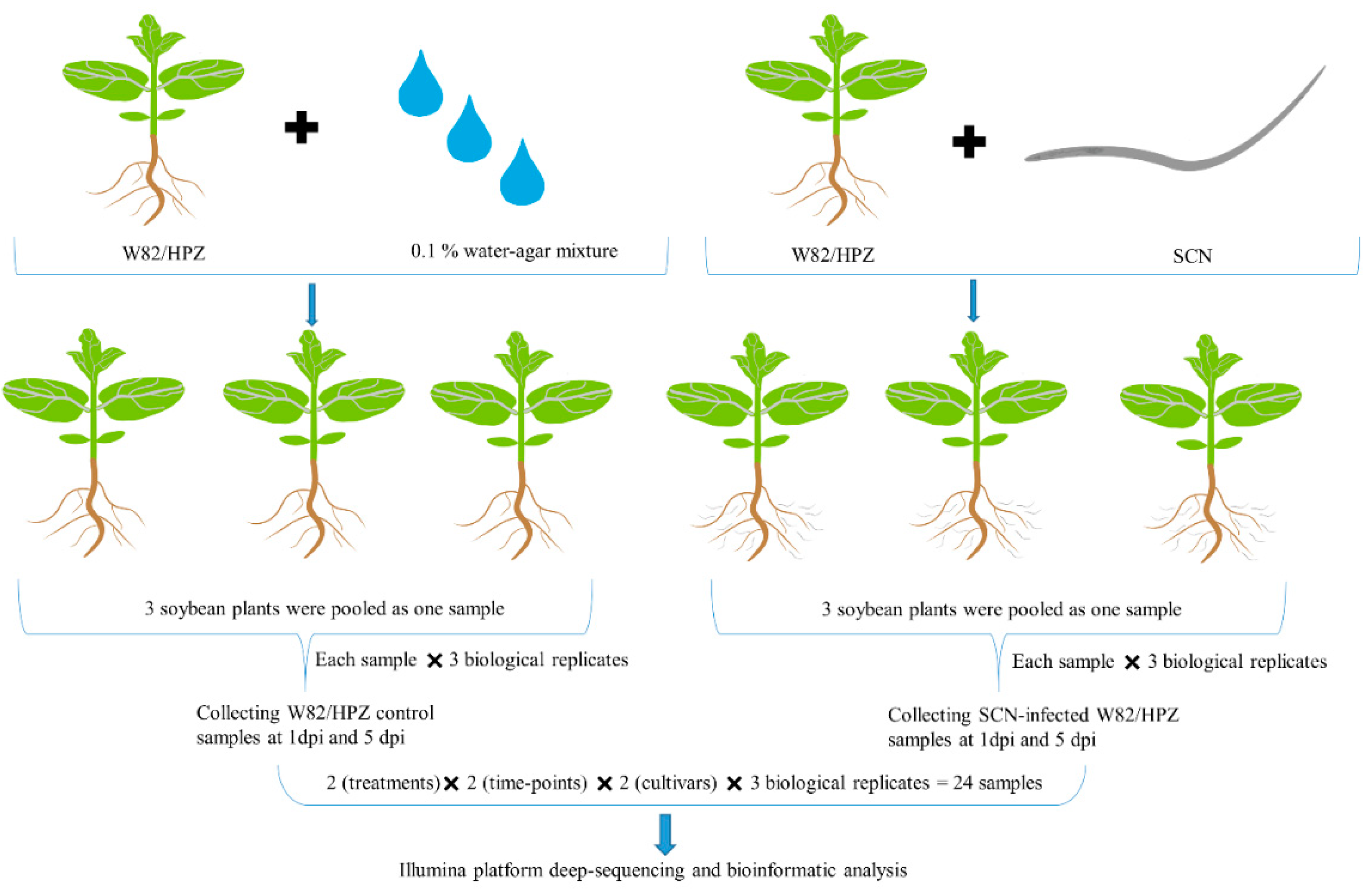
4.3. Sequencing Sample Preparation
4.4. RNA Isolation, Construction of Small RNA Libraries, and Sequencing
4.5. Analysis of Sequencing Data, Differential miRNA Expression Profiling, and Target Prediction
4.6. Quantitative Real-Time PCR (qRT-PCR) Analysis to Validate miRNA Expression
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| SCN | soybean cyst nematode |
| miRNA | microRNA |
| qRT-PCR | quantitative real-time PCR |
References
- Whitham, S.A.; Qi, M.; Innes, R.W.; Ma, W.; Lopescaitar, V.S.; Hewezi, T. Molecular Soybean-Pathogen Interactions. Annu. Rev. Phytopathol. 2016, 54, 443–468. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, H.; Chu, D. Genetic structure analysis of populations of the soybean cyst nematode, Heterodera glycines, from north China. Nematology 2015, 17, 591–600. [Google Scholar] [CrossRef]
- Koenning, S.R.; Wrather, J.A. Suppression of Soybean Yield Potential in the Continental United States by Plant Diseases from 2006 to 2009. Plant Health Progress. 2010, 11, 5. [Google Scholar] [CrossRef]
- Concibido, V.C.; Diers, B.W.; Arelli, P.R. A decade of QTL mapping for cyst nematode resistance in soybean. Crop Sci. 2004, 44, 1121–1131. [Google Scholar] [CrossRef]
- Mitchum, M.G. Soybean resistance to the soybean cyst nematode Heterodera glycines: An update. Phytopathology 2016, 106, 1444–1450. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.E.; Lee, T.G.; Guo, X.; Melito, S.; Wang, K.; Bayless, A.M.; Wang, J.; Hughes, T.J.; Willis, D.K.; Clemente, T.E. Copy Number Variation of Multiple Genes at Rhg1 Mediates Nematode Resistance in Soybean. Science 2012, 338, 1206–1209. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Chen, X. Small RNAs and Their Roles in Plant Development. Annu. Rev. Cell Dev. Biol. 2009, 25, 21–44. [Google Scholar] [CrossRef] [PubMed]
- Bologna, N.G.; Voinnet, O. The Diversity, Biogenesis, and Activities of Endogenous Silencing Small RNAs in Arabidopsis. Annu. Rev. Plant Biol. 2014, 65, 473–503. [Google Scholar] [CrossRef] [PubMed]
- Chitwood, D.H.; Sinha, N.R. Plant Development: Small RNAs and the Metamorphosis of Leaves. Curr. Biol. 2014, 24, R1087–R1089. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Balyan, S.C.; Mutum, R.D.; Kansal, S.; Kumar, S.; Raghuvanshi, S. Insights into the small RNA-mediated networks in response to abiotic stress in plants. In Elucidation of Abiotic Stress Signaling in Plants; Springer: New York, NY, USA, 2015; pp. 45–91. [Google Scholar]
- Curaba, J.; Singh, M.B.; Bhalla, P.L. miRNAs in the crosstalk between phytohormone signalling pathways. J. Exp. Bot. 2014, 65, 1425–1438. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Chen, L.; Li, W.N.; Wang, L.L.; Xie, H.Y. Plant MicroRNAs Responsive to Fungal Infection. Adv. Mater. Res. 2014, 941–944, 1141–1145. [Google Scholar] [CrossRef]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Carrington, J.C. microRNA-Directed Phasing during Trans-Acting siRNA Biogenesis in Plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Achkar, N.P.; Cambiagno, D.A.; Manavella, P.A. miRNA Biogenesis: A Dynamic Pathway. Trends Plant Sci. 2016, 21, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- Park, M.Y.; Wu, G.; Gonzalezsulser, A.; Vaucheret, H.; Poethig, R.S. Nuclear processing and export of microRNAs in Arabidopsis. Proc. Natl. Acad. Sci. USA 2005, 102, 3691–3696. [Google Scholar] [CrossRef] [PubMed]
- German, M.A.; Pillay, M.; Jeong, D.; Hetawal, A.; Luo, S.; Janardhanan, P.E.; Kannan, V.; Rymarquis, L.A.; Nobuta, K.; German, R. Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nat. Biotechnol. 2008, 26, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Navarro, L.; Dunoyer, P.; Jay, F.; Arnold, B.C.; Dharmasiri, N.; Estelle, M.; Voinnet, O.; Jones, J.D.G. A plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Science 2006, 312, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Fahlgren, N.; Howell, M.D.; Kasschau, K.D.; Chapman, E.J.; Sullivan, C.M.; Cumbie, J.S.; Givan, S.A.; Law, T.F.; Grant, S.R.; Dangl, J.L. High-throughput sequencing of Arabidopsis microRNAs: Evidence for frequent birth and death of MIRNA genes. PLoS ONE 2007, 2, e219. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Fang, Y.; Feng, L.; Guo, H. Characterization of conserved and novel microRNAs and their targets, including a TuMV-induced TIR–NBS–LRR class R gene-derived novel miRNA in Brassica. FEBS Lett. 2008, 582, 2445–2452. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhao, Y.; Zhao, J.; Wang, S.; Jin, Y.; Chen, Z.; Fang, Y.; Hua, C.; Ding, S.; Guo, H. Cotton plants export microRNAs to inhibit virulence gene expression in a fungal pathogen. Nat. Plants 2016, 2, 16153. [Google Scholar] [CrossRef] [PubMed]
- Medina, C.; Rocha, M.D.; Magliano, M.; Ratpopoulo, A.; Revel, B.; Marteu, N.; Magnone, V.; Lebrigand, K.; Cabrera, J.; Barcala, M. Characterization of microRNAs from Arabidopsis galls highlights a role for miR159 in the plant response to the root-knot nematode Meloidogyne incognita. New Phytol. 2017, 216, 882–896. [Google Scholar] [CrossRef] [PubMed]
- Combier, J.; Frugier, F.; De Billy, F.; Boualem, A.; Elyahyaoui, F.; Moreau, S.; Vernie, T.; Ott, T.; Gamas, P.; Crespi, M. MtHAP2-1 is a key transcriptional regulator of symbiotic nodule development regulated by microRNA169 in Medicago truncatula. Genes Dev. 2006, 20, 3084–3088. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xue, W.; Shaopeng, Z.; Dawei, L.; Yuxi, D.; Wei, D.; Baohong, Z. Identification of Soybean MicroRNAs Involved in Soybean Cyst Nematode Infection by Deep Sequencing. PLoS ONE 2012, 7, e39650. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Li, Y.; Zhang, Q.; Xu, T.; Qiu, L.; Fan, Y.; Wang, L. Novel miRNA and phasiRNA biogenesis networks in soybean roots from two sister lines that are resistant and susceptible to SCN race 4. PLoS ONE 2014, 9, e110051. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Shichen, W.; Todd, T.C.; Johnson, C.D.; Tang, G.; Trick, H.N. Genome-wide identification of soybean microRNA responsive to soybean cyst nematodes infection by deep sequencing. BMC Genomics 2017, 18, 572. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Gao, L.; Yang, Y.; Zhai, J.; Arikit, S.; Yu, Y.; Duan, S.; Chan, V.; Xiong, Q.; Yan, J. Roles of small RNAs in soybean defense against Phytophthora sojae infection. Plant J. 2014, 79, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, Y.; Zhu, X.; Wang, Y.; Jung, K.-H.; Chen, L.; Xuan, Y.; Duan, Y. The transcriptomic changes of Huipizhi Heidou(Glycine max), a nematode-resistant black soybean during Heterodera glycines race 3 infection. J. Plant Physiol. 2018, 220, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Riggs, R.D.; Kim, K.S. Structural Changes Associated with Resistance of Soybean to Heterodera glycines. J. Nematol. 1987, 19, 177–187. [Google Scholar] [PubMed]
- Handoo, Z.A.; Anand, S.C. Biological manifestation of resistance to soybean cyst nematode development in ‘Hartwig’ soybean. Crop Prot. 1993, 12, 371–372. [Google Scholar] [CrossRef]
- Riggs, R.D. Ultrastructural Changes in Peking Soybeans Infected With Heterodera glycines. Phytopathology 1973, 63, 76. [Google Scholar] [CrossRef]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef]
- Griffithsjones, S.; Grocock, R.J.; Dongen, S.V.; Bateman, A.; Enright, A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Xia, R.; Chen, H.; He, Y. TBtools, a Toolkit for Biologists integrating various HTS-data handling tools with a user-friendly interface. bioRxiv 2018. [Google Scholar] [CrossRef]
- Jiang, N.; Meng, J.; Cui, J.; Sun, G.; Luan, Y. Function identification of miR482b, a negative regulator during tomato resistance to Phytophthora infestans. Hortic. Res. 2018, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Tan, J.; Zhou, C.; Yang, X.; Yang, F.; Zhang, S.; Sun, S.; Miao, X.; Shi, Z. The OsmiR396-OsGRF8-OsF3H-flavonoid pathway mediates resistance to the brown planthopper in rice (Oryza sativa). Plant Biotechnol. J. 2019, 17, 1657–1669. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Ma, Y.; Liu, N.; Xu, J.; Hu, Q.; Li, Y.; Wu, Y.; Xie, S.; Zhu, L.; Min, L. microRNAs involved in auxin signalling modulate male sterility under high-temperature stress in cotton (Gossypium hirsutum). Plant J. 2017, 91, 977–994. [Google Scholar] [CrossRef] [PubMed]
- Hewezi, T.; Howe, P.; Maier, T.; Baum, T.J. Arabidopsis small RNAs and their targets during cyst nematode parasitism. Mol. Plant-Microbe Interact. 2008, 21, 1622–1634. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, J.; Barcala, M.; Garcia, A.; Riomachin, A.; Medina, C.; Jaubertpossamai, S.; Favery, B.; Maizel, A.; Ruizferrer, V.; Fenoll, C. Differentially expressed small RNAs in Arabidopsis galls formed by Meloidogyne javanica: A functional role for miR390 and its TAS3-derived tasiRNAs. New Phytol. 2016, 209, 1625–1640. [Google Scholar] [CrossRef] [PubMed]
- Koter, M.D.; Świecicka, M.; Matuszkiewicz, M.; Pacak, A.; Derebecka, N.; Filipecki, M. The miRNAome dynamics during developmental and metabolic reprogramming of tomato root infected with potato cyst nematode. Plant Sci. 2018, 268, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Shukla, N.; Joshi, G.; Vijayakumar, C.; Kumar, A. Genome-wide identification and characterization of miRNAome from tomato (Solanum lycopersicum) roots and root-knot nematode (Meloidogyne incognita) during susceptible interaction. PLoS ONE 2017, 12, e0175178. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Meiling, Y.; Lu, L.; Xiaoming, Z. Diverse Functions of Small RNAs in Different Plant–Pathogen Communications. Front. Microbiol. 2016, 7, 1552. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Steffens, J.C. Overexpression of polyphenol oxidase in transgenic tomato plants results in enhanced bacterial disease resistance. Planta 2002, 215, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Masato, K.; Akiko, O.O.; Yutaka, A.; Takanobu, Y.; Koji, A.; Tohru, T.; Tsutomu, A. GMC oxidoreductase, a highly expressed protein in a potent biocontrol agent Fusarium oxysporum Cong:1-2, is dispensable for biocontrol activity. J. Gen. Appl. Microbiol. 2011, 57, 207–217. [Google Scholar]
- Hsiao, Y.M.; Liu, Y.F.; Lee, P.Y.; Hsu, P.C.; Pan, Y.C. Functional Characterization of copA Gene Encoding Multicopper Oxidase in Xanthomonas campestris pv. campestris. J. Appl. Microbiol. 2011, 59, 9290–9302. [Google Scholar]
- Zhang, W.; Gao, S.; Zhou, X.; Chellappan, P.; Chen, Z.; Zhou, X.; Zhang, X.; Fromuth, N.; Coutino, G.; Coffey, M. Bacteria-responsive microRNAs regulate plant innate immunity by modulating plant hormone networks. Plant Mol. Biol. 2011, 75, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Sudisha, J.; WenYing, Z.; Mostafa, A.; Gui, F.J. Spatio-temporal expression of miRNA159 family members and their GAMYB target gene during the modulation of gibberellin-induced grapevine parthenocarpy. J. Exp. Bot. 2018, 69, 3639–3650. [Google Scholar]
- Guan, Q.; Lu, X.; Zeng, H.; Zhang, Y.; Zhu, J. Heat stress induction of miR398 triggers a regulatory loop that is critical for thermotolerance in Arabidopsis. Plant J. 2013, 74, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Meng, Y.; Wise, R.P. Mla- and Rom1- mediated control of microRNA398 and chloroplast copper/zinc superoxide dismutase regulates cell death in response to the barley powdery mildew fungus. New Phytol. 2014, 201, 1396–1412. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Kapoor, A.; Zhu, J. Posttranscriptional Induction of Two Cu/Zn Superoxide Dismutase Genes in Arabidopsis Is Mediated by Downregulation of miR398 and Important for Oxidative Stress Tolerance. Plant Cell. 2006, 18, 2051–2065. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.J.; Chen, X.J.; Wu, X.L.; Ling, J.Q.; Xu, P. Overexpression of the AP2/EREBP transcription factor OPBP1 enhances disease resistance and salt tolerance in tobacco. Plant Mol. Biol. 2004, 55, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.A.; Jones, J.D.G. The Role of Leucine-Rich Repeat Proteins in Plant Defences. Adv. Bot. Res. 1997, 24, 89–167. [Google Scholar]
- Reinprecht, Y.; Yadegari, Z.; Perry, G.E.; Siddiqua, M.; Wright, L.C.; McClean, P.E.; Pauls, K.P. In silico comparison of genomic regions containing genes coding for enzymes and transcription factors for the phenylpropanoid pathway in Phaseolus vulgaris L. and Glycine max L. Front. Plant Sci. 2013, 4, 317. [Google Scholar] [CrossRef] [PubMed]
- Siddique, S.; Grundler, F.M.W. Metabolism in Nematode Feeding Sites. Adv. Bot. Res. 2015, 73, 119–138. [Google Scholar]
- Díaz-Manzano, F.E.; Cabrera, J.; Ripoll, J.J.; del Olmo, I.; Andrés, M.F.; Silva, A.C.; Barcala, M.; Sánchez, M.; Ruíz-Ferrer, V.; de Almeida-Engler, J. A role for the gene regulatory module microRNA172/TARGET OF EARLY ACTIVATION TAGGED 1/FLOWERING LOCUS T (miRNA 172/TOE 1/FT) in the feeding sites induced by Meloidogyne javanica in Arabidopsis thaliana. New Phytol. 2018, 217, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Li, Z.; Fan, J.; Hu, C.; Yang, R.; Qi, X.; Chen, H.; Zhao, F.; Wang, S. Identification of jasmonic acid-associated microRNAs and characterization of the regulatory roles of the miR319/TCP4 module under root-knot nematode stress in tomato. J. Exp. Bot. 2015, 66, 4653–4667. [Google Scholar] [CrossRef] [PubMed]
- Jaubertpossamai, S.; Noureddine, Y.; Favery, B. MicroRNAs, new players in the plant-nematode interaction. Front. Plant Sci. 2019, 10, 1180. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Weiberg, A.; Dellota, E.; Yamane, D.; Jin, H. Botrytis small RNA Bc-siR37 suppresses plant defense genes by cross-kingdom RNAi. RNA Biol. 2017, 14, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Gu, Y.; Jia, X.; Kang, W.; Pan, S.; Tang, X.; Chen, X.; Tang, G. Effective Small RNA Destruction by the Expression of a Short Tandem Target Mimic in Arabidopsis. Plant Cell. 2012, 24, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Nizampatnam, N.R.; Schreier, S.J.; Damodaran, S.; Adhikari, S. microRNA160 dictates stage-specific auxin and cytokinin sensitivities and directs soybean nodule development. Plant J. 2015, 84, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Hu, Z.; Chen, R.; Jiang, Q.; Song, G.; Zhang, H.; Xi, Y. Targeted mutagenesis in soybean using the CRISPR-Cas9 system. Sci. Rep. 2015, 5, 10342. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, R.; Knap, H.T.; Lewis, S.A. Inoculation Method for Studying Early Responses of Glycine max to Heterodera glycines. J. Nematol. 1998, 30, 237–240. [Google Scholar] [PubMed]
- Faghihi, J.; Ferris, J.M. An efficient new device to release eggs from Heterodera glycines. J. Nematol. 2000, 32, 411–413. [Google Scholar] [PubMed]
- Bybd, D.W.; Kirkpatrick, T.E.; Barker, K.R. An Improved Technique for Clearing and Staining Plant Tissues for Detection of Nematodes. J. Nematol. 1983, 15, 142–143. [Google Scholar] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.Y.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ruotti, V.; Stewart, R.; Thomson, J.A.; Dewey, C.N. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 2010, 26, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence ID | Raw Reads | Clean Reads | Q30 (%) |
|---|---|---|---|
| W1C_1 | 27,715,234 | 20,070,645 | 96.82 |
| W1C_2 | 27,484,434 | 21,474,884 | 96.78 |
| W1C_3 | 26,580,279 | 20,491,433 | 94.98 |
| W1N_1 | 30,925,791 | 19,880,712 | 96.79 |
| W1N_2 | 31,646,226 | 22,819,271 | 95.36 |
| W1N_3 | 42,045,429 | 23,714,940 | 97.49 |
| H1C_1 | 48,930,291 | 33,148,086 | 94.54 |
| H1C_2 | 43,635,613 | 19,622,055 | 94.57 |
| H1C_3 | 58,984,552 | 31,856,657 | 94.94 |
| H1N_1 | 55,931,293 | 36,612,046 | 96.78 |
| H1N_2 | 44,159,489 | 28,178,164 | 96.89 |
| H1N_3 | 44,233,050 | 27,250,451 | 96.87 |
| W5C_1 | 47,456,852 | 32,505,320 | 96.94 |
| W5C_2 | 46,321,364 | 21,402,542 | 96.90 |
| W5C_3 | 37,909,448 | 23,830,730 | 96.84 |
| W5N_1 | 39,218,867 | 20,669,157 | 94.60 |
| W5N_2 | 51,829,668 | 38,402,767 | 94.18 |
| W5N_3 | 51,434,403 | 34,331,810 | 94.77 |
| H5C_1 | 43,331,598 | 32,478,323 | 94.71 |
| H5C_2 | 46,365,046 | 35,279,570 | 94.27 |
| H5C_3 | 50,242,257 | 32,787,129 | 94.90 |
| H5N_1 | 33,241,813 | 25,374,292 | 96.39 |
| H5N_2 | 42,384,481 | 20,954,527 | 96.90 |
| H5N_3 | 48,968,205 | 20,332,827 | 96.97 |
| Total reads | 1,020,975,683 | 643,468,338 |
| Sequence ID | Known-miRNAs | Novel-miRNAs | Total |
|---|---|---|---|
| W1C_1 | 481 | 230 | 711 |
| W1C_2 | 489 | 232 | 721 |
| W1C_3 | 521 | 242 | 763 |
| W1N_1 | 471 | 214 | 685 |
| W1N_2 | 475 | 227 | 702 |
| W1N_3 | 494 | 213 | 707 |
| H1C_1 | 547 | 243 | 790 |
| H1C_2 | 514 | 241 | 755 |
| H1C_3 | 552 | 242 | 794 |
| H1N_1 | 540 | 245 | 785 |
| H1N_2 | 508 | 238 | 746 |
| H1N_3 | 512 | 235 | 747 |
| W5C_1 | 552 | 240 | 792 |
| W5C_2 | 495 | 238 | 733 |
| W5C_3 | 525 | 249 | 774 |
| W5N_1 | 502 | 239 | 741 |
| W5N_2 | 541 | 249 | 790 |
| W5N_3 | 555 | 249 | 804 |
| H5C_1 | 536 | 244 | 780 |
| H5C_2 | 529 | 247 | 776 |
| H5C_3 | 534 | 248 | 782 |
| H5N_1 | 519 | 232 | 751 |
| H5N_2 | 517 | 240 | 757 |
| H5N_3 | 507 | 244 | 751 |
| Total | 634 | 252 | 886 |
| DE Set | Total DE miRNA | Upregulated | Downregulated |
|---|---|---|---|
| W1C_1_W1C_2_W1C_3 vs W1N_1_W1N_2_W1N_3 | 11 | 9 | 2 |
| H1C_1_H1C_2_H1C_3 vs H1N_1_H1N_2_H1N_3 | 7 | 2 | 5 |
| W5C_1_W5C_2_W5C_3 vs W5N_1_W5N_2_W5N_3 | 3 | 3 | 0 |
| H5C_1_H5C_2_H5C_3 vs H5N_1_H5N_2_H5N_3 | 3 | 3 | 0 |
| W1N_1_W1N_2_W1N_3 vs W5N_1_W5N_2_W5N_3 | 11 | 3 | 8 |
| H1N_1_H1N_2_H1N_3 vs H5N_1_H5N_2_H5N_3 | 9 | 6 | 3 |
| W1N_1_W1N_2_W1N_3 vs H1N_1_H1N_2_H1N_3 | 10 | 3 | 7 |
| W5N_1_W5N_2_W5N_3 vs H5N_1_H5N_2_H5N_3 | 9 | 0 | 9 |
| DE Set | miRNA | Target Gene | Function Annotation |
|---|---|---|---|
| W1C vs W1N | gma-miR408a-5p | Glyma.04G248700.Wm82.a2.v1 | Xylanase inhibitor N-terminal |
| gma-miR408a-5p | Glyma.06G114200.Wm82.a2.v1 | Xylanase inhibitor N-terminal | |
| gma-miR408a-5p | Glyma.07G103400.Wm82.a2.v1 | Protein tyrosine kinase | |
| gma-miR408a-5p | Glyma.09G174000.Wm82.a2.v1 | Protein tyrosine kinase | |
| gma-miR408a-5p | Glyma.10G282000.Wm82.a2.v1 | Ubiquitin-conjugating enzyme | |
| gma-miR408a-5p | Glyma.20G107300.Wm82.a2.v1 | Ubiquitin-conjugating enzyme | |
| gma-miR4415a-3p | Glyma.13G076900.Wm82.a2.v1 | Multicopper oxidase | |
| gma-miR4415a-3p | Glyma.14G041300.Wm82.a2.v1 | Multicopper oxidase | |
| gma-miR4415a-3p | Glyma.20G051700.Wm82.a2.v1 | Multicopper oxidase | |
| gma-miR4415a-3p | Glyma.20G051900.Wm82.a2.v1 | Multicopper oxidase | |
| gma-miR4415a-3p | Glyma.20G051600.Wm82.a2.v1 | Multicopper oxidase | |
| gma-miR4415a-3p | Glyma.20G051700.Wm82.a2.v1 | Multicopper oxidase | |
| gma-miR4415a-3p | Glyma.20G051900.Wm82.a2.v1 | Multicopper oxidase | |
| H1C vs H1N | novel_miR_63 | Glyma.05G145000.Wm82.a2.v1 | ABC transporter |
| novel_miR_63 | Glyma.08G101500.Wm82.a2.v1 | ABC transporter | |
| novel_miR_63 | Glyma.08G149300.Wm82.a2.v1 | Glycosyl hydrolases family 28 | |
| novel_miR_63 | Glyma.10G231100.Wm82.a2.v1 | Glycosyl hydrolases family 28 | |
| novel_miR_63 | Glyma.15G269400.Wm82.a2.v1 | Glycosyl hydrolases family 28 | |
| novel_miR_237 | Glyma.05G145000.Wm82.a2.v1 | ABC transporter | |
| novel_miR_237 | Glyma.08G101500.Wm82.a2.v1 | ABC transporter | |
| novel_miR_237 | Glyma.08G149300.Wm82.a2.v1 | Glycosyl hydrolases family 28 | |
| novel_miR_237 | Glyma.10G231100.Wm82.a2.v1 | Glycosyl hydrolases family 28 | |
| novel_miR_237 | Glyma.15G269400.Wm82.a2.v1 | Glycosyl hydrolases family 28 | |
| gma-miR3522 | Glyma.04G121700.Wm82.a2.v1 | Polyphenol oxidase middle domain | |
| gma-miR3522 | Glyma.07G193300.Wm82.a2.v1 | Polyphenol oxidase middle domain | |
| gma-miR3522 | Glyma.07G193500.Wm82.a2.v1 | Polyphenol oxidase middle domain | |
| gma-miR3522 | Glyma.13G183200.Wm82.a2.v1 | Polyphenol oxidase middle domain | |
| gma-miR3522 | Glyma.15G071200.Wm82.a2.v1 | Polyphenol oxidase middle domain | |
| gma-miR3522 | Glyma.05G167100.Wm82.a2.v1 | Neprosin activation peptide | |
| gma-miR3522 | Glyma.08G125400.Wm82.a2.v1 | Neprosin activation peptide | |
| W5C vs W5N | gma-miR408a-3p | Glyma.02G231600.Wm82.a2.v1 | Multicopper oxidase |
| gma-miR408a-3p | Glyma.02G261600.Wm82.a2.v1 | Multicopper oxidase | |
| gma-miR408a-3p | Glyma.11G164000.Wm82.a2.v1 | Multicopper oxidase | |
| gma-miR408a-3p | Glyma.11G233400.Wm82.a2.v1 | Multicopper oxidase | |
| gma-miR408a-3p | Glyma.14G056100.Wm82.a2.v1 | Multicopper oxidase | |
| gma-miR408a-3p | Glyma.14G198900.Wm82.a2.v1 | Multicopper oxidase | |
| gma-miR408a-3p | Glyma.18G023600.Wm82.a2.v1 | Multicopper oxidase | |
| gma-miR408a-3p | Glyma.18G057200.Wm82.a2.v1 | Multicopper oxidase | |
| gma-miR408a-3p | Glyma.03G189800.Wm82.a2.v1 | Leucine Rich repeats | |
| gma-miR408a-3p | Glyma.06G142500.Wm82.a2.v1 | Leucine Rich repeats | |
| gma-miR408a-3p | Glyma.19G190200.Wm82.a2.v1 | Leucine rich repeat | |
| novel_miR_178 | Glyma.04G103900.Wm82.a2.v1 | AP2 domain | |
| novel_miR_178 | Glyma.20G224000.Wm82.a2.v1 | Myb-like DNA-binding domain | |
| H5C vs H5N | novel_miR_106 | Glyma.01G031500.Wm82.a2.v1 | Aldehyde dehydrogenase family |
| novel_miR_106 | Glyma.02G034000.Wm82.a2.v1 | Aldehyde dehydrogenase family | |
| novel_miR_106 | Glyma.16G168700.Wm82.a2.v1 | Leucine rich repeat | |
| novel_miR_106 | Glyma.15G245900.Wm82.a2.v1 | Leucine rich repeat | |
| novel_miR_106 | Glyma.17G250800.Wm82.a2.v1 | Leucine rich repeat | |
| novel_miR_106 | Glyma.09G184300.Wm82.a2.v1 | Serine hydroxymethyltransferase | |
| novel_miR_34 | Glyma.01G031800.Wm82.a2.v1 | K+ potassium transporter | |
| novel_miR_34 | Glyma.02G033600.Wm82.a2.v1 | K+ potassium transporter | |
| novel_miR_34 | Glyma.04G200600.Wm82.a2.v1 | Auxin response factor | |
| novel_miR_34 | Glyma.06G164900.Wm82.a2.v1 | Auxin response factor | |
| W1N vs W5N | gma-miR159a-5p | Glyma.01G183300.Wm82.a2.v1 | NB-ARC domain |
| gma-miR159a-5p | Glyma.15G230900.Wm82.a2.v1 | NB-ARC domain | |
| gma-miR159a-5p | Glyma.06G134200.Wm82.a2.v1 | Protein kinase domain | |
| gma-miR159a-5p | Glyma.06G258300.Wm82.a2.v1 | Protein kinase domain | |
| gma-miR5037c | Glyma.01G005400.Wm82.a2.v1 | Phosphofructokinase | |
| gma-miR5037c | Glyma.04G139400.Wm82.a2.v1 | Plant calmodulin-binding domain | |
| gma-miR5037c | Glyma.20G154800.Wm82.a2.v1 | GMC oxidoreductase | |
| gma-miR5037c | Glyma.U040400.Wm82.a2.v1 | GMC oxidoreductase | |
| gma-miR5037c | Glyma.08G275900.Wm82.a2.v1 | mTERF | |
| gma-miR5037c | Glyma.08G306000.Wm82.a2.v1 | mTERF | |
| gma-miR5225 | Glyma.15G252700.Wm82.a2.v1 | Protein tyrosine kinase | |
| gma-miR5225 | Glyma.19G130400.Wm82.a2.v1 | VQ motif | |
| novel_miR_13 | Glyma.05G232000.Wm82.a2.v1 | Leucine rich repeat | |
| novel_miR_13 | Glyma.08G039400.Wm82.a2.v1 | Leucine rich repeat | |
| novel_miR_13 | Glyma.07G103400.Wm82.a2.v1 | Protein tyrosine kinase | |
| W1N vs H1N | novel_miR_120 | Glyma.01G181900.Wm82.a2.v1 | Cytochrome P450 |
| novel_miR_120 | Glyma.09G279100.Wm82.a2.v1 | Cytochrome P450 | |
| novel_miR_142 | Glyma.04G135400.Wm82.a2.v1 | Myb-like DNA-binding domain | |
| novel_miR_142 | Glyma.18G159300.Wm82.a2.v1 | Leucine rich repeat | |
| novel_miR_200 | Glyma.20G046100.Wm82.a2.v1 | NB-ARC domain | |
| novel_miR_200 | Glyma.20G046200.Wm82.a2.v1 | NB-ARC domain | |
| novel_miR_200 | Glyma.01G112600.Wm82.a2.v1 | Multicopper oxidase | |
| novel_miR_200 | Glyma.07G133900.Wm82.a2.v1 | Multicopper oxidase | |
| W5N vs H5N | gma-miR398d | Glyma.05G055000.Wm82.a2.v1 | Copper/zinc superoxide dismutase (SODC) |
| gma-miR398d | Glyma.11G236800.Wm82.a2.v1 | Multicopper oxidase | |
| gma-miR862b | Glyma.01G062400.Wm82.a2.v1 | Copper amine oxidase | |
| gma-miR862b | Glyma.01G063700.Wm82.a2.v1 | SBP domain | |
| gma-miR862b | Glyma.02G121300.Wm82.a2.v1 | SBP domain | |
| gma-miR862b | Glyma.04G254300.Wm82.a2.v1 | Serine hydroxymethyltransferase | |
| gma-miR862b | Glyma.06G107800.Wm82.a2.v1 | Serine hydroxymethyltransferase | |
| gma-miR862b | Glyma.10G036700.Wm82.a2.v1 | AP2 domain | |
| gma-miR862b | Glyma.13G123100.Wm82.a2.v1 | AP2 domain | |
| gma-miR862b | Glyma.11G166300.Wm82.a2.v1 | Glutaredoxin | |
| gma-miR862b | Glyma.11G232300.Wm82.a2.v1 | Glutaredoxin | |
| novel_miR_105 | Glyma.01G043300.Wm82.a2.v1 | WRKY DNA -binding domain | |
| novel_miR_105 | Glyma.13G365600.Wm82.a2.v1 | Glycosyl hydrolases family 17 | |
| novel_miR_105 | Glyma.15G007600.Wm82.a2.v1 | Glycosyl hydrolases family 17 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, P.; Han, B.; Wang, Y.; Zhu, X.; Xuan, Y.; Liu, X.; Fan, H.; Chen, L.; Duan, Y. Identification of MicroRNAs That Respond to Soybean Cyst Nematode Infection in Early Stages in Resistant and Susceptible Soybean Cultivars. Int. J. Mol. Sci. 2019, 20, 5634. https://doi.org/10.3390/ijms20225634
Lei P, Han B, Wang Y, Zhu X, Xuan Y, Liu X, Fan H, Chen L, Duan Y. Identification of MicroRNAs That Respond to Soybean Cyst Nematode Infection in Early Stages in Resistant and Susceptible Soybean Cultivars. International Journal of Molecular Sciences. 2019; 20(22):5634. https://doi.org/10.3390/ijms20225634
Chicago/Turabian StyleLei, Piao, Bing Han, Yuanyuan Wang, Xiaofeng Zhu, Yuanhu Xuan, Xiaoyu Liu, Haiyan Fan, Lijie Chen, and Yuxi Duan. 2019. "Identification of MicroRNAs That Respond to Soybean Cyst Nematode Infection in Early Stages in Resistant and Susceptible Soybean Cultivars" International Journal of Molecular Sciences 20, no. 22: 5634. https://doi.org/10.3390/ijms20225634
APA StyleLei, P., Han, B., Wang, Y., Zhu, X., Xuan, Y., Liu, X., Fan, H., Chen, L., & Duan, Y. (2019). Identification of MicroRNAs That Respond to Soybean Cyst Nematode Infection in Early Stages in Resistant and Susceptible Soybean Cultivars. International Journal of Molecular Sciences, 20(22), 5634. https://doi.org/10.3390/ijms20225634