The Hypervariable Region of K-Ras4B Governs Molecular Recognition and Function

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
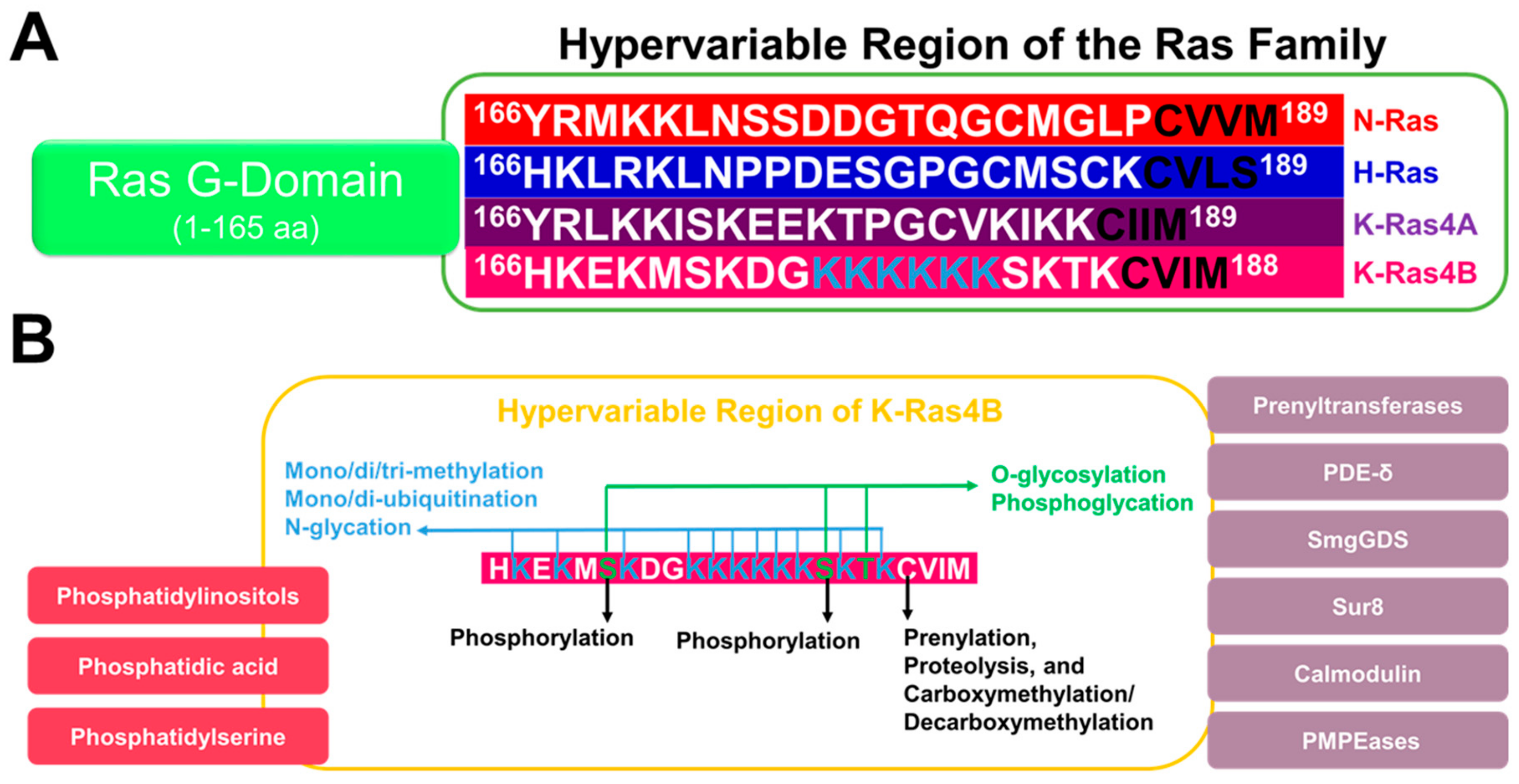
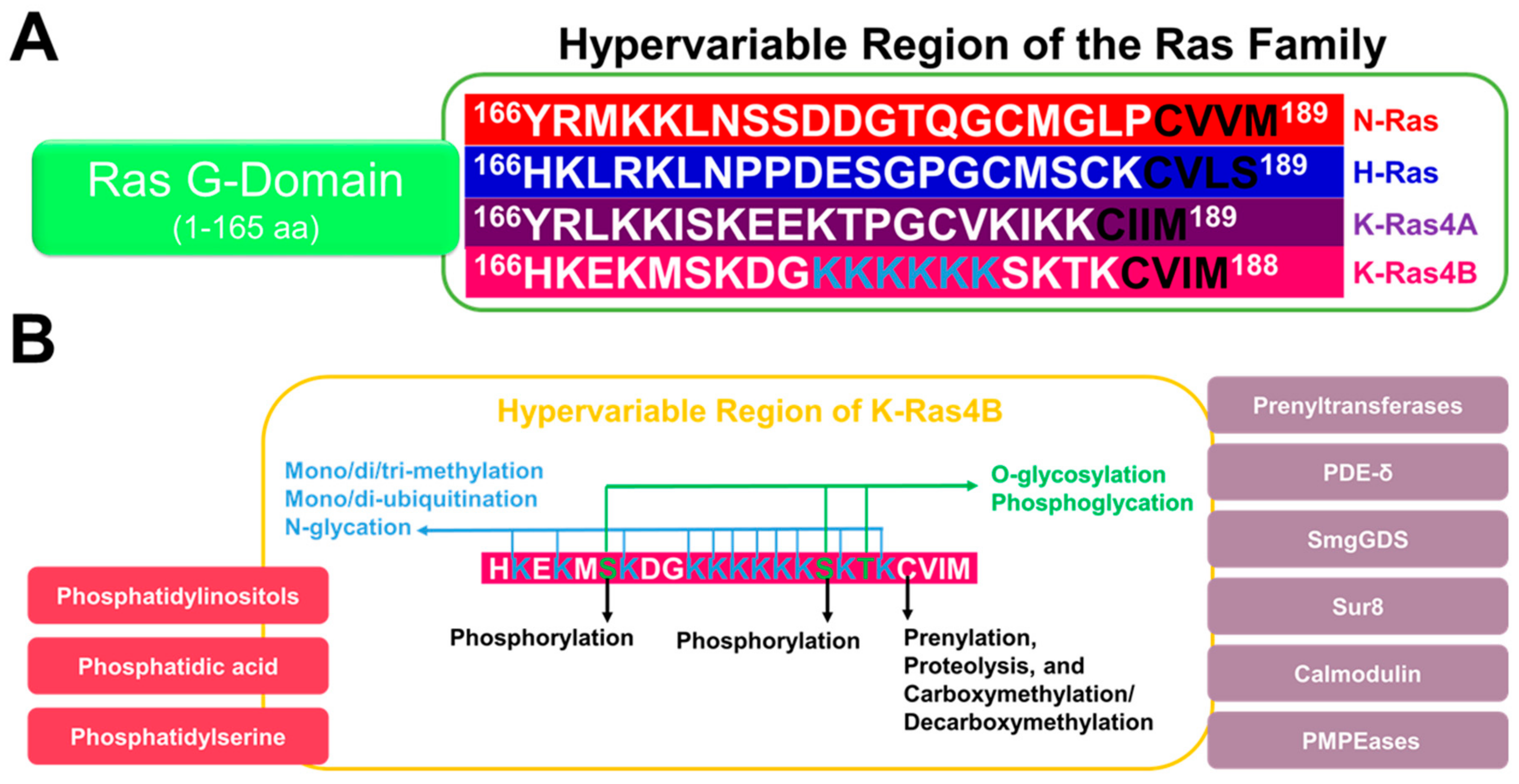{kind=link}
1. Introduction
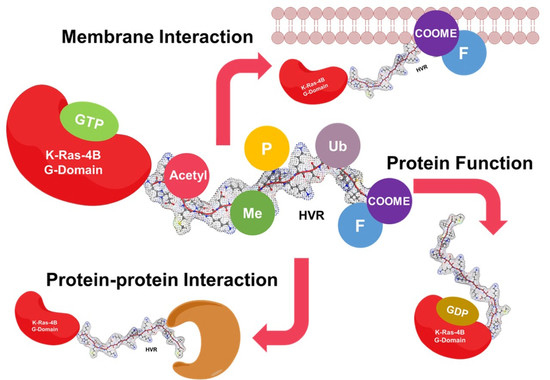
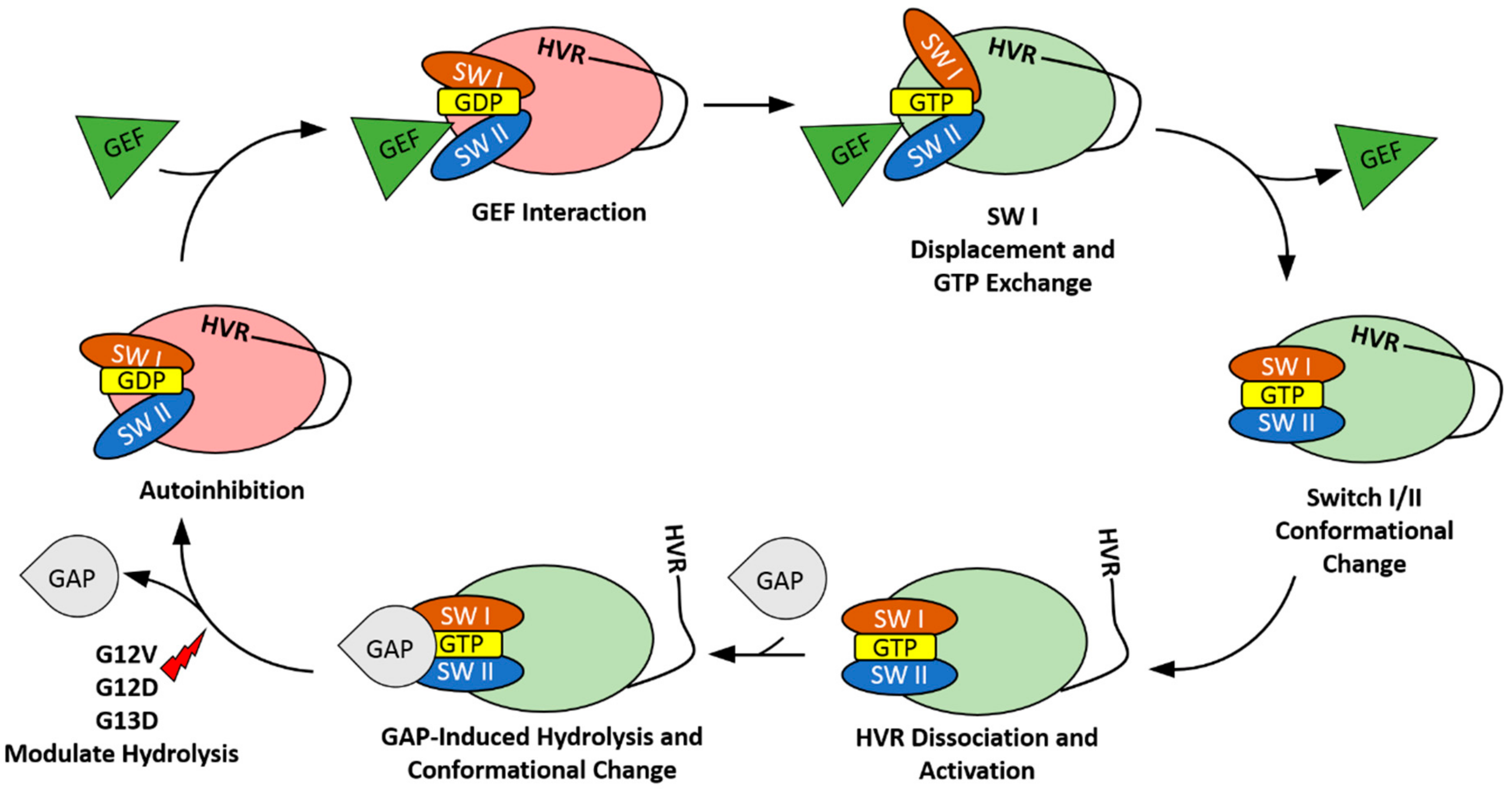
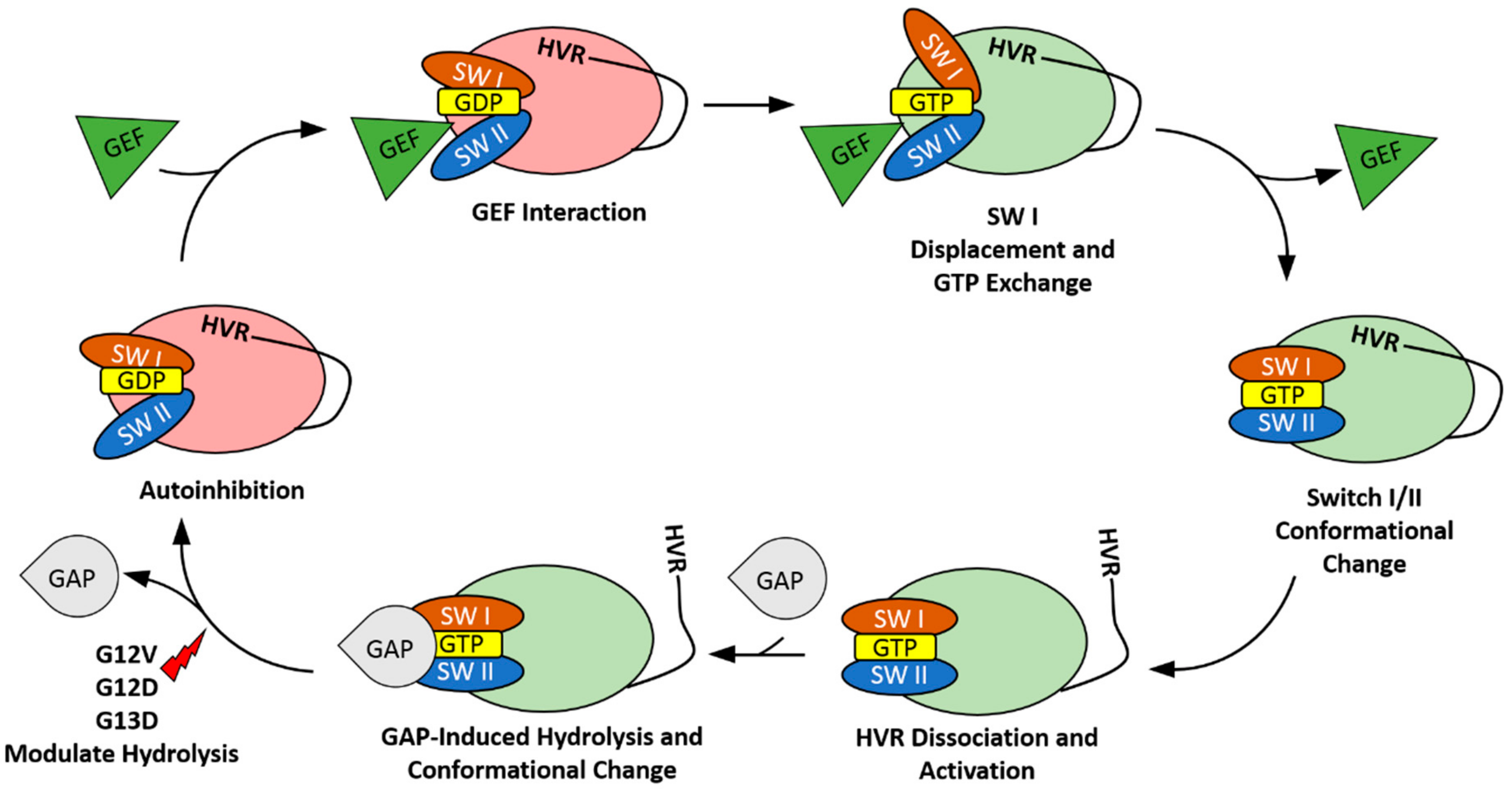
2. The HVR Interacts with the G-domain
3. The HVR Participates in Protein–Protein Interactions
4. The HVR Binds to the Plasma Membrane
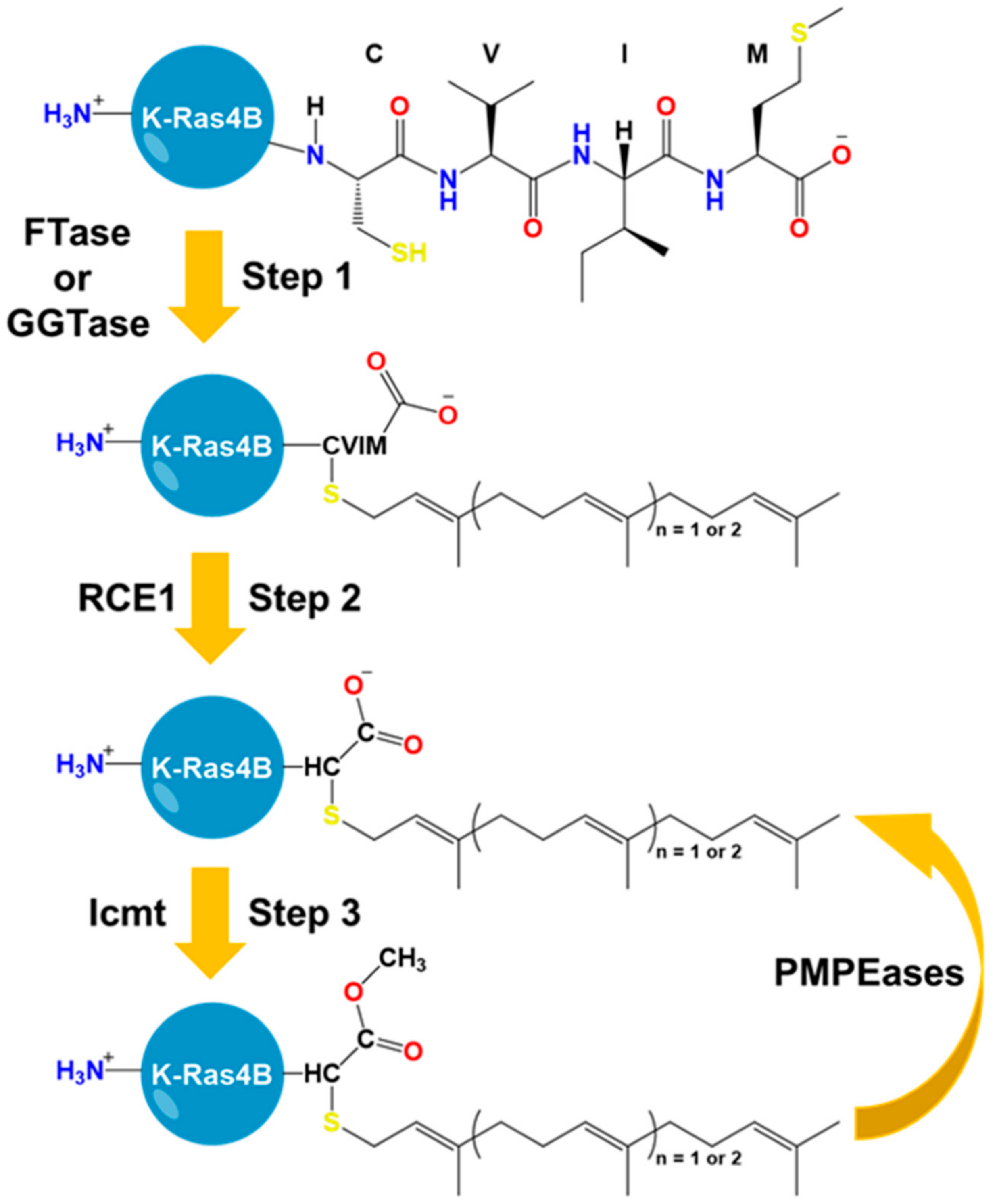
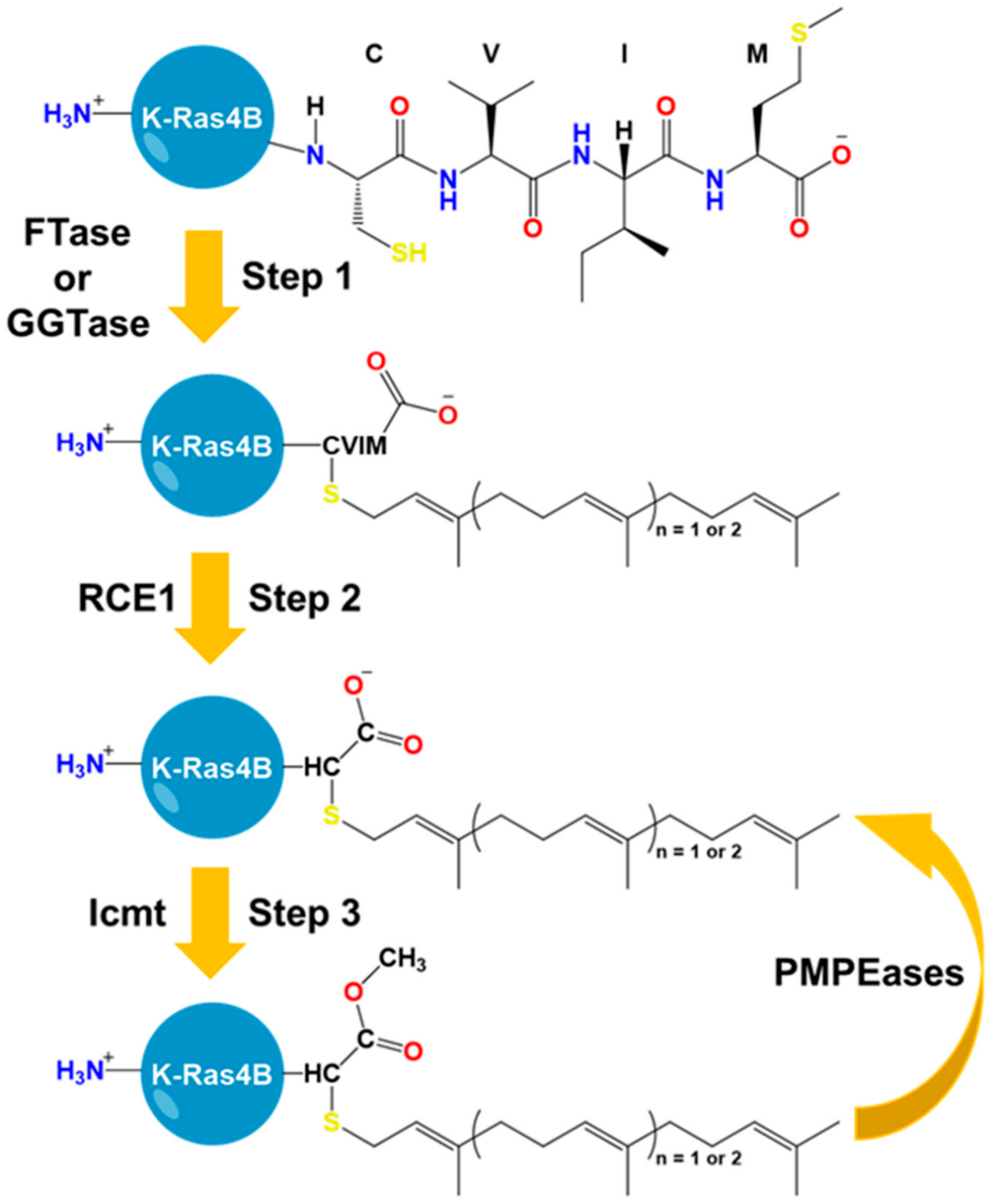
5. The HVR Undergoes Post-Translational Modifications
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| K-Ras4B | Kirsten ras oncogene homolog B |
| HVR | Hypervariable region |
| PDE-δ | Phosphodiesterase δ |
| PKC | Protein kinase C |
| PKA | Protein kinase A |
| GDP | Guanosine diphosphate |
| GTP | Guanosine triphosphate |
| GEF | Guanine nucleotide exchange factor |
| GAP | GTPase-activating protein |
| PI3K | Phosphatidylinositol 3-kinases |
| aa | Amino acids |
| SWI | Switch I region |
| SWII | Switch II region |
| SmgGDS | A chaperone and a guanine nucleotide exchange factor |
| RASSF | Ras association domain-containing protein |
| IQGAP1 | Q motif GTPase activating protein 1 |
| PIP2 | Phosphotidylinositol 4,5-Bisphosphate |
| PIP3 | Phosphatidylinositol-trisphosphate |
| PA | Phosphatidic acid |
| PtDSer | Phosphatidylserine |
| PTMs | Post-translational modifications |
| FTAse | Farnesyltrasferase |
| GGTase | Geranylgeranyltransferase |
| GTPase | Guanosine triphosphate hydrolyzing proteins |
| RCE1 | Ras-converting enzyme 1 |
| Icmt | Isoprenylcysteine carboxyl methyltransferase |
| PMPEases | Prenylated/polyisoprenylated methylated protein methyl esterases |
| FTIs | FTase inhibitors |
| PKG2 | Cyclic GMP-dependent protein kinase 2 |
| GMP | Guanosine monophosphate |
| pK-Ras4B | Phosphorylated K-Ras4B |
| CUL3 | Cullin-Based Ubiquitin Ligase 3 |
| LZTR1 | Leucine zipper-like transcription regulator 1 |
| MAPK | Mitogen-activated protein kinase |
References
- Keul, N.D.; Oruganty, K.; Schaper Bergman, E.T.; Beattie, N.R.; McDonald, W.E.; Kadirvelraj, R.; Gross, M.L.; Phillips, R.S.; Harvey, S.C.; Wood, Z.A. The entropic force generated by intrinsically disordered segments tunes protein function. Nature 2018, 563, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Pleasance, E.D.; Cheetham, R.K.; Stephens, P.J.; McBride, D.J.; Humphray, S.J.; Greenman, C.D.; Varela, I.; Lin, M.L.; Ordonez, G.R.; Bignell, G.R.; et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2010, 463, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed]
- Stephen, A.G.; Esposito, D.; Bagni, R.K.; McCormick, F. Dragging ras back in the ring. Cancer Cell 2014, 25, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.B.; Sass, P.M. Post-translational processing of purified human K-ras in Xenopus oocytes. Cancer Commun. 1991, 3, 383–388. [Google Scholar] [CrossRef]
- Prior, I.A.; Hancock, J.F. Compartmentalization of Ras proteins. J. Cell Sci. 2001, 114, 1603–1608. [Google Scholar]
- Cox, A.D.; Der, C.J.; Philips, M.R. Targeting RAS Membrane Association: Back to the Future for Anti-RAS Drug Discovery? Clin. Cancer Res. 2015, 21, 1819–1827. [Google Scholar] [CrossRef]
- Ye, N.; Xu, Q.; Li, W.; Wang, P.; Zhou, J. Recent Advances in Developing K-Ras Plasma Membrane Localization Inhibitors. Curr. Top. Med. Chem. 2019. [Google Scholar] [CrossRef]
- Chavan, T.S.; Jang, H.; Khavrutskii, L.; Abraham, S.J.; Banerjee, A.; Freed, B.C.; Johannessen, L.; Tarasov, S.G.; Gaponenko, V.; Nussinov, R.; et al. High-Affinity Interaction of the K-Ras4B Hypervariable Region with the Ras Active Site. Biophys. J. 2015, 109, 2602–2613. [Google Scholar] [CrossRef]
- Long, S.B.; Casey, P.J.; Beese, L.S. The basis for K-Ras4B binding specificity to protein farnesyltransferase revealed by 2 A resolution ternary complex structures. Structure 2000, 8, 209–222. [Google Scholar] [CrossRef]
- Chen, Z.; Otto, J.C.; Bergo, M.O.; Young, S.G.; Casey, P.J. The C-terminal polylysine region and methylation of K-Ras are critical for the interaction between K-Ras and microtubules. J. Biol. Chem. 2000, 275, 41251–41257. [Google Scholar] [CrossRef] [PubMed]
- Dharmaiah, S.; Bindu, L.; Tran, T.H.; Gillette, W.K.; Frank, P.H.; Ghirlando, R.; Nissley, D.V.; Esposito, D.; McCormick, F.; Stephen, A.G.; et al. Structural basis of recognition of farnesylated and methylated KRAS4b by PDEdelta. Proc. Natl. Acad. Sci. USA 2016, 113, E6766–E6775. [Google Scholar] [CrossRef]
- Abraham, S.J.; Nolet, R.P.; Calvert, R.J.; Anderson, L.M.; Gaponenko, V. The hypervariable region of K-Ras4B is responsible for its specific interactions with calmodulin. Biochemistry 2009, 48, 7575–7583. [Google Scholar] [CrossRef] [PubMed]
- Saikumar, P.; Ulsh, L.S.; Clanton, D.J.; Huang, K.P.; Shih, T.Y. Novel phosphorylation of c-ras p21 by protein kinases. Oncogene Res. 1988, 3, 213–222. [Google Scholar]
- Bivona, T.G.; Quatela, S.E.; Bodemann, B.O.; Ahearn, I.M.; Soskis, M.J.; Mor, A.; Miura, J.; Wiener, H.H.; Wright, L.; Saba, S.G.; et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol. Cell 2006, 21, 481–493. [Google Scholar] [CrossRef]
- Jang, H.; Abraham, S.J.; Chavan, T.S.; Hitchinson, B.; Khavrutskii, L.; Tarasova, N.I.; Nussinov, R.; Gaponenko, V. Mechanisms of membrane binding of small GTPase K-Ras4B farnesylated hypervariable region. J. Biol. Chem. 2015, 290, 9465–9477. [Google Scholar] [CrossRef]
- Lopez-Alcala, C.; Alvarez-Moya, B.; Villalonga, P.; Calvo, M.; Bachs, O.; Agell, N. Identification of essential interacting elements in K-Ras/calmodulin binding and its role in K-Ras localization. J. Biol. Chem. 2008, 283, 10621–10631. [Google Scholar] [CrossRef]
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res. 2015, 13, 1325–1335. [Google Scholar] [CrossRef]
- Bandaru, P.; Kondo, Y.; Kuriyan, J. The Interdependent Activation of Son-of-Sevenless and Ras. Cold Spring Harb. Perspect. Med. 2019, 9, a031534. [Google Scholar] [CrossRef]
- Lu, S.; Banerjee, A.; Jang, H.; Zhang, J.; Gaponenko, V.; Nussinov, R. GTP Binding and Oncogenic Mutations May Attenuate Hypervariable Region (HVR)-Catalytic Domain Interactions in Small GTPase K-Ras4B, Exposing the Effector Binding Site. J. Biol. Chem. 2015, 290, 28887–28900. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Jang, H.; Nussinov, R.; Gaponenko, V. The disordered hypervariable region and the folded catalytic domain of oncogenic K-Ras4B partner in phospholipid binding. Curr. Opin. Struct. Biol. 2016, 36, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Mazhab-Jafari, M.T.; Marshall, C.B.; Smith, M.J.; Gasmi-Seabrook, G.M.; Stathopulos, P.B.; Inagaki, F.; Kay, L.E.; Neel, B.G.; Ikura, M. Oncogenic and RASopathy-associated K-RAS mutations relieve membrane-dependent occlusion of the effector-binding site. Proc. Natl. Acad. Sci. USA 2015, 112, 6625–6630. [Google Scholar] [CrossRef]
- Janosi, L.; Li, Z.; Hancock, J.F.; Gorfe, A.A. Organization, dynamics, and segregation of Ras nanoclusters in membrane domains. Proc. Natl. Acad. Sci. USA 2012, 109, 8097–8102. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Prakash, P.; Liang, H.; Cho, K.J.; Gorfe, A.A.; Hancock, J.F. Lipid-Sorting Specificity Encoded in K-Ras Membrane Anchor Regulates Signal Output. Cell 2017, 168, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Banerjee, A.; Chavan, T.S.; Lu, S.; Zhang, J.; Gaponenko, V.; Nussinov, R. The higher level of complexity of K-Ras4B activation at the membrane. FASEB J. 2016, 30, 1643–1655. [Google Scholar] [CrossRef]
- Muratcioglu, S.; Chavan, T.S.; Freed, B.C.; Jang, H.; Khavrutskii, L.; Freed, R.N.; Dyba, M.A.; Stefanisko, K.; Tarasov, S.G.; Gursoy, A.; et al. GTP-Dependent K-Ras Dimerization. Structure 2015, 23, 1325–1335. [Google Scholar] [CrossRef]
- Ambrogio, C.; Kohler, J.; Zhou, Z.W.; Wang, H.; Paranal, R.; Li, J.; Capelletti, M.; Caffarra, C.; Li, S.; Lv, Q.; et al. KRAS Dimerization Impacts MEK Inhibitor Sensitivity and Oncogenic Activity of Mutant KRAS. Cell 2018, 172, 857–868. [Google Scholar] [CrossRef]
- Nancy, V.; Callebaut, I.; El Marjou, A.; de Gunzburg, J. The delta subunit of retinal rod cGMP phosphodiesterase regulates the membrane association of Ras and Rap GTPases. J. Biol. Chem. 2002, 277, 15076–15084. [Google Scholar] [CrossRef]
- Sidhu, R.S.; Clough, R.R.; Bhullar, R.P. Ca2+/calmodulin binds and dissociates K-RasB from membrane. Biochem. Biophys. Res. Commun. 2003, 304, 655–660. [Google Scholar] [CrossRef]
- Sperlich, B.; Kapoor, S.; Waldmann, H.; Winter, R.; Weise, K. Regulation of K-Ras4B Membrane Binding by Calmodulin. Biophys J. 2016, 111, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Lakshman, B.; Messing, S.; Schmid, E.M.; Clogston, J.D.; Gillette, W.K.; Esposito, D.; Kessing, B.; Fletcher, D.A.; Nissley, D.V.; McCormick, F.; et al. Quantitative biophysical analysis defines key components modulating recruitment of the GTPase KRAS to the plasma membrane. J. Biol. Chem. 2019, 294, 2193–2207. [Google Scholar] [CrossRef] [PubMed]
- Villalonga, P.; Lopez-Alcala, C.; Bosch, M.; Chiloeches, A.; Rocamora, N.; Gil, J.; Marais, R.; Marshall, C.J.; Bachs, O.; Agell, N. Calmodulin binds to K-Ras, but not to H- or N-Ras, and modulates its downstream signaling. Mol. Cell Biol. 2001, 21, 7345–7354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agamasu, C.; Ghirlando, R.; Taylor, T.; Messing, S.; Tran, T.H.; Bindu, L.; Tonelli, M.; Nissley, D.V.; McCormick, F.; Stephen, A.G. KRAS Prenylation Is Required for Bivalent Binding with Calmodulin in a Nucleotide-Independent Manner. Biophys. J. 2019, 116, 1049–1063. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.; Banerjee, A.; Marcus, K.; Makowski, L.; Mattos, C.; Gaponenko, V.; Nussinov, R. The Structural Basis of the Farnesylated and Methylated KRas4B Interaction with Calmodulin. Structure 2019. [Google Scholar] [CrossRef]
- Garrido, E.; Lazaro, J.; Jaumot, M.; Agell, N.; Rubio-Martinez, J. Modeling and subtleties of K-Ras and Calmodulin interaction. PLoS Comput. Biol. 2018, 14, e1006552. [Google Scholar] [CrossRef] [Green Version]
- Fivaz, M.; Meyer, T. Reversible intracellular translocation of KRas but not HRas in hippocampal neurons regulated by Ca2+/calmodulin. J. Cell Biol. 2005, 170, 429–441. [Google Scholar] [CrossRef]
- Agell, N.; Bachs, O.; Rocamora, N.; Villalonga, P. Modulation of the Ras/Raf/MEK/ERK pathway by Ca2+, and calmodulin. Cell Signal. 2002, 14, 649–654. [Google Scholar] [CrossRef]
- Wang, M.T.; Holderfield, M.; Galeas, J.; Delrosario, R.; To, M.D.; Balmain, A.; McCormick, F. K-Ras Promotes Tumorigenicity through Suppression of Non-canonical Wnt Signaling. Cell 2015, 163, 1237–1251. [Google Scholar] [CrossRef] [Green Version]
- Nussinov, R.; Wang, G.; Tsai, C.J.; Jang, H.; Lu, S.; Banerjee, A.; Zhang, J.; Gaponenko, V. Calmodulin and PI3K Signaling in KRAS Cancers. Trends Cancer 2017, 3, 214–224. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.L. The polybasic region of Ras and Rho family small GTPases: A regulator of protein interactions and membrane association and a site of nuclear localization signal sequences. Cell Signal. 2003, 15, 1071–1080. [Google Scholar] [CrossRef]
- Williams, C.L. A new signaling paradigm to control the prenylation and trafficking of small GTPases. Cell Cycle 2013, 12, 2933–2934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terrell, E.M.; Durrant, D.E.; Ritt, D.A.; Sealover, N.E.; Sheffels, E.; Spencer-Smith, R.; Esposito, D.; Zhou, Y.; Hancock, J.F.; Kortum, R.L.; et al. Distinct Binding Preferences between Ras and Raf Family Members and the Impact on Oncogenic Ras Signaling. Mol. Cell 2019. [Google Scholar] [CrossRef] [PubMed]
- Sieburth, D.S.; Sun, Q.; Han, M. SUR-8, a conserved Ras-binding protein with leucine-rich repeats, positively regulates Ras-mediated signaling in C. elegans. Cell 1998, 94, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Han, M.; Guan, K.L. The leucine-rich repeat protein SUR-8 enhances MAP kinase activation and forms a complex with Ras and Raf. Genes Dev. 2000, 14, 895–900. [Google Scholar]
- Inder, K.L.; Hill, M.M.; Hancock, J.F. Nucleophosmin and nucleolin regulate K-Ras signaling. Commun. Integr. Biol. 2010, 3, 188–190. [Google Scholar] [CrossRef]
- Elad-Sfadia, G.; Haklai, R.; Balan, E.; Kloog, Y. Galectin-3 augments K-Ras activation and triggers a Ras signal that attenuates ERK but not phosphoinositide 3-kinase activity. J. Biol. Chem. 2004, 279, 34922–34930. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Cao, J.; Miller, S.P.; Jing, H.; Lin, H. Comparative Nucleotide-Dependent Interactome Analysis Reveals Shared and Differential Properties of KRas4a and KRas4b. ACS Cent. Sci. 2018, 4, 71–80. [Google Scholar] [CrossRef]
- Adhikari, H.; Counter, C.M. Interrogating the protein interactomes of RAS isoforms identifies PIP5K1A as a KRAS-specific vulnerability. Nat. Commun. 2018, 9, 3646. [Google Scholar] [CrossRef]
- Donninger, H.; Vos, M.D.; Clark, G.J. The RASSF1A tumor suppressor. J. Cell Sci. 2007, 120, 3163–3172. [Google Scholar] [CrossRef] [Green Version]
- Vos, M.D.; Ellis, C.A.; Elam, C.; Ulku, A.S.; Taylor, B.J.; Clark, G.J. RASSF2 is a novel K-Ras-specific effector and potential tumor suppressor. J. Biol. Chem. 2003, 278, 28045–28051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckfeld, K.; Hesson, L.; Vos, M.D.; Bieche, I.; Latif, F.; Clark, G.J. RASSF4/AD037 is a potential ras effector/tumor suppressor of the RASSF family. Cancer Res. 2004, 64, 8688–8693. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, H.; Kubota, K.; Inoue, T.; Isono, F.; Ando, O. IQGAP1 selectively interacts with K-Ras but not with H-Ras and modulates K-Ras function. Biochem. Biophys. Res. Commun. 2014, 444, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.L.; Buck, M. Computational Modeling Reveals that Signaling Lipids Modulate the Orientation of K-Ras4A at the Membrane Reflecting Protein Topology. Structure 2017, 25, 679–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, W.D.; Inoue, T.; Park, W.S.; Kim, M.L.; Park, B.O.; Wandless, T.J.; Meyer, T. PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science 2006, 314, 1458–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.H.; Laurent, G.; Bause, A.S.; Spang, R.; German, N.; Haigis, M.C.; Haigis, K.M. HDAC6 and SIRT2 regulate the acetylation state and oncogenic activity of mutant K-RAS. Mol. Cancer Res. MCR 2013, 11, 1072–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.H.; Nickerson, S.; Kim, E.T.; Liot, C.; Laurent, G.; Spang, R.; Philips, M.R.; Shan, Y.; Shaw, D.E.; Bar-Sagi, D.; et al. Regulation of RAS oncogenicity by acetylation. Proc. Natl. Acad. Sci. USA 2012, 109, 10843–10848. [Google Scholar] [CrossRef] [Green Version]
- Arozarena, I.; Calvo, F.; Crespo, P. Ras, an actor on many stages: Posttranslational modifications, localization, and site-specified events. Genes Cancer 2011, 2, 182–194. [Google Scholar] [CrossRef] [Green Version]
- Ahearn, I.M.; Haigis, K.; Bar-Sagi, D.; Philips, M.R. Regulating the regulator: Post-translational modification of RAS. Nat. Rev. Mol. Cell Biol. 2011, 13, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Jang, H.; Gu, S.; Zhang, J.; Nussinov, R. Drugging Ras GTPase: A comprehensive mechanistic and signaling structural view. Chem. Soc. Rev. 2016, 45, 4929–4952. [Google Scholar] [CrossRef] [Green Version]
- Ahearn, I.; Zhou, M.; Philips, M.R. Posttranslational Modifications of RAS Proteins. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Wright, L.P.; Philips, M.R. Thematic review series: Lipid posttranslational modifications. CAAX modification and membrane targeting of Ras. J. Lipid Res. 2006, 47, 883–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancock, J.F.; Magee, A.I.; Childs, J.E.; Marshall, C.J. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell 1989, 57, 1167–1177. [Google Scholar] [CrossRef]
- Ntai, I.; Fornelli, L.; DeHart, C.J.; Hutton, J.E.; Doubleday, P.F.; LeDuc, R.D.; van Nispen, A.J.; Fellers, R.T.; Whiteley, G.; Boja, E.S.; et al. Precise characterization of KRAS4b proteoforms in human colorectal cells and tumors reveals mutation/modification cross-talk. Proc. Natl. Acad. Sci. USA 2018, 115, 4140–4145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duverna, R.; Ablordeppey, S.Y.; Lamango, N.S. Biochemical and docking analysis of substrate interactions with polyisoprenylated methylated protein methyl esterase. Curr. Cancer Drug Targets 2010, 10, 634–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oboh, O.T.; Lamango, N.S. Liver prenylated methylated protein methyl esterase is the same enzyme as Sus scrofa carboxylesterase. J. Biochem. Mol. Toxicol. 2008, 22, 51–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamango, N.S.; Duverna, R.; Zhang, W.; Ablordeppey, S.Y. Porcine Liver Carboxylesterase Requires Polyisoprenylation for High Affinity Binding to Cysteinyl Substrates. Open Enzym. Inhib. J. 2009, 2, 12–27. [Google Scholar] [CrossRef] [Green Version]
- Amissah, F.; Duverna, R.; Aguilar, B.J.; Poku, R.A.; Kiros, G.E.; Lamango, N.S. Polyisoprenylated methylated protein methyl esterase overexpression and hyperactivity promotes lung cancer progression. Am. J. Cancer Res. 2014, 4, 116–134. [Google Scholar]
- Poku, R.A.; Amissah, F.; Duverna, R.; Aguilar, B.J.; Kiros, G.E.; Lamango, N.S. Polyisoprenylated methylated protein methyl esterase as a putative drug target for androgen-insensitive prostate cancer. Ecancermedicalscience 2014, 8, 459. [Google Scholar] [CrossRef]
- Nkembo, A.T.; Amissah, F.; Ntantie, E.; Poku, R.A.; Salako, O.O.; Ikpatt, O.F.; Lamango, N.S. Polyisoprenylated cysteinyl amide inhibitors deplete K-Ras and induce caspase-dependent apoptosis in lung cancer cells. Curr. Cancer Drug Targets 2019. [Google Scholar] [CrossRef]
- Poku, R.A.; Salako, O.O.; Amissah, F.; Nkembo, A.T.; Ntantie, E.; Lamango, N.S. Polyisoprenylated cysteinyl amide inhibitors induce caspase 3/7- and 8-mediated apoptosis and inhibit migration and invasion of metastatic prostate cancer cells. Am. J. Cancer Res. 2017, 7, 1515–1527. [Google Scholar] [PubMed]
- Ntantie, E.; Fletcher, J.; Amissah, F.; Salako, O.O.; Nkembo, A.T.; Poku, R.A.; Ikpatt, F.O.; Lamango, N.S. Polyisoprenylated cysteinyl amide inhibitors disrupt actin cytoskeleton organization, induce cell rounding and block migration of non-small cell lung cancer. Oncotarget 2017, 8, 31726–31744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nkembo, A.T.; Salako, O.; Poku, R.A.; Amissah, F.; Ntantie, E.; Flores-Rozas, H.; Lamango, N.S. Disruption of actin filaments and suppression of pancreatic cancer cell viability and migration following treatment with polyisoprenylated cysteinyl amides. Am. J. Cancer Res. 2016, 6, 2532–2546. [Google Scholar]
- Aguilar, B.J.; Nkembo, A.T.; Duverna, R.; Poku, R.A.; Amissah, F.; Ablordeppey, S.Y.; Lamango, N.S. Polyisoprenylated methylated protein methyl esterase: A putative biomarker and therapeutic target for pancreatic cancer. Eur. J. Med. Chem. 2014, 81, 323–333. [Google Scholar] [CrossRef] [Green Version]
- Amissah, F.; Duverna, R.; Aguilar, B.J.; Poku, R.A.; Lamango, N.S. Polyisoprenylated methylated protein methyl esterase is both sensitive to curcumin and overexpressed in colorectal cancer: Implications for chemoprevention and treatment. Biomed. Res. Int. 2013, 2013, 416534. [Google Scholar] [CrossRef] [Green Version]
- Konstantinopoulos, P.A.; Karamouzis, M.V.; Papavassiliou, A.G. Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat. Rev. Drug Discov. 2007, 6, 541–555. [Google Scholar] [CrossRef]
- Novotny, C.J.; Hamilton, G.L.; McCormick, F.; Shokat, K.M. Farnesyltransferase-Mediated Delivery of a Covalent Inhibitor Overcomes Alternative Prenylation to Mislocalize K-Ras. ACS Chem. Biol. 2017, 12, 1956–1962. [Google Scholar] [CrossRef]
- Alvarez-Moya, B.; Lopez-Alcala, C.; Drosten, M.; Bachs, O.; Agell, N. K-Ras4B phosphorylation at Ser181 is inhibited by calmodulin and modulates K-Ras activity and function. Oncogene 2010, 29, 5911–5922. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.J.; Casteel, D.E.; Prakash, P.; Tan, L.; van der Hoeven, D.; Salim, A.A.; Kim, C.; Capon, R.J.; Lacey, E.; Cunha, S.R.; et al. AMPK and Endothelial Nitric Oxide Synthase Signaling Regulates K-Ras Plasma Membrane Interactions via Cyclic GMP-Dependent Protein Kinase 2. Mol. Cell Biol. 2016, 36, 3086–3099. [Google Scholar] [CrossRef] [Green Version]
- Barcelo, C.; Paco, N.; Morell, M.; Alvarez-Moya, B.; Bota-Rabassedas, N.; Jaumot, M.; Vilardell, F.; Capella, G.; Agell, N. Phosphorylation at Ser-181 of oncogenic KRAS is required for tumor growth. Cancer Res. 2014, 74, 1190–1199. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Moya, B.; Barcelo, C.; Tebar, F.; Jaumot, M.; Agell, N. CaM interaction and Ser181 phosphorylation as new K-Ras signaling modulators. Small GTPases 2011, 2, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.Y.; Sperlich, B.; Li, F.Y.; Al-Ayoubi, S.; Chen, H.X.; Zhao, Y.F.; Li, Y.M.; Weise, K.; Winter, R.; Chen, Y.X. Phosphorylation Weakens but Does Not Inhibit Membrane Binding and Clustering of K-Ras4B. ACS Chem. Biol. 2017, 12, 1703–1710. [Google Scholar] [CrossRef]
- Barcelo, C.; Paco, N.; Beckett, A.J.; Alvarez-Moya, B.; Garrido, E.; Gelabert, M.; Tebar, F.; Jaumot, M.; Prior, I.; Agell, N. Oncogenic K-ras segregates at spatially distinct plasma membrane signaling platforms according to its phosphorylation status. J. Cell Sci. 2013, 126, 4553–4559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kollar, P.; Rajchard, J.; Balounova, Z.; Pazourek, J. Marine natural products: Bryostatins in preclinical and clinical studies. Pharm. Biol. 2014, 52, 237–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigenzahn, J.W.; Collu, G.M.; Kartnig, F.; Pieraks, M.; Vladimer, G.I.; Heinz, L.X.; Sedlyarov, V.; Schischlik, F.; Fauster, A.; Rebsamen, M.; et al. LZTR1 is a regulator of RAS ubiquitination and signaling. Science 2018, 362, 1171–1177. [Google Scholar] [CrossRef]
- Steklov, M.; Pandolfi, S.; Baietti, M.F.; Batiuk, A.; Carai, P.; Najm, P.; Zhang, M.; Jang, H.; Renzi, F.; Cai, Y.; et al. Mutations in LZTR1 drive human disease by dysregulating RAS ubiquitination. Science 2018, 362, 1177–1182. [Google Scholar] [CrossRef]
- Darling, A.L.; Uversky, V.N. Intrinsic Disorder and Posttranslational Modifications: The Darker Side of the Biological Dark Matter. Front. Genet. 2018, 9, 158. [Google Scholar] [CrossRef]
- Dunker, A.K.; Brown, C.J.; Lawson, C.J.D.; Iakoucheva-Sebat, L.M.; Vucetic, S.; Obradovic, Z. The Protein Trinity: Structure/Function Relationships That Include Intrinsic Disorder. Sci. World J. 2002, 2, 49–50. [Google Scholar] [CrossRef] [Green Version]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradovic, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Vucetic, S.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Obradovic, Z.; Uversky, V.N. Functional anthology of intrinsic disorder. 3. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J. Proteome Res. 2007, 6, 1917–1932. [Google Scholar] [CrossRef] [Green Version]
- Jackson, J.H.; Li, J.W.; Buss, J.E.; Der, C.J.; Cochrane, C.G. Polylysine domain of K-ras 4B protein is crucial for malignant transformation. Proc. Natl. Acad. Sci. USA 1994, 91, 12730–12734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orren, D.K.; Machwe, A. Lysine Acetylation of Proteins and Its Characterization in Human Systems. Methods Mol. Biol. 2019, 1983, 107–130. [Google Scholar] [CrossRef] [PubMed]
- Lanouette, S.; Mongeon, V.; Figeys, D.; Couture, J.F. The functional diversity of protein lysine methylation. Mol. Syst. Biol. 2014, 10, 724. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. Glycation research in amino acids: A place to call home. Amino Acids 2012, 42, 1087–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattiroli, F.; Sixma, T.K. Lysine-targeting specificity in ubiquitin and ubiquitin-like modification pathways. Nat. Struct. Mol. Biol. 2014, 21, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Hancock, J.F.; Paterson, H.; Marshall, C.J. A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell 1990, 63, 133–139. [Google Scholar] [CrossRef]
- Zhou, Y.; Hancock, J.F. A novel prenyl-polybasic domain code determines lipid-binding specificity of the K-Ras membrane anchor. Small GTPases 2018, 1–5. [Google Scholar] [CrossRef]
- Cadwallader, K.A.; Paterson, H.; Macdonald, S.G.; Hancock, J.F. N-terminally myristoylated Ras proteins require palmitoylation or a polybasic domain for plasma membrane localization. Mol. Cell Biol. 1994, 14, 4722–4730. [Google Scholar] [CrossRef] [Green Version]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [Green Version]
- Kessler, D.; Gmachl, M.; Mantoulidis, A.; Martin, L.J.; Zoephel, A.; Mayer, M.; Gollner, A.; Covini, D.; Fischer, S.; Gerstberger, T.; et al. Drugging an undruggable pocket on KRAS. Proc. Natl. Acad. Sci. USA 2019, 116, 15823–15829. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Du, G.; Skowronek, K.; Frohman, M.A.; Bar-Sagi, D. Phospholipase D2-generated phosphatidic acid couples EGFR stimulation to Ras activation by Sos. Nat. Cell Biol. 2007, 9, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Prakash, P.; Gorfe, A.A.; Hancock, J.F. Ras and the Plasma Membrane: A Complicated Relationship. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussinov, R.; Tsai, C.J.; Jang, H. Oncogenic Ras Isoforms Signaling Specificity at the Membrane. Cancer Res. 2018, 78, 593–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuld, N.J.; Vervacke, J.S.; Lorimer, E.L.; Simon, N.C.; Hauser, A.D.; Barbieri, J.T.; Distefano, M.D.; Williams, C.L. The chaperone protein SmgGDS interacts with small GTPases entering the prenylation pathway by recognizing the last amino acid in the CAAX motif. J. Biol. Chem. 2014, 289, 6862–6876. [Google Scholar] [CrossRef] [Green Version]
- Diver, M.M.; Pedi, L.; Koide, A.; Koide, S.; Long, S.B. Atomic structure of the eukaryotic intramembrane RAS methyltransferase ICMT. Nature 2018, 553, 526–529. [Google Scholar] [CrossRef]
- Wang, D.; Kon, N.; Lasso, G.; Jiang, L.; Leng, W.; Zhu, W.G.; Qin, J.; Honig, B.; Gu, W. Acetylation-regulated interaction between p53 and SET reveals a widespread regulatory mode. Nature 2016, 538, 118–122. [Google Scholar] [CrossRef] [Green Version]
- Ansari, N.A.; Moinuddin Mir, A.R.; Habib, S.; Alam, K.; Ali, A.; Khan, R.H. Role of early glycation Amadori products of lysine-rich proteins in the production of autoantibodies in diabetes type 2 patients. Cell Biochem. Biophys. 2014, 70, 857–865. [Google Scholar] [CrossRef]
- Lea, M.A.; Luke, A.; Martinson, C.; Velazquez, O. Influence of carbamoylation on some analytical properties of basic polypeptides. Int. J. Pept. Protein Res. 1986, 27, 251–260. [Google Scholar] [CrossRef]
- Colon-Bolea, P.; Crespo, P. Lysine methylation in cancer: SMYD3-MAP3K2 teaches us new lessons in the Ras-ERK pathway. Bioessays 2014, 36, 1162–1169. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelkarim, H.; Banerjee, A.; Grudzien, P.; Leschinsky, N.; Abushaer, M.; Gaponenko, V. The Hypervariable Region of K-Ras4B Governs Molecular Recognition and Function. Int. J. Mol. Sci. 2019, 20, 5718. https://doi.org/10.3390/ijms20225718
Abdelkarim H, Banerjee A, Grudzien P, Leschinsky N, Abushaer M, Gaponenko V. The Hypervariable Region of K-Ras4B Governs Molecular Recognition and Function. International Journal of Molecular Sciences. 2019; 20(22):5718. https://doi.org/10.3390/ijms20225718
Chicago/Turabian StyleAbdelkarim, Hazem, Avik Banerjee, Patrick Grudzien, Nicholas Leschinsky, Mahmoud Abushaer, and Vadim Gaponenko. 2019. "The Hypervariable Region of K-Ras4B Governs Molecular Recognition and Function" International Journal of Molecular Sciences 20, no. 22: 5718. https://doi.org/10.3390/ijms20225718