1. Introduction
Colorectal cancer (CRC) is initiated when the colonic epithelial loses the function of the adenomatous polyposis coli (APC) pathway which is essentially a part of Wnt signaling pathway [
1,
2]. Loss of APC function results in nuclear accumulation of β-catenin which triggers a series of modulations to several transcription factors; an important one being TCf-4, which in turn upregulates several target genes including the cyclin D1 proto oncogene [
3]. Ultimately, APC mutations affect the G1 to S transition of the cell cycle causing cell growth dysregulation and initiating the formation of intestinal polyps [
4]. The APC mutant mice developed many adenomas in their small intestines and a few in the large intestine that rarely progress to invasive adenocarcinoma [
1]. To overcome the drawbacks, Henoi et al. [
5] developed mice (CPC; APC) carrying CDX2PNLS Cre recombinase transgene and a loxP-targeted APC allele that led to the development of primarily colorectal tumor in 17% of the mice when followed for 300 days. These CPC; APC animals resembled human colorectal lesions with biallelic APC inactivation, β-catenin dysregulation, global DNA hypomethylation, and aneuploidy. A current view is that most colorectal cancers arise from adenomatous precursors and accumulate gain-of-function mutations in proto-oncogenes and loss-of-function mutations in tumor suppressor genes resulting in the initiation of adenomatous lesions and progression to carcinoma [
6,
7]. The development of neoplasia is a multistep process involving the inactivation of a variety of tumor-suppressor and DNA-repair genes and simultaneous activation of certain oncogenes.
Numerous mouse models of CRC have been developed, providing insights into pathogenesis mechanisms, tools for discovery, validation of novel therapeutic targets, and a predictive platform in which to test new prevention and therapeutic strategies [
1]. The orthotropic and syngeneic mice models are commonly used as a surrogate of CRC where mouse CRC cell lines harboring the KRAS mutation are used to generate a KRAS mutated CRC mimicry.
The fact that CRC patients harboring the KRAS mutation in their neoplastic tissues have detrimental effect upon administration of monoclonal antibodies like cetuximab and panitumumab, which has otherwise improved the median survival of KRAS wild type patients, has prompted the necessity to precisely understand the molecular events caused by KRAS mutation in the CRC microenvironment. To achieve this goal a genetic mouse model of KRAS mutated CRC is essential.
Moreover, based on the current knowledge, 80% of the sporadic CRC population harbor APC mutation while 45% of all CRC patients have KRAS mutation [
6]. This indicates that at least 25% of CRC patients have both APC and KRAS mutations. Thus, constructing a model that carries both APC and KRAS mutations is also important for thorough molecular evaluation of the cancer. Though, it is relevant to mention that attempts to generate such models with APC inactivation and KRAS mutation has proven to be embryonically lethal thus making it impossible to have a model of KRAS mutated CRC.
In the current study we used simple strategies to develop conditional genetic mouse models where both the alleles, namely floxed APC and mutated KRAS, can be simultaneously expressed by tamoxifen, which activates the large intestine specific transcription factor CDX2 through CRE recombinase fused to mutated estrogen receptor ERT2 [
8]. The more efficient and specific CRE-ERT2 can only gain access to the nuclear compartment after exposure to tamoxifen. When these mice are bred with mice containing loxP-flanked sequence, tamoxifen-inducible, Cre-mediated recombination will result in deletion of the floxed sequences in the Cre-expressing cells of the offspring. Following this strategy, we have successfully generated and molecularly characterized the model to establish resemblance to human KRAS mutated CRC. This model is thus far the most robust model to investigate the KRAS mutated CRC.
3. Discussion
Animal models for human colon cancer can be useful for studying the mechanism of cancer development and for testing cancer prevention and treatment approaches. However, several recent reviews clearly show that the available models are flawed and the cancer that develops in these models is often significantly different from human colon cancer in terms of latency, intestinal location, or molecular signature [
22]. Several animal models have been developed in the past decades namely APC
min (adenomatous polyposis coli), AOM (azoxymethane), MNU (
N-methyl-
N-nitrosourea), MMR (mismatch repair), TGF-β, (transforming growth factor-beta) xenograft, and the non-murine zebrafish models of human colorectal cancer (CRC) disease [
23]. Further, the orthotopic model has the advantage of closely mimicking human CRC including tumor microenvironment [
24,
25,
26]. It recapitulates all of the critical components of the tumor microenvironment, as well as all of the angiogenic factors, growth factors, and cytokines, thus finely mimicking the human CRC in terms of both metastasis and microenvironments, allowing evaluation of the alterations in and modulations of the microenvironment on tumorigenesis and progression [
24]. Recently one of the orthotopic models has been extended and refined into a sophisticated GEMM-derived orthotopic transplant model of KRAS-mutant colorectal cancer for high-throughput drug discovery screening and candidate drug validation [
27]. This model has its own drawbacks wherein it is difficult to recapitulate the adenoma carcinoma transition to understand initiation and inception of neoplasia.
Our genetic model represents KRAS mutation and APC gene truncation, which is genetically identical to KRAS mutated CRC patients. As the onset of the cancer is tightly controlled under tamoxifen inducible CDX2 Cre, the changing etiology of the disease can be monitored in a regulated fashion which would be helpful in determining the changes at the different time points of disease progression. APC and KRAS are the most frequent mutations encountered in CRC [
28] and our model can express both the mutation simultaneously along with definitive development of neoplasm.
Development of a preclinical animal model that mimics KRAS mutated colorectal cancer is urgent. Colorectal cancer patients with tumors harboring KRAS mutation are excluded from receiving the otherwise beneficial therapy of anti-EGFR monoclonal antibodies, and currently have no alternate FDA-approved treatment options available. Ongoing research requires validations of different modalities on a robust preclinical study platform that will resemble the disease phenotype and genetic profile. Heterozygous mutation of KRAS oncogene along with deletion mutation of adenomatous polyposis coli (APC) gene is embryonically lethal in animal models. In this context we have developed a viable mouse model of colorectal cancer bearing KRAS mutation that authentically serves the purpose of a preclinical model for the human CRC disease.
The final model was generated by the cross of the KRAS
+/− colony and CDX2Cre APC
f/f colony when the desirable APC
f/f KRAS
+/− CDX2-Cre-ERT2 was developed, genotyped, and characterized. The model animals were under a tamoxifen inducible molecular switch. Micro PET scan was utilized to detect and measure tumor volume and standard uptake value (SUV). H&E staining was done to establish neoplasm and immunohistochemistry was performed to determine histological similarities with human FFPE biopsies. The MSI/MSS status was also determined. Our model tends to resemble MSS characteristics, which is prevalent in about 85% of the human CRC population. Although the human CMS3 subgroup includes patients with mutated KRAS and are characterized by a mixed MSI status [
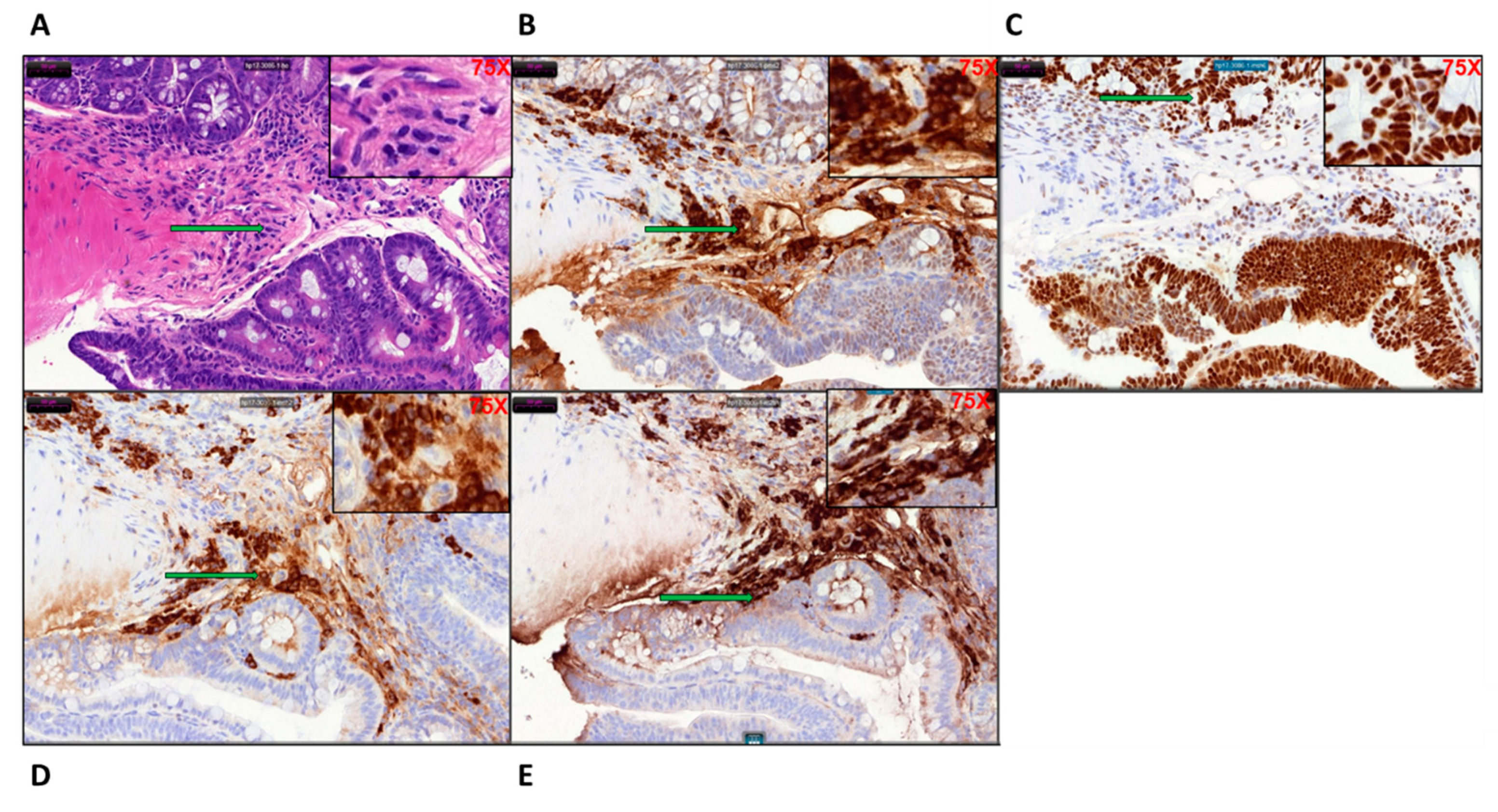
29], this group possibly represents a transition phenotype or intra-tumoral heterogeneity. Our data show strong staining for MSH6 and PMS2 but a lesser degree of staining for MLH1 and MSH2, which is probably an indication of this transition phenotype.
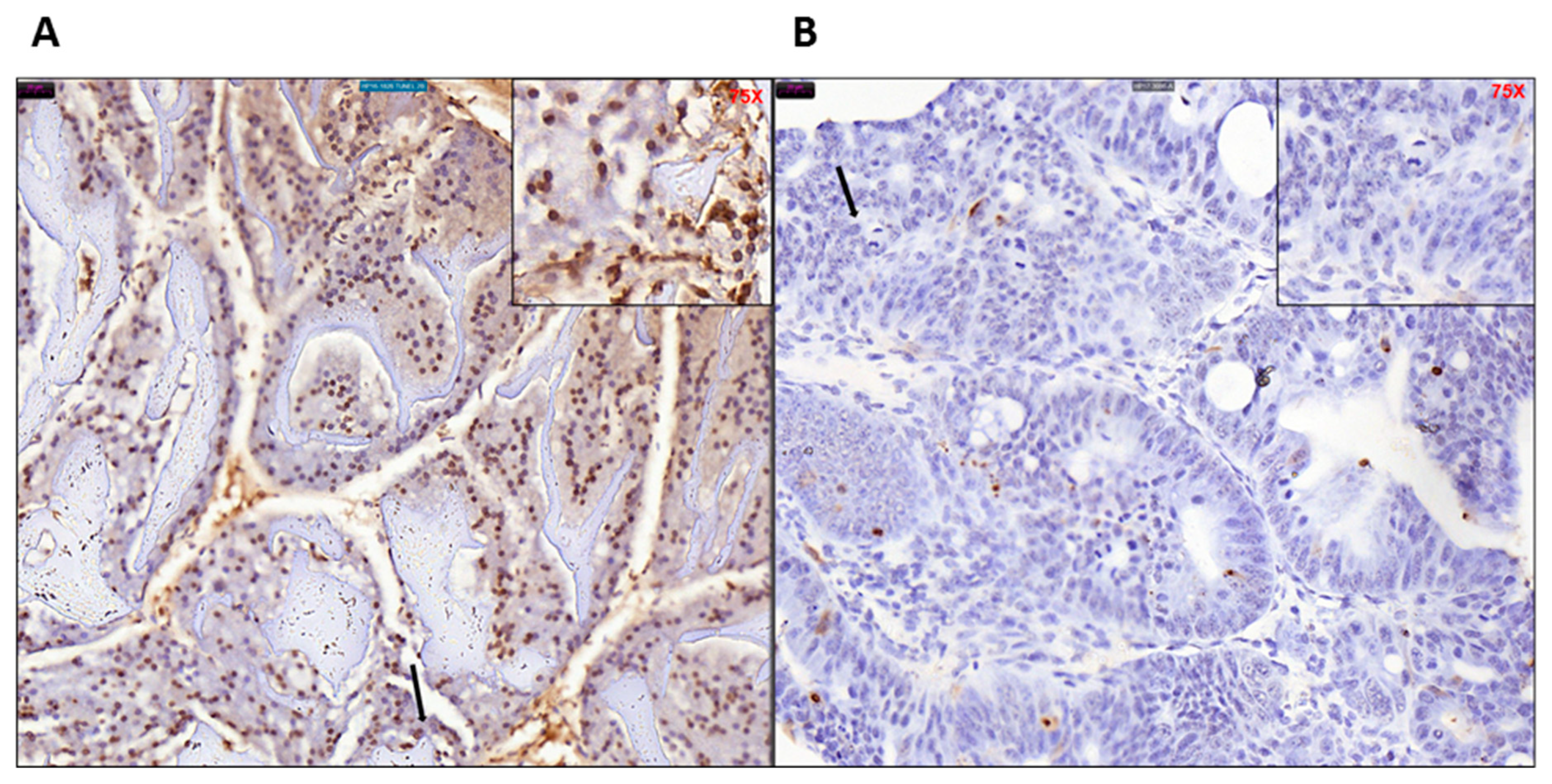
Due to the CDX2 tissue specific promoter characteristics, the onset of focal lesion was found to be initiated at the starting of the large bowel right at the ileocecal junction. In the early stages inflammation and focal lesion were most prominent in the cecum region and progressed to the entire colon. The rectum did not show any anomaly during the entire study. We have clearly established that the neoplasm developed in our KPC: APC model closely resembles the human CRC with distinct upregulation of HOX B6 and HDAC 3 and suppression of the expression of cyclin Kinase inhibitor P21. Similarly, the downregulation of mediators of programmed cell death that includes the initiator caspases (caspase 2, 8, 9), and executioner caspases (caspase 3 and 7) clearly support the transformed nature of the tamoxifen-induced tumors. The prominent upregulation of BCl2 and downregulation of BAD further confirms the fact. Furthermore, this model also provides the flexibility of inducing the disease at a definite time point and age fashion. Prior to induction, the animals are phenotypically normal. The CDX2-ERT2 molecular switch is very tight that we have not encountered a single animal that developed a tumor or disease phenotype without tamoxifen induction. It has not been possible to phenotypically distinguish between the positive, negative, and the experimental group without tamoxifen induction. Hence during later studies, we compared the tamoxifen treated and untreated animal within the experimental group only. The two parental strains which initially served as our positive and negative control showed limited effects with tamoxifen induction where the negative control did not develop tumors or mutant KRAS expression at the maximum dosage of tamoxifen (TAM) (1 mg/20 g body weight i.p. injection for five consecutive days) and the positive control developed a tumor with very low SUV with a prolonged period of latency (approx. 85–100 days) post tamoxifen administration at the highest dose. On the contrary our model rapidly developed aggressive KRAS mutated high SUV tumors at a very low (single injection of 100 μg) tamoxifen injection. The aggressiveness and the latency period were dependent on the concentration of tamoxifen making this model a unique tool for elucidation of the molecular mechanism of CRC. Additionally, unlike the human population, these animals have identical genetic backgrounds and can efficiently serve to analyze the complex cancer genomics under various treatment conditions similar to studies described [
28] in the human population and this will further our knowledge of the disease etiology. Our model includes mutations in APC and KRAS, and therefore represents the most common genetic alterations detected in human colorectal cancer specimens. A survey of the c-bio portal dataset across seven studies encompassing 3475 samples revealed that the alterations observed included APC (61%), TP53 (54%), KRAS (35%), PIK3CA (19%), BRAF (11%), and NRAS (5%). Among these, the KRAS, NRAS, and BRAF have clinical significance.
Induction of biallelic APC loss and heterozygous KRAS mutation in our mice led to the development of cancers in the proximal colon including the cecum. A survey of the SEER dataset [
30], among 180,605 patients, 31% of patients had a proximal cancer including the appendix, cecum, or ascending colon. Induction of more distal tumors including the sigmoid colon, recto sigmoid, and the rectum represent 50% of incidence in humans continues to remain a challenge. For example, in the APC: min model of biallelic APC knockout mice, the tumors observed are benign small bowel adenomas. It is fair to state, that there is a biological preference in mice to develop tumors proximally rather than distally. The early development of proximal tumors forces intervention to maximize the experimental output in the now limited life expectancy of the animal. The key may lie in gene manipulation such that the carcinogenesis is controlled at a rate that will facilitate more distal tumor development. In keeping with the above hypothesis, the introduction of a KRAS mutation combined with the controlled induction of biallelic APC loss in our model has moved the development of the tumor more distally, from the small bowel to the proximal colon, while simultaneously changing the histology from a benign adenoma to a malignant adenocarcinoma. Thus, the KPC: APC model will serve as an advanced preclinical platform for better understanding the disease mechanism at the molecular level as well as for evaluation of various therapeutics.
Our model provides a robust platform to study the intricacies of oncogenic KRAS mutation that complicates cancers like that of the colon. We have successfully developed a viable animal model of KRAS mutated colorectal cancer with tamoxifen inducible tumors harboring KRAS and APC mutations. The animal model tumors mimic the human disease type both genetically, immunohistochemically, and molecularly. This model can serve as an authentic preclinical platform for understanding the disease mechanism at the molecular level as well for evaluation of various therapeutic interventions.
4. Methods
4.1. Animal Colonies
All experimental procedures were performed after approval from the Albert Einstein College of Medicine, Animal Welfare Committee on Use and Care of Animals (IACUC, 2017206, on 13 March 2019) and according to the New York State and U.S. federal regulations. All the mice were housed in specific pathogen-free conditions within the barrier facility.
4.2. Animal Breeding
The final model mice with tamoxifen-inducible KRAS G12D expression (KPC APC) was derived by breeding a Cre
+/− APC
f/f with KRAS
+/−Apc
f/f mice. Genotyping for Cre
+/−, KRAS G12D mutation, and APC
f/f genes was done by TransnetYX using the primer probes found in
Table S2. The breeding was initiated by crossing C57 BL/6 mice with truncated APC floxed allele and heterozygous KRAS floxed outbred mice to generate an APC
f/f KRAS
+/f mouse colony. In another set of breeding APC floxed mice were crossed with CDX2-Cre-ERT2 mice and selected for APC
f/f CDX2-Cre-ERT2 after a second round of inbreeding. The final model (APC
f/f KRAS
+/f CDX2-Cre-ERT2) was generated by crossing the two parental strains APC
f/f KRAS
+/f and APC
f/f CDX2-Cre-ERT2.
4.3. Tumor Generation
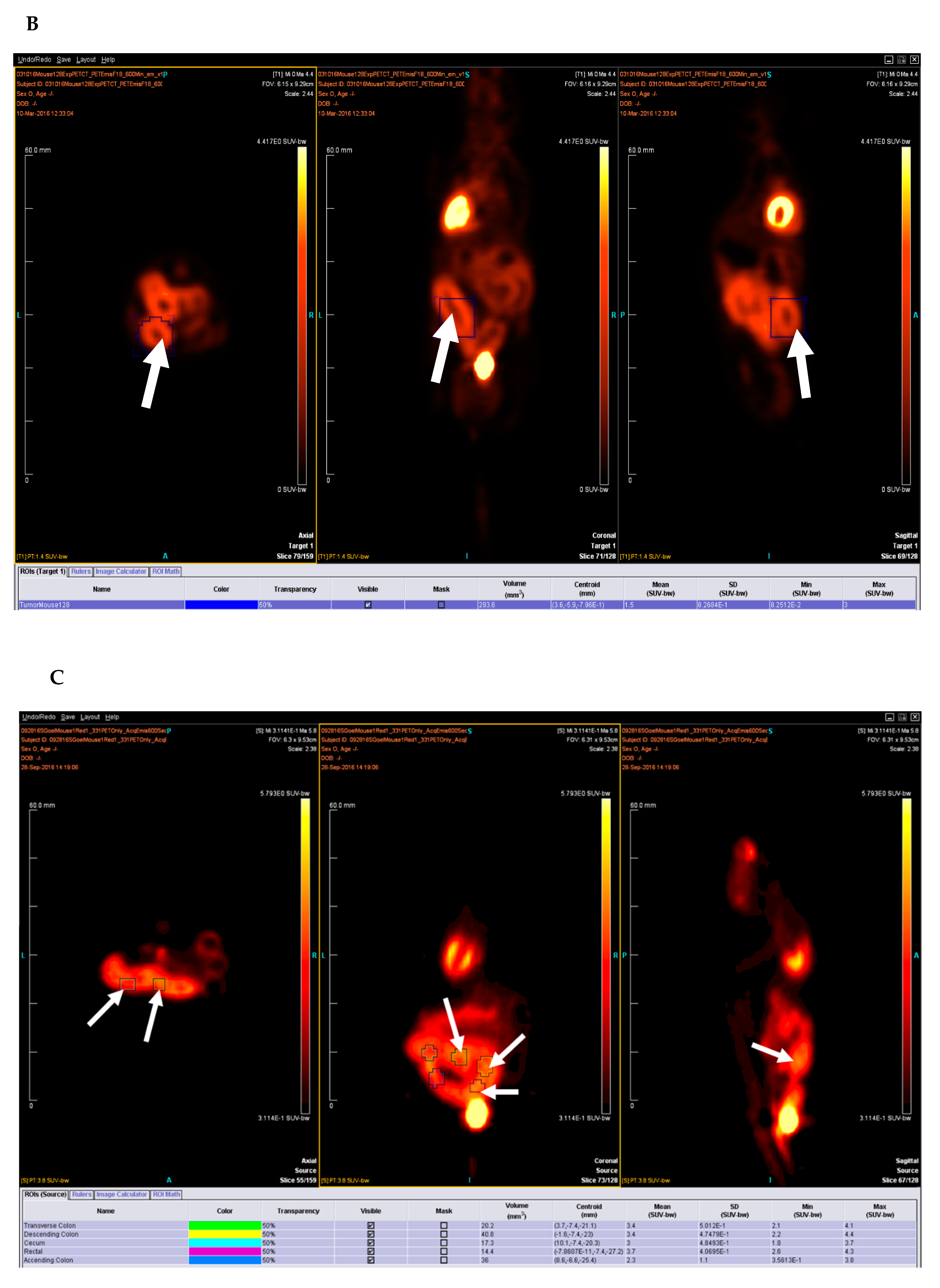
Eight to 10-week old APCf/f KRAS+/f CDX2-Cre-ERT2 mice were injected with 0.1 mg of tamoxifen (TAM) dissolved in corn oil (Sigma-Aldrich, St. Louis, MO, USA # T5648 and #C8267) per 20 g body weight via intraperitoneal route (i.p.) and was monitored for cancer development. Mice that reached critical condition reported by ≥10% weight loss and focal growth with standard uptake value (SUV) > 2.5 as determined by PET/CT scan, as well as the remaining mice in the experimental cohort were then sacrificed for tissue collection. Mice were euthanized using isoflurane. The cecum and the inflamed colon tissue were excised, washed in sterile double distilled water, followed by sterile 1× PBS, frozen in liquid nitrogen, and preserved at −80 °C until further analysis. The optimal dose of tamoxifen necessary to induce a tumor was determined by titrating the animals with a single dose of intraperitoneal (i.p.) tamoxifen injection at various concentrations ranging from 1 mg/20 g body weight to 1 μg/20 g body weight.
4.4. PET/CT Scan
4.4.1. Positron Emission Tomography (PET) and Computed Tomography (CT) Methods in the Micro-PET Imaging Facility
PET/CT scans are commonly used in humans for diagnostic imaging of many conditions including cancer and inflammation [
31]. Micro-PET extends this tool to small animal imaging, allowing for research applications, mostly in mice and rats [
32]. [18F] fluoro-2-deoxyglucose (FDG) is a radioactive positron emitting radiotracer used in this study.
4.4.2. Animal Preparation
All mice were imaged after 8–12 h of fasting, secured to the imaging palette with a breathing tube over their snout to supply 1.5% isoflurane-oxygen mixture anesthesia to continue through the imaging portion of the procedure. Each mouse was placed on a heating pad before and during scanning to maintain normal body temperature. Mice were injected with 300–400 uCi (12–15 MBq) in 0.1 mL normal saline [18F] fluoro-2-deoxyglucose (FDG) via the tail vein and imaging began 1 h after injection.
4.4.3. Image Acquisition
Imaging was performed on an Inveon Multimodality scanner (Siemens Medical Solutions USA, Malvern, PA, USA), in which CT rays are generated by 80 kV peak voltage difference between cathode and tungsten target at 0.5 mA current and 200 milli second exposure time. The CT field of view was 5.5 cm by 8.5 cm with an overall resolution without magnification of 50 microns. After each acquisition, data were sorted into 3D sinograms, and images were reconstructed using a 2D-ordered subset expectation maximization algorithm. Data were corrected for deadtime counting losses, random coincidences, and the measured non-uniformity of detector response.
4.4.4. Image and Data Analysis
Analysis is performed using either ASIPRO or IRW (both Siemens Medical Solutions USA, Malvern, PA, USA) dedicated software. All image studies are inspected visually in a rotating 3D projection display to examine for interpretability and image artifact. Manual regions of interest (ROI) are defined around areas of pathologic uptake and compared with co-registered CT image data. Successive scrolling through 2D slices (each 1.2 mm thick in the axial images) permits measurement of a radioactivity within defined volumes. The activity concentration within this volume divided by the activity per gram of body mass of total injected radioactivity determines the standardized uptake value (SUV).
4.5. Gross Anatomy Upon Dissection
The animals were euthanized by carbon dioxide asphyxiation placed on a paraffin tray in ventral position and the limbs were stretched and pinned. Incision was made vertically along the peritoneal wall to expose the abdomen. Starting at the rectum/colon, the coiled intestines was unzipped by pulling gently, using scissors and the surface of the entire intestine was photographed. Colectomy of the inflamed colon was performed and longitudinally cut open to visualize the internal morphology of the mucosa.
4.6. Histopathology
The dissected and opened colon was fixed in neutral buffered formalin and subjected to paraffin embedding by Swiss roll method [
33]. The paraffin embedded untreated and tamoxifen treated, mouse colon sections were deparaffinized and hydrated by transferring them through the following solutions: twice in xylene for 5 min, twice in 96% ethanol, 90% ethanol, 80% ethanol, and finally double distilled water (DDW), for 3 min. Routine hematoxylin and eosin staining was performed, and histopathological evaluation of neoplastic tumor was confirmed by a pathologist (Q.L.).
Immunohistochemical staining was performed for pan-cytokeratin A1/3 (abcam Cambridge, UK # AB27988) to establish the epithelial origin of the poorly differentiated tumor tissues. Primary monoclonal antibodies against MLH1 (clone ES05, Novocastra, Leica Biosystems Newcastle Ltd., Newcastle Upon Tyne, UK, 1:30), MSH2 (clone FE11, Calbiochem, Merck KGaA, Darmstadt, Germany, 1:50), MSH6 (clone EPR3945, Epitomics Inc, Burlingame, CA, USA, 1:200), and PMS2 (clone EPR3947, Epitomics Inc, Burlingame, CA, USA, 1:200) were applied to 5-μm-thick 10% formalin-fixed, paraffin-embedded tissue sections. The sections were deparaffinized in xylene three times, 10 min each, and subsequently rehydrated through graded alcohols to distilled water. Antigen heat retrieval was performed in 1 mM EDTA (pH 9.0) for 10 min (PMS2 15 min) using a microwave oven. Next, the sections were cooled down in room temperature for 1.5 h. After rinsing in distilled water and TBS successively, sections were incubated with specific monoclonal antigen at 4 °C for 2 h. Finally, the sections were covered with streptavidin peroxidase (Dako, Santa Barbara, CA) diluted 1:100 in PBS, incubated for 30 min at 37 °C, washed three times in PBS, and stained with 3,3′-diaminobenzidine as a substrate for the peroxidase for approximately 30 min at 37 °C. Counter staining was performed using Mayer’s hematoxylin. Cells were viewed under 40× magnification in a light microscope.
4.7. Morphometry
Slides were scanned on the 3DHistech P250 slide scanner (SIG #1S10OD019961-01) (Budapest, Hungary). Brown stain analysis was completed on the whole piece of tissue on every slide with 3DHistech Quant Center, using the DensitoQuant module. In this module, brown stain pixels were distinguished from the rest of the tissue by color thresh-holding. The analysis of blue versus brown areas of tissue was completed in ImageJ, using the color threshold module.
4.8. RNA Sequence Analysis
High-throughput mRNA sequencing (RNA-seq) offers the ability to quantify genes, analyze transcriptomes, and measure transcript expression in a single assay as described elsewhere [
30]. Essentially cDNA was synthesized and put to high-throughput transcriptome sequencing for the 56 CRC cell lines analyzed for this study. The 56 cells lines were separated based on KRAS status. The constitutive expression of gene of interest was computed in the two groups and the mean and standard error of mean was determined. The detailed methodology of the high throughput sequencing and the cutoff and threshold are detailed in the following reference [
9].
4.9. Protein Isolation and Western Blot
The whole tissue lysate was collected by mincing tissue into fine pieces then grinding with a pestle and incubating for 40 min in RIPA buffer containing protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA # P8340). The lysate was then centrifuged at >12,000 rpm for 1 h to remove tissue debris. For immunoblots, the protein estimation was performed by Bradford assay and 50 µg of protein lysate was loaded into each lane. Protein was resolved by SDS-PAGE electrophoresis and transferred to PVDF(polyvinylidene difluoride) membranes then probed with primary antibodies (
Table S3). The blot was then incubated with HRP-conjugated secondary antibodies (
Table S3) and visualized by chemiluminescence using Clarity Western ECL (enhanced luminol-based chemiluminescent) substrate (Bio-Rad, Hercules, CA, USA# 170-5061). Visualization and densitometry were done using Li-Cor Odyssey Fc imaging system. For densitometry, the band intensity was quantified using Image Studio Lite software (Li-Cor Corporate, Lincoln, NE, USA). The quantified intensity of each sample was normalized to the intensity of its respective housekeeping protein: B-actin or GAPDH.
4.10. Active KRAS Pull Down Assay
For active KRAS pull down assay, the tissue lysate was obtained by mincing and grinding the cecum tissue for 2 min in NP-40 lysis buffer (Cell biolabs, Inc, San Diego, CA, USA# STA-400-K) containing protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA, cat. no. P8340), then centrifuged for 2 min at 14,000 rpm to removed tissue debris. Then, 4 mg of tissue lysate were used for the pull down assay according to the kit protocol (Cell biolabs, Inc, San Diego, CA, USA # STA-400-K).
4.11. mRNA Isolation and RT-qPCR Analyses
Total RNA was isolated and purified using Qiagen’s RNeasy Mini kit (Qiagen, Hilden, Germany#74106). RNA concentration was estimated using Nanodrop. Then, 500 ng of RNA was used to synthesize cDNA with iScript Reverse Transcription Supermix (Bio-Rad, Hercules, CA, USA# 1001708841). For qPCR, the stock of standard sample was made from a pool of cDNA collected from 17 mice. cDNA was diluted 1:25 times with sterile double distilled water and mixed with primers (
Table S4) Power Up SYBR Green mix (Thermo Fisher, Waltham, MA, USA # A25777), then amplified using the cycle and condition detailed in
Table S5. The amplification was performed using Bio-Rad CFX96 Real Time System (Hercules, CA, USA). The primers used in the experiment were obtained from Millipore Sigma (Burlington, MA, USA). The GAPDH primer (
Table S4) was amplified and used as a housekeeping gene for normalization.
4.12. TUNEL Assay
Mouse colon sections were stained in duplicate using ApopTag Plus Peroxidase in Situ Apoptosis Detection Kit (Millipore, Burlington, MA, USA# S7101). The paraffin embedded mouse colon sections were deparaffinized and hydrated by transferring them through xylene to ethanol washings. Samples were pretreated with proteinase K (20 μg/mL), quenched with hydrogen peroxide, and incubated with equilibration buffer. After washing, samples were incubated with TdT enzyme at 37 °C for 1 h, then incubated with anti-digoxigenin conjugate for 30 min at room temperature and washed 4 times with PBS for 2 min each afterward. After staining, samples were developed with peroxidase substrate, washed, counterstained, then mounted onto glass slides.
4.13. Statistical Analysis
Mean of data collected from 3 mice was generated for the tamoxifen treated group and untreated group in Western blot densitometry and RT-qPCR analysis. Two tailed t-test was used to determine statistical significance (p < 0.05). Statistics were calculated using Microsoft Office Excel.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









