Pyruvate Dehydrogenase and Tricarboxylic Acid Cycle Enzymes Are Sensitive Targets of Traumatic Brain Injury Induced Metabolic Derangement


 ,
,  ,
,  ,
,  ,
,  , ,
, , 
 and
and
Abstract
1. Introduction
2. Results
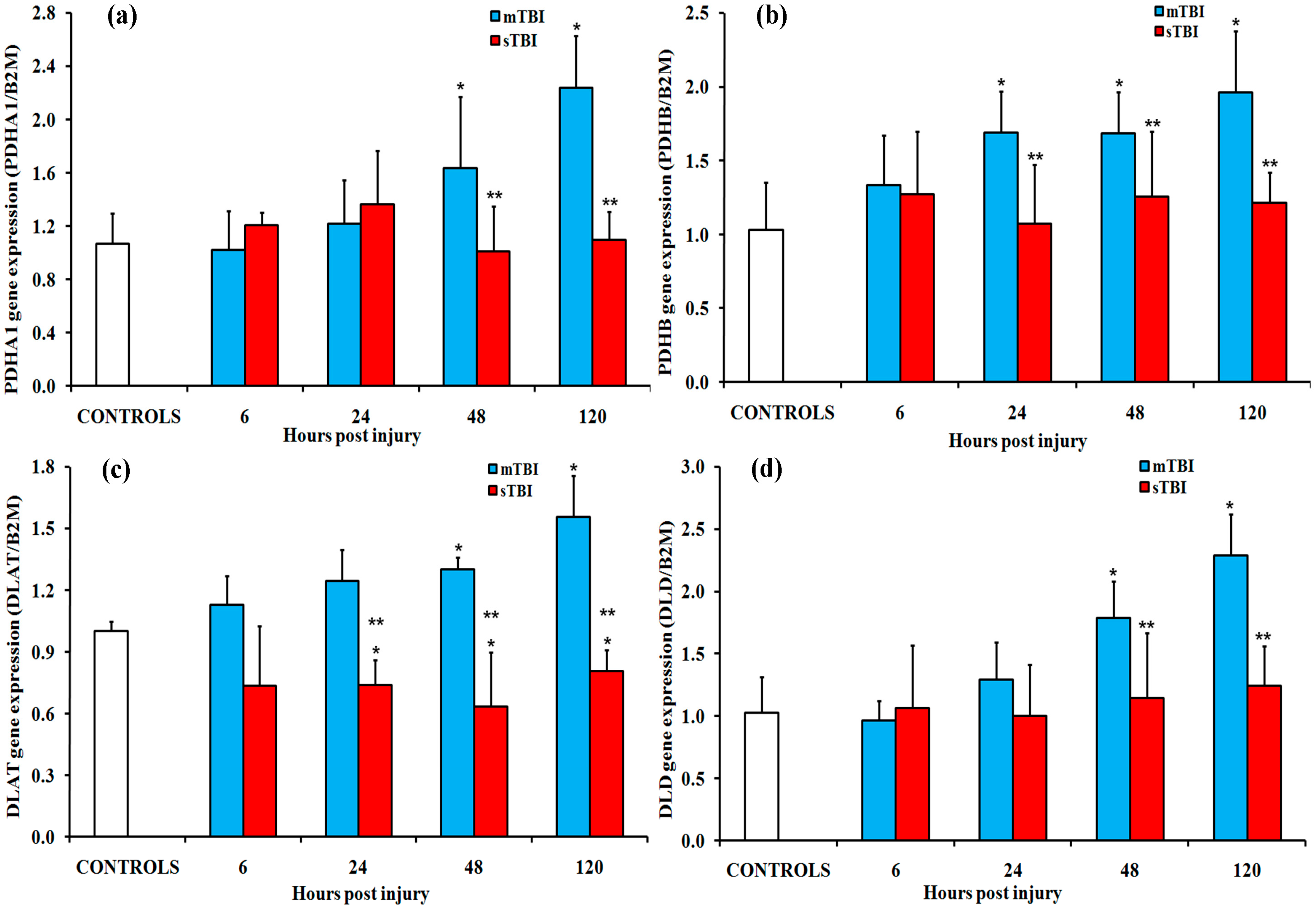
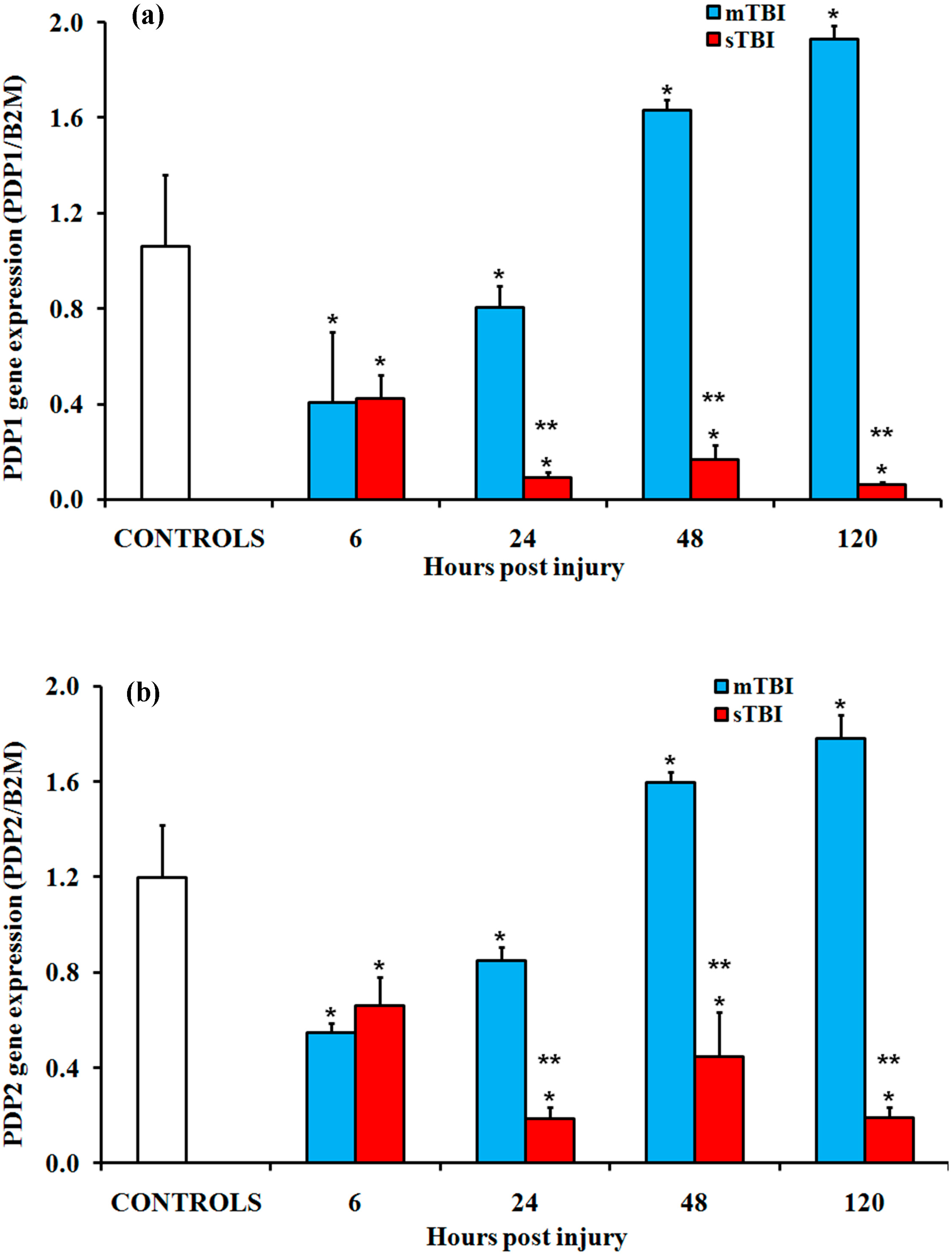
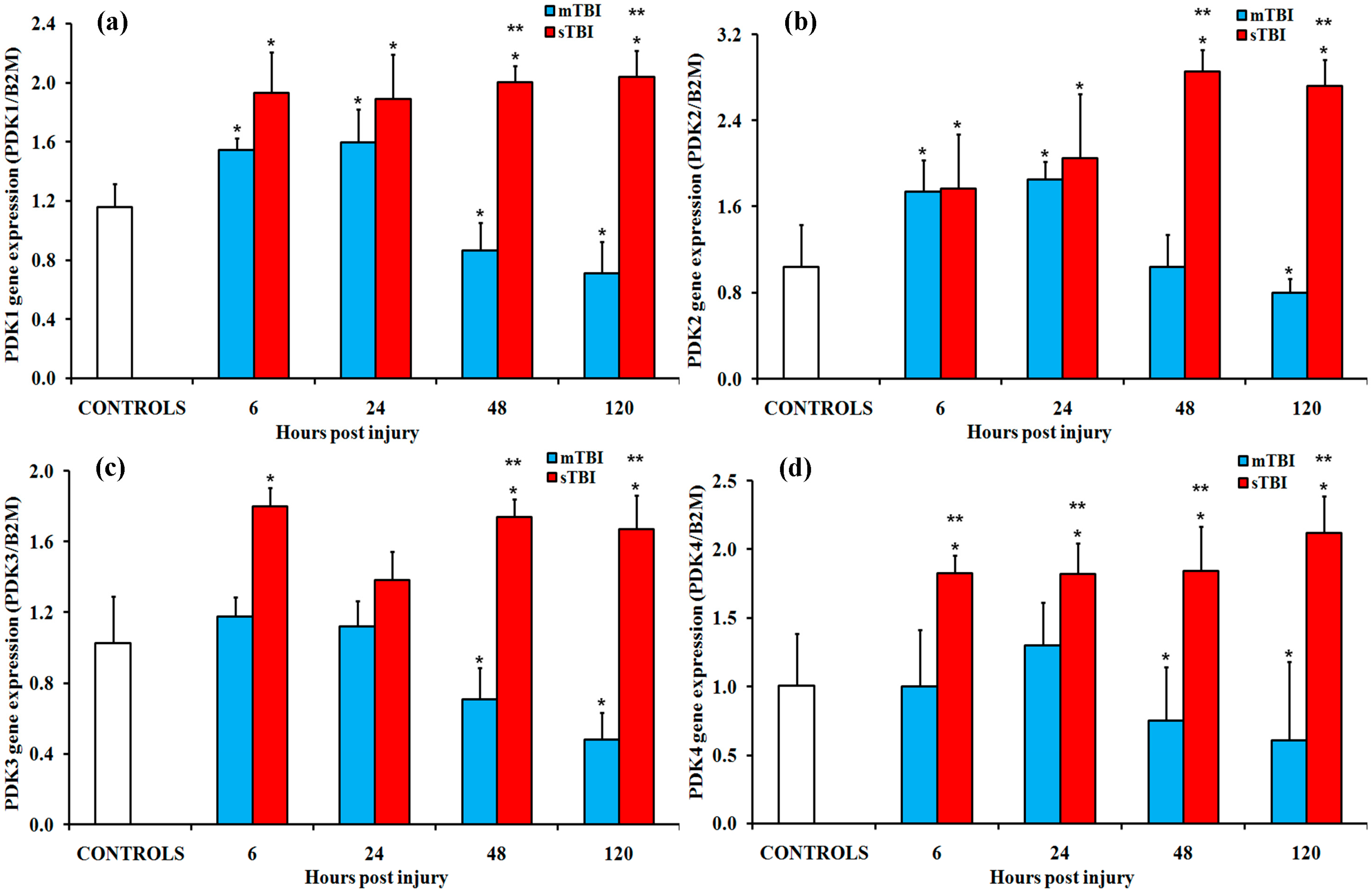
2.1. Gene Expressions of the PDH Complex and Its Regulatory Enzymes after Graded TBI
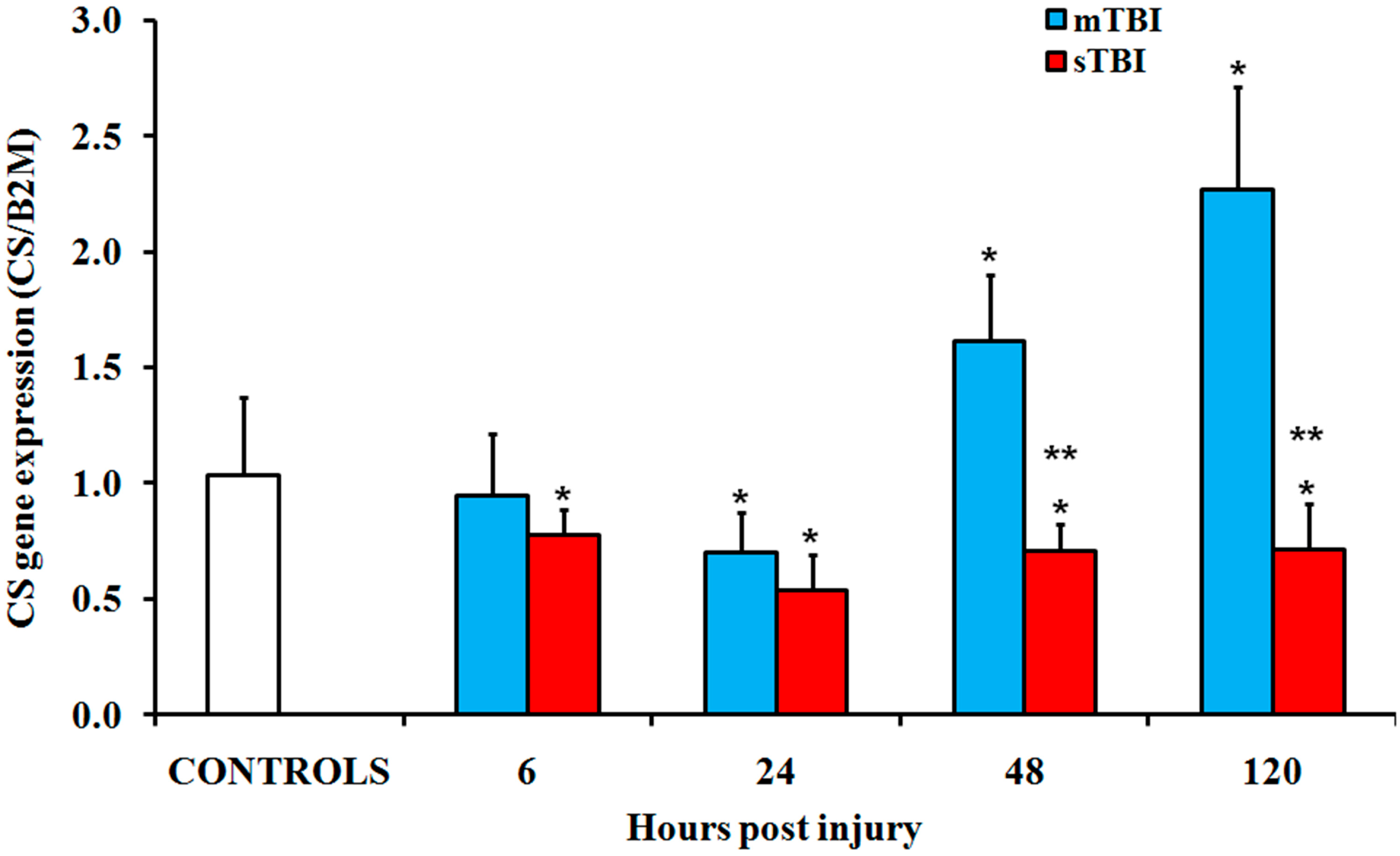
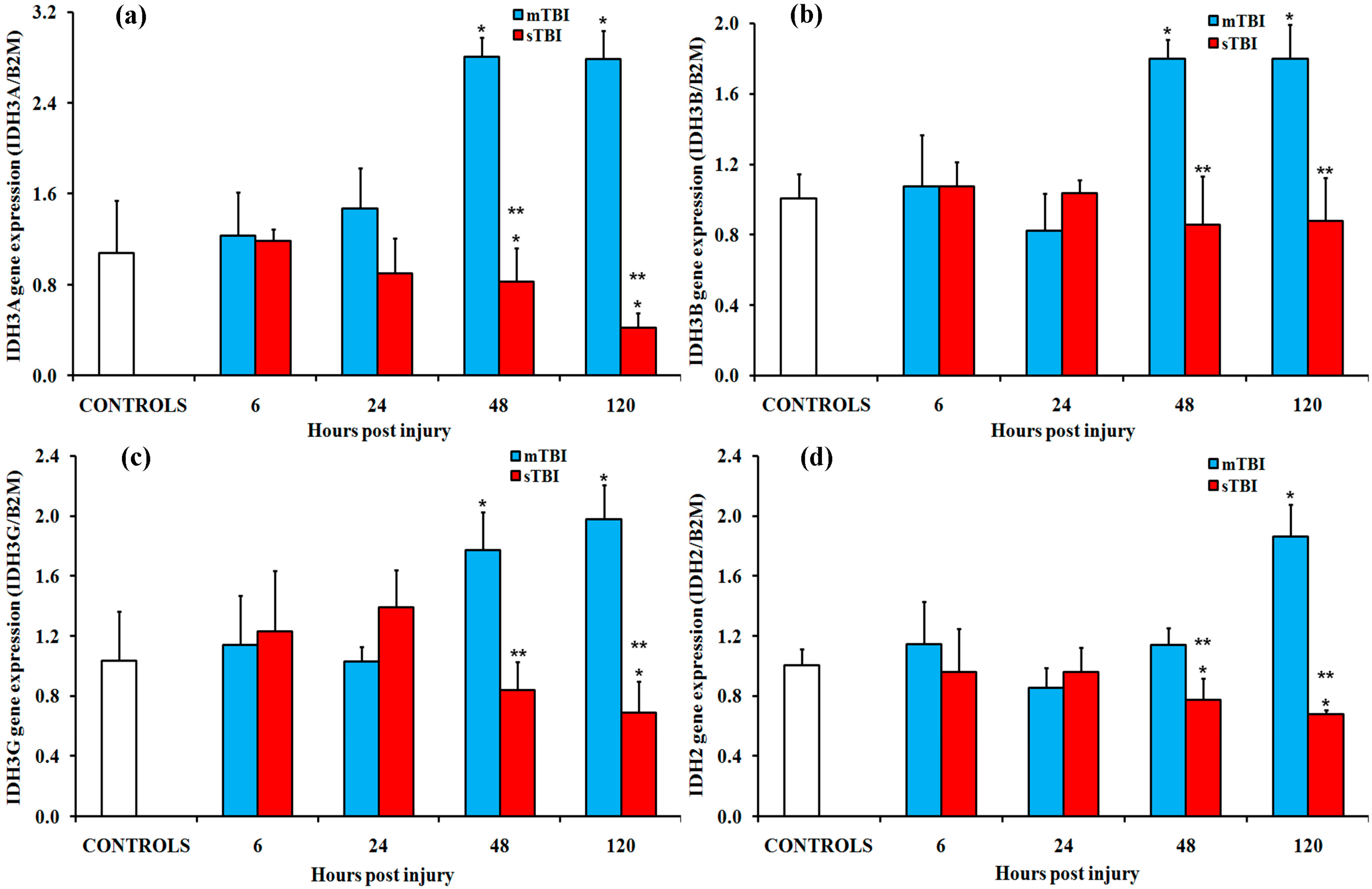
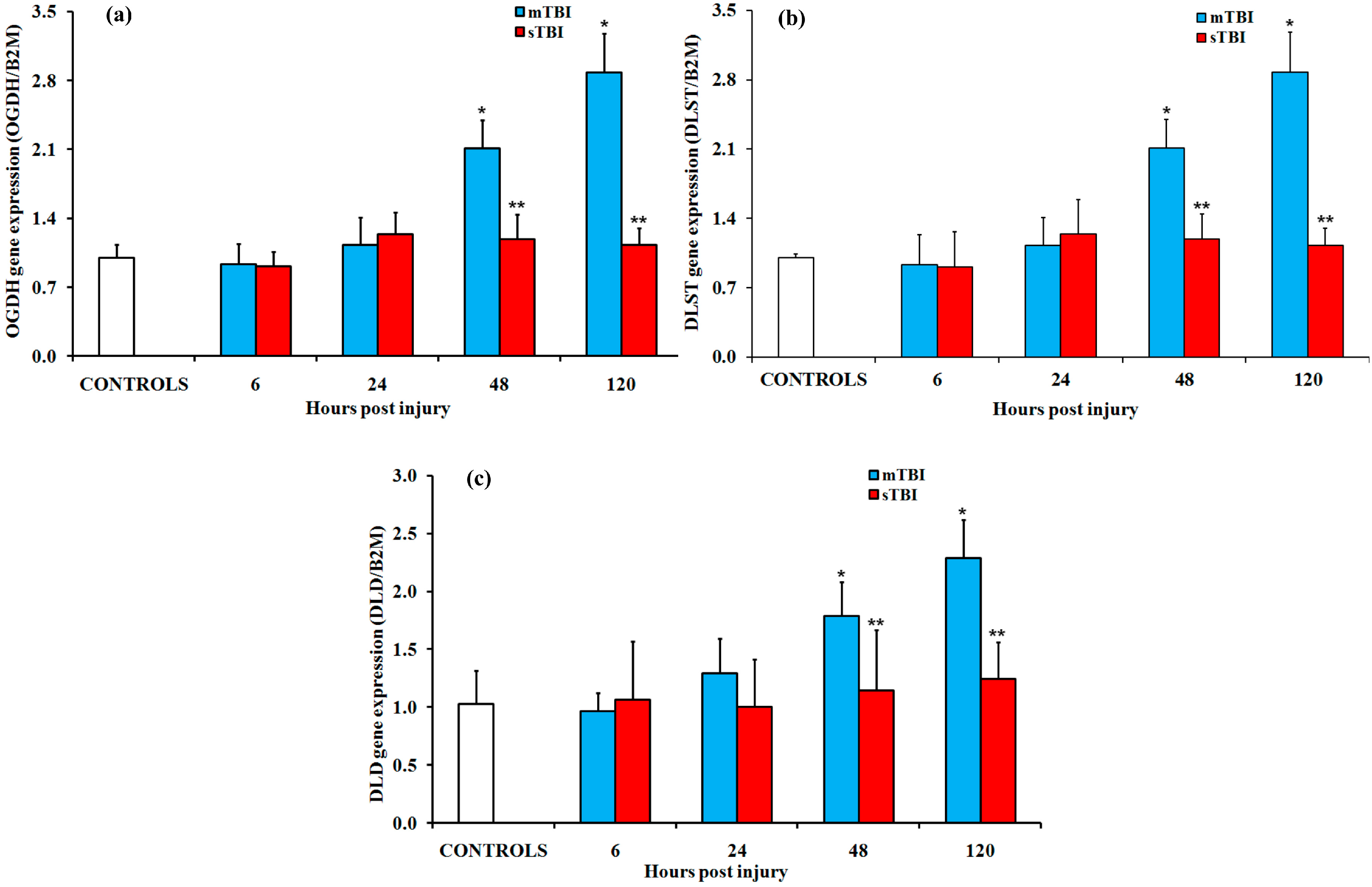
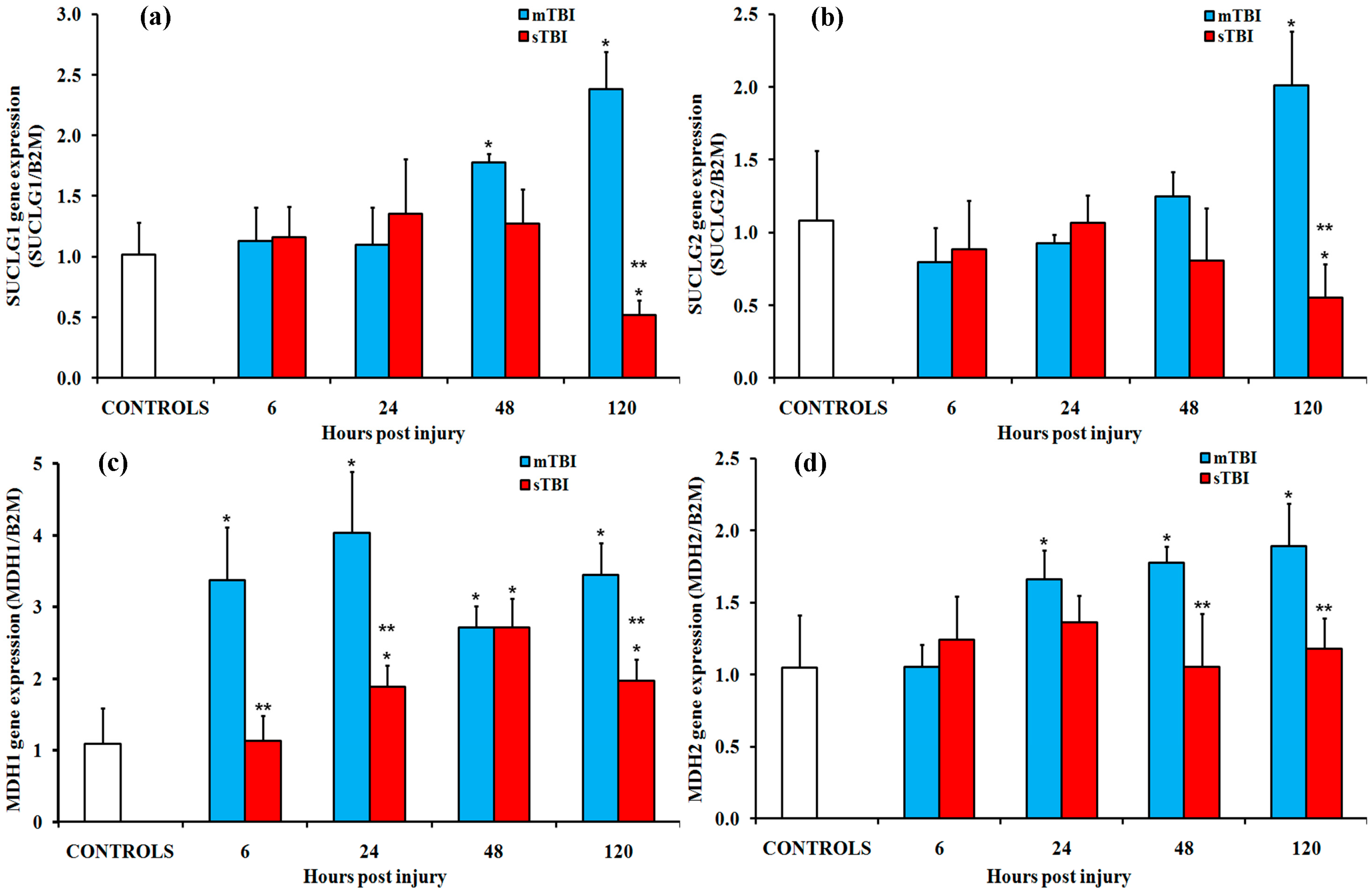
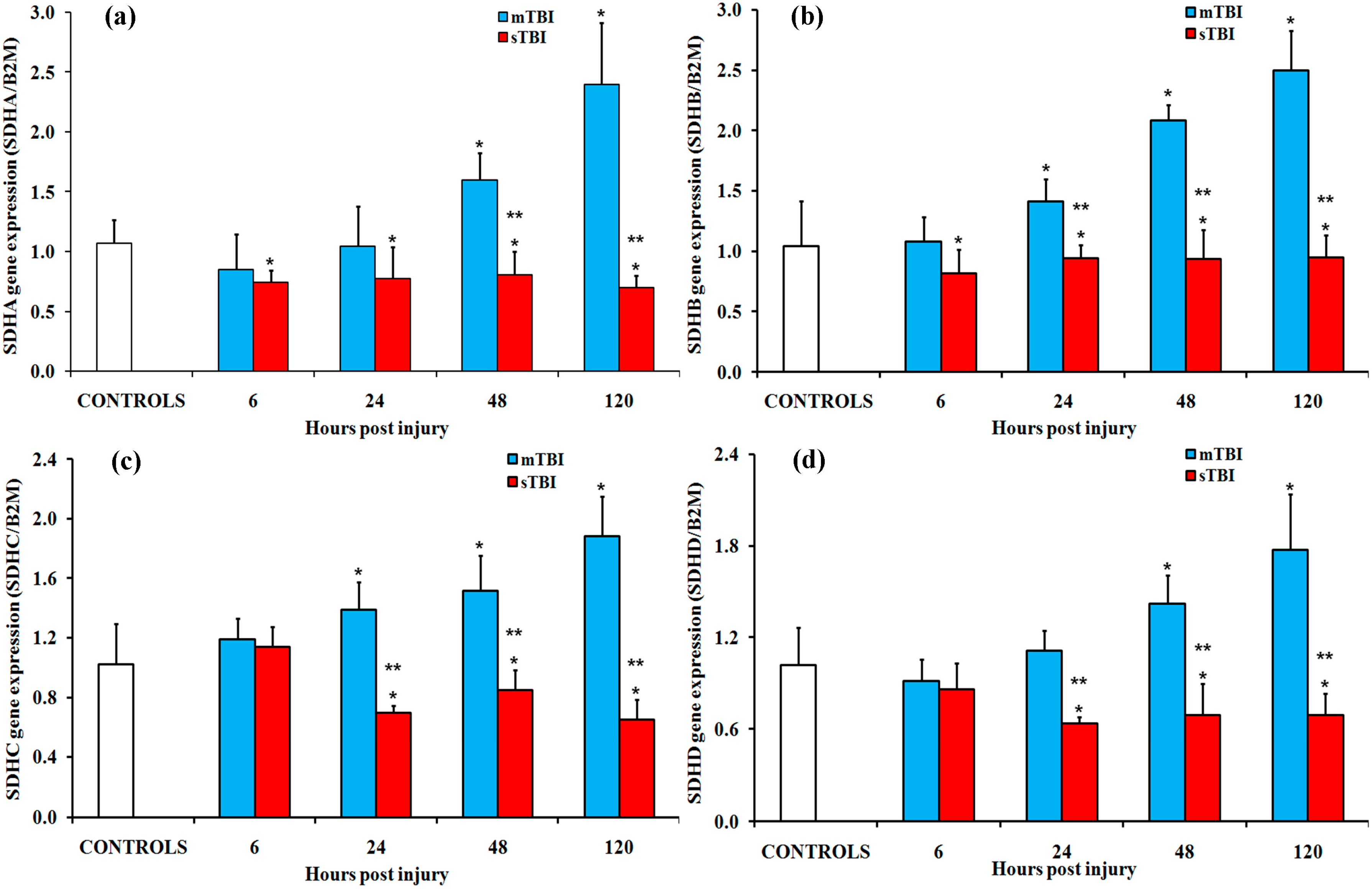
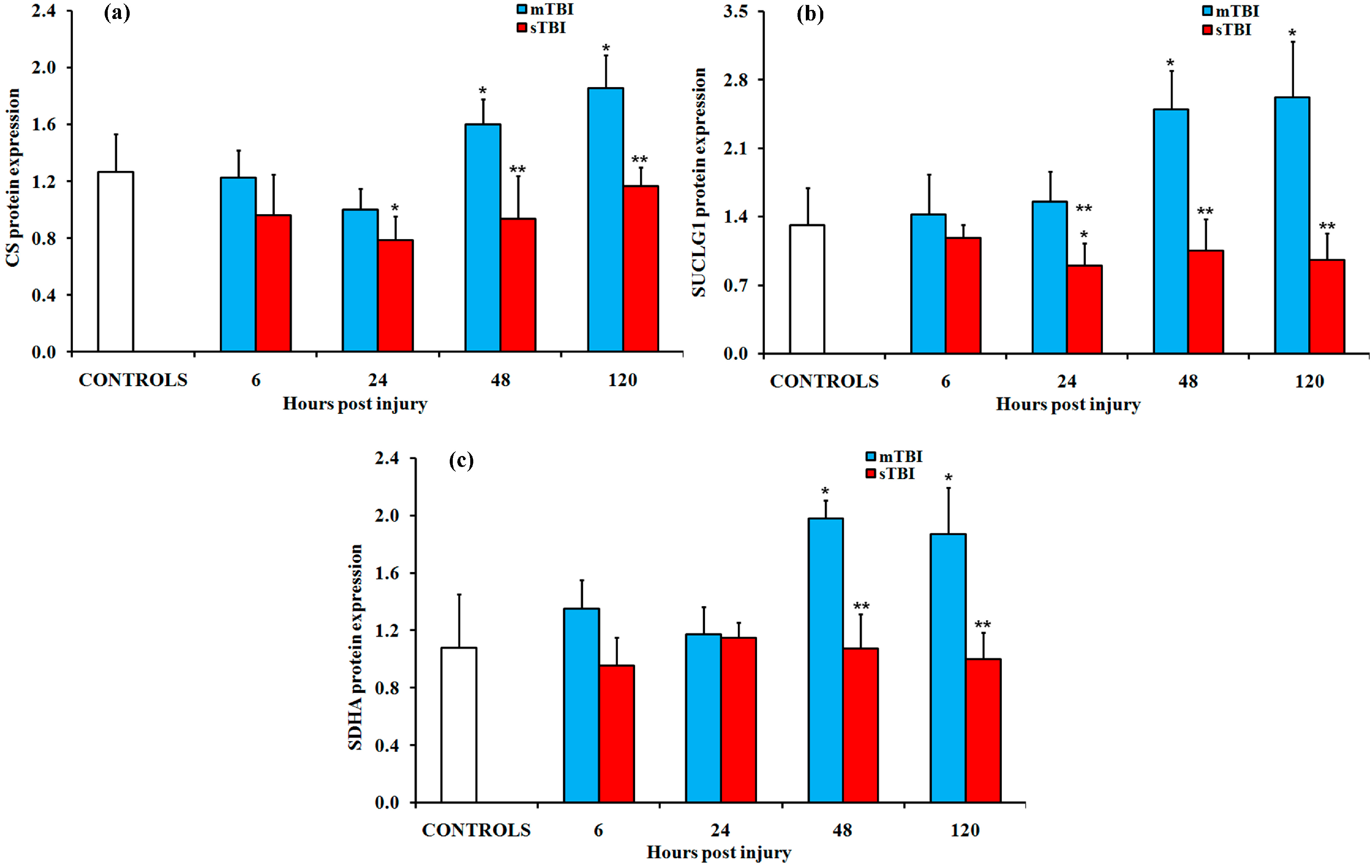
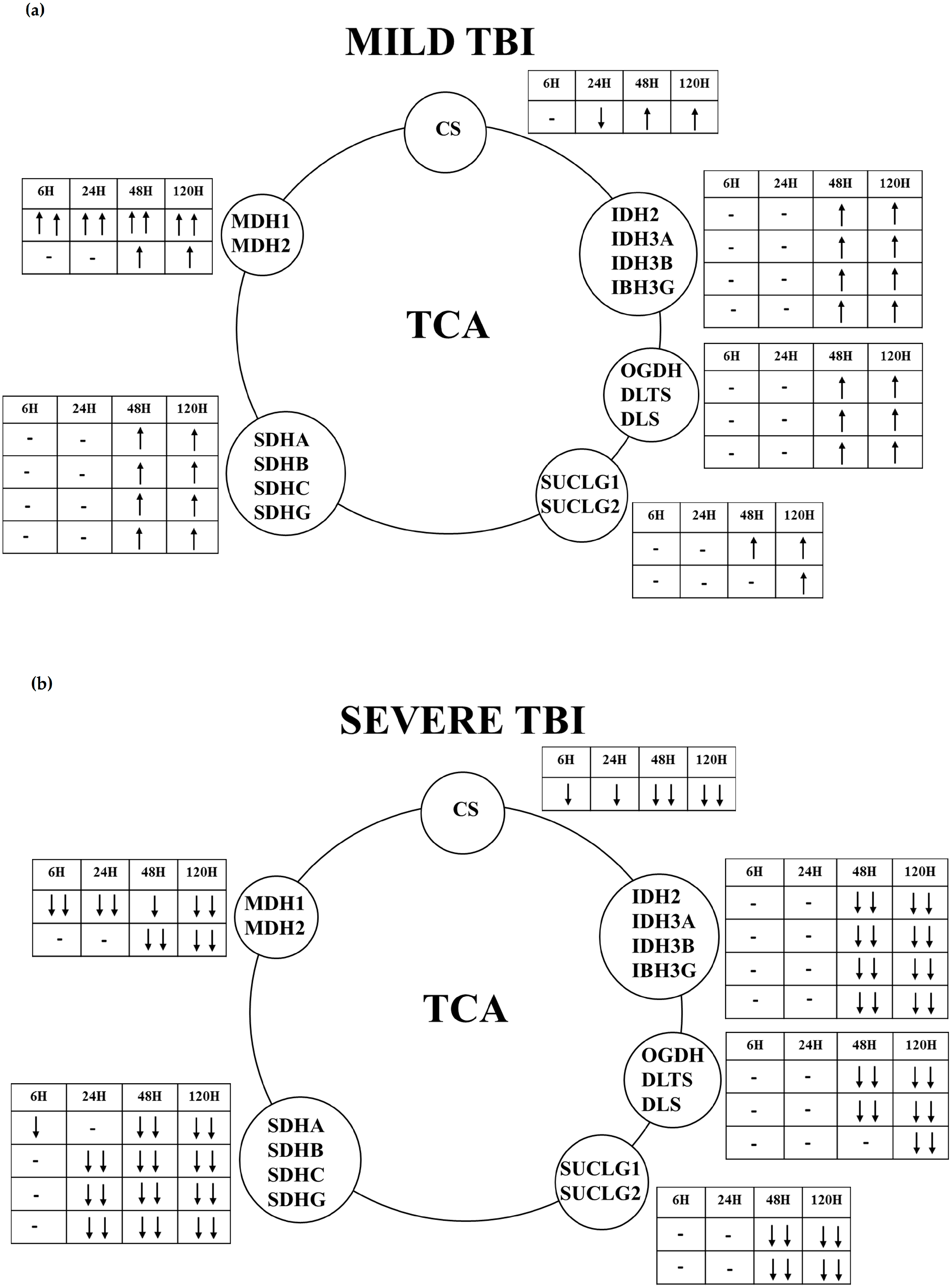
2.2. Gene and Protein Expressions of the Main Enzymes Regulating the TCA Cycle after Graded TBI
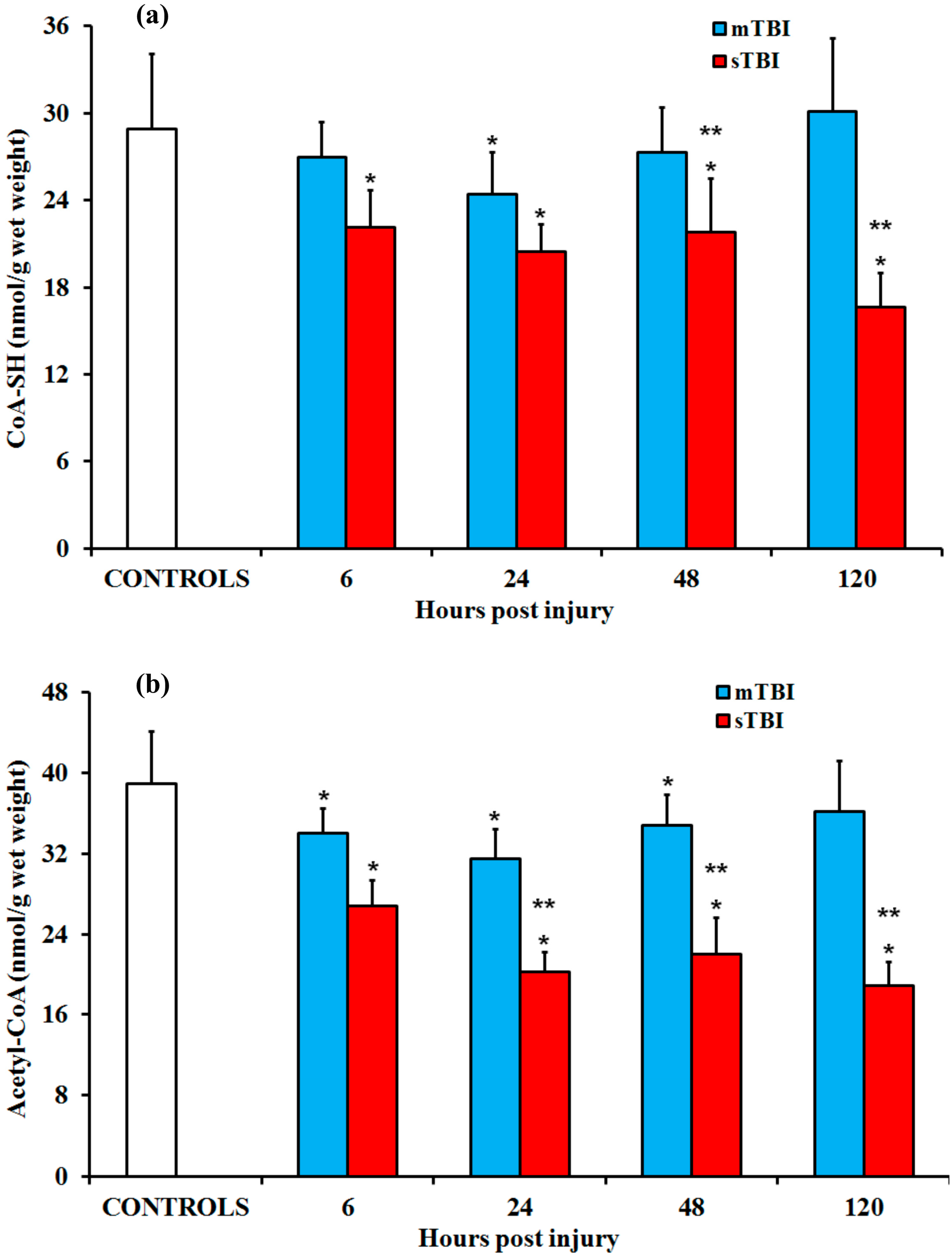
2.3. CoA-SH and Acetyl-CoAafter Graded TBI
3. Discussion
4. Materials and Methods
4.1. Animal and TBI Induction
4.2. Tissue Preparation for the Determination of Metabolites, Genes and Protein Expressions
4.3. Determination of Gene Expressions
4.4. Determination of Protein Expressions
4.5. Analysis of Metabolites
4.6. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Vakifahmetoglu-Norberg, H.; Ouchida, A.T.; Norberg, E. The role of mitochondria in metabolism and cell death. Biochem. Biophys. Res. Commun. 2017, 482, 426–431. [Google Scholar] [CrossRef]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Lapel, M.; Weston, P.; Strassheim, D.; Karoor, V.; Burns, N.; Lyubchenko, T.; Paucek, P.; Stenmark, K.R.; Gerasimovskaya, E.V. Glycolysis and oxidative phosphorylation are essential for purinergic receptor-mediated angiogenic responses in vasa vasorum endothelial cells. Am. J. Physiol. Cell Physiol. 2017, 312, C56–C70. [Google Scholar] [CrossRef] [PubMed]
- Caravas, J.; Wildman, D.E. A genetic perspective on glucose consumption in the cerebral cortex during human development. Diabetes Obes. Metab. 2014, 16 (Suppl. 1), 21–25. [Google Scholar] [CrossRef] [PubMed]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef]
- Ye, C.Y.; Lei, Y.; Tang, X.C.; Zhang, H.Y. Donepezil attenuates Aβ-associated mitochondrial dysfunction and reduces mitochondrial Aβ accumulation in vivo and in vitro. Neuropharmacology 2015, 95, 29–36. [Google Scholar] [CrossRef]
- Lehmann, S.; Martins, L.M. Insights into mitochondrial quality control pathways and Parkinson’s disease. J. Mol. Med. 2013, 91, 665–671. [Google Scholar] [CrossRef]
- Lazzarino, G.; Amorini, A.M.; Petzold, A.; Gasperini, C.; Ruggieri, S.; Quartuccio, M.E.; Lazzarino, G.; Di Stasio, E.; Tavazzi, B. Serum Compounds of energy metabolism impairment are related to disability, disease course and neuroimaging in multiple sclerosis. Mol. Neurobiol. 2017, 54, 7520–7533. [Google Scholar] [CrossRef]
- Walczak, J.; Dębska-Vielhaber, G.; Vielhaber, S.; Szymański, J.; Charzyńska, A.; Duszyński, J.; Szczepanowska, J. Distinction of sporadic and familial forms of ALS based on mitochondrial characteristics. FASEB J. 2019, 33, 4388–4403. [Google Scholar] [CrossRef]
- Yang, M.; Xu, Y.; Heisner, J.S.; Sun, J.; Stowe, D.F.; Kwok, W.M.; Camara, A.K.S. Peroxynitrite nitrates adenine nucleotide translocase and voltage-dependent anion channel 1 and alters their interactions and association with hexokinase II in mitochondria. Mitochondrion 2018, 46, 380–392. [Google Scholar] [CrossRef]
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2018, 1, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Vagnozzi, R.; Tavazzi, B.; Signoretti, S.; Amorini, A.M.; Belli, A.; Cimatti, M.; Delfini, R.; Di Pietro, V.; Finocchiaro, A.; Lazzarino, G. Temporal window of metabolic brain vulnerability to concussions: Mitochondrial-related impairment—Part I. Neurosurgery 2007, 61, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Vagnozzi, R.; Signoretti, S.; Tavazzi, B.; Floris, R.; Ludovici, A.; Marziali, S.; Tarascio, G.; Amorini, A.M.; Di Pietro, V.; Delfini, R.; et al. Temporal window of metabolic brain vulnerability to concussion: A pilot 1H-magnetic resonance spectroscopic study in concussed athletes—Part III. Neurosurgery 2008, 62, 1286–1295. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.L.; Kulbe, J.R.; Singh, I.N.; Wang, J.A.; Hall, E.D. Synaptic mitochondria are more susceptible to traumatic brain injury-induced oxidative damage and respiratory dysfunction than non-synaptic mitochondria. Neuroscience 2018, 386, 265–283. [Google Scholar] [CrossRef]
- Tavazzi, B.; Signoretti, S.; Lazzarino, G.; Amorini, A.M.; Delfini, R.; Cimatti, M.; Marmarou, A.; Vagnozzi, R. Cerebral oxidative stress and depression of energy metabolism correlate with severity of diffuse brain injury in rats. Neurosurgery 2005, 56, 582–589. [Google Scholar] [CrossRef]
- Di Pietro, V.; Amorini, A.M.; Tavazzi, B.; Vagnozzi, R.; Logan, A.; Lazzarino, G.; Signoretti, S.; Lazzarino, G.; Belli, A. The molecular mechanisms affecting N-acetylaspartate homeostasis following experimental graded traumatic brain injury. Mol. Med. 2014, 20, 147–157. [Google Scholar] [CrossRef]
- Signoretti, S.; Di Pietro, V.; Vagnozzi, R.; Lazzarino, G.; Amorini, A.M.; Belli, A.; D’Urso, S.; Tavazzi, B. Transient alterations of creatine, creatine phosphate, N-acetylaspartate and high-energy phosphates after mild traumatic brain injury in the rat. Mol. Cell Biochem. 2010, 333, 269–277. [Google Scholar] [CrossRef]
- Amorini, A.M.; Lazzarino, G.; Di Pietro, V.; Signoretti, S.; Lazzarino, G.; Belli, A.; Tavazzi, B. Severity of experimental traumatic brain injury modulates changes in concentrations of cerebral free amino acids. J. Cell Mol. Med. 2017, 21, 530–542. [Google Scholar] [CrossRef]
- Di Pietro, V.; Lazzarino, G.; Amorini, A.M.; Signoretti, S.; Hill, L.J.; Porto, E.; Tavazzi, B.; Lazzarino, G.; Belli, A. Fusion or fission: The destiny of mitochondria in traumatic brain injury of different severities. Sci. Rep. 2017, 7, 9189. [Google Scholar] [CrossRef]
- Tavazzi, B.; Vagnozzi, R.; Signoretti, S.; Amorini, A.M.; Belli, A.; Cimatti, M.; Delfini, R.; Di Pietro, V.; Finocchiaro, A.; Lazzarino, G. Temporal window of metabolic brain vulnerability to concussions: Oxidative and nitrosative stresses—Part II. Neurosurgery 2007, 61, 390–395. [Google Scholar] [CrossRef]
- Bhowmick, S.; D’Mello, V.; Ponery, N.; Abdul-Muneer, P.M. Neurodegeneration and sensorimotor deficits in the mouse model of traumatic brain injury. Brain Sci. 2018, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Jalloh, I.; Carpenter, K.L.; Grice, P.; Howe, D.J.; Mason, A.; Gallagher, C.N.; Helmy, A.; Murphy, M.P.; Menon, D.K.; Carpenter, T.A.; et al. Glycolysis and the pentose phosphate pathway after human traumatic brain injury: Microdialysis studies using 1,2-(13)C2 glucose. J. Cereb. Blood Flow Metab. 2015, 35, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; McArthur, D.L.; Alger, J.R.; Etchepare, M.; Hovda, D.A.; Glenn, T.C.; Huang, S.; Dinov, I.; Vespa, P.M. Early nonischemic oxidative metabolic dysfunction leads to chronic brain atrophy in traumatic brain injury. J. Cereb. Blood Flow Metab. 2010, 30, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Stovell, M.G.; Yan, J.L.; Sleigh, A.; Mada, M.O.; Carpenter, T.A.; Hutchinson, P.J.A.; Carpenter, K.L.H. Assessing metabolism and injury in acute human traumatic brain injury with magnetic resonance spectroscopy: Current and future applications. Front. Neurol. 2017, 8, 426. [Google Scholar] [CrossRef] [PubMed]
- Amorini, A.M.; Lazzarino, G.; Di Pietro, V.; Signoretti, S.; Lazzarino, G.; Belli, A.; Tavazzi, B. Metabolic, enzymatic and gene involvement in cerebral glucose dysmetabolism after traumatic brain injury. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2016, 1862, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Cheng, H.L.; Huang, R.Q.; Zhuang, Z.; Shi, J.X. Quantitative detection of the expression of mitochondrial cytochrome c oxidase subunits mRNA in the cerebral cortex after experimental traumatic brain injury. Brain Res. 2009, 1251, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Xing, G.; Barry, E.S.; Benford, B.; Grunberg, N.E.; Li, H.; Watson, W.D.; Sharma, P. Impact of repeated stress on traumatic brain injury-induced mitochondrial electron transport chain expression and behavioral responses in rats. Front. Neurol. 2013, 4, 196. [Google Scholar] [CrossRef][Green Version]
- Xu, Z.; Lv, X.A.; Dai, Q.; Lu, M.; Jin, Z. Exogenous BDNF increases mitochondrial pCREB and alleviates neuronal metabolic defects following mechanical injury in a MPTP-dependent way. Mol. Neurobiol. 2018, 55, 3499–3512. [Google Scholar] [CrossRef]
- Wu, Q.; Luo, C.L.; Tao, L.Y. Dynamin-related protein 1 (Drp1) mediating mitophagy contributes to the pathophysiology of nervous system diseases and brain injury. Histol. Histopathol. 2017, 32, 551–559. [Google Scholar]
- Fischer, T.D.; Hylin, M.J.; Zhao, J.; Moore, A.N.; Waxham, M.N.; Dash, P.K. Altered mitochondrial dynamics and TBI pathophysiology. Front. Syst. Neurosci. 2016, 10, 29. [Google Scholar] [CrossRef]
- Üçal, M.; Kraitsy, K.; Weidinger, A.; Paier-Pourani, J.; Patz, S.; Fink, B.; Molcanyi, M.; Schäfer, U. Comprehensive profiling of modulation of nitric oxide levels and mitochondrial activity in the injured brain: An experimental study based on the fluid percussion injury model in rats. J. Neurotrauma 2017, 34, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Carteri, R.B.; Kopczynski, A.; Rodolphi, M.S.; Strogulski, N.R.; Sartor, M.; Feldmann, M.; De Bastiani, M.A.; Duval Wannmacher, C.M.; de Franceschi, I.D.; Hansel, G.; et al. Testosterone administration after traumatic brain injury reduces mitochondrial dysfunction and neurodegeneration. J. Neurotrauma 2019, 36, 2246–2259. [Google Scholar] [CrossRef] [PubMed]
- Deng-Bryant, Y.; Prins, M.L.; Hovda, D.A.; Harris, N.G. Ketogenic diet prevents alterations in brain metabolism in young but not adult rats after traumatic brain injury. J. Neurotrauma 2011, 28, 1813–1825. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.J.; Attwell, D. The energetics of CNS white matter. J. Neurosci. 2012, 32, 356–371. [Google Scholar] [CrossRef]
- Xing, G.; Ren, M.; Watson, W.D.; O’Neill, J.T.; Verma, A. Traumatic brain injury-induced expression and phosphorylation of pyruvate dehydrogenase: A mechanism of dysregulated glucose metabolism. Neurosci. Lett. 2009, 454, 38–42. [Google Scholar] [CrossRef]
- Zhou, J.; Burns, M.P.; Huynh, L.; Villapol, S.; Taub, D.D.; Saavedra, J.M.; Blackman, M.R. Temporal changes in cortical and hippocampal expression of genes important for brain glucose metabolism following controlled cortical impact injury in mice. Front. Endocrinol. 2017, 8, 231. [Google Scholar] [CrossRef]
- Opii, W.O.; Nukala, V.N.; Sultana, R.; Pandya, J.D.; Day, K.M.; Merchant, M.L.; Klein, J.B.; Sullivan, P.G.; Butterfield, D.A. Proteomic identification of oxidized mitochondrial proteins following experimental traumatic brain injury. J. Neurotrauma 2007, 24, 772–789. [Google Scholar] [CrossRef]
- Xing, G.; Ren, M.; O’Neill, J.T.; Verma, A.; Watson, W.D. Controlled cortical impact injury and craniotomy result in divergent alterations of pyruvate metabolizing enzymes in rat brain. Exp. Neurol. 2012, 234, 31–38. [Google Scholar] [CrossRef]
- Xing, G.; Ren, M.; O’Neill, J.T.; Verma, A.; Watson, W.D. Pyruvate dehydrogenase phosphatase1 mRNA expression is divergently and dynamically regulated between rat cerebral cortex, hippocampus and thalamus after traumatic brain injury: A potential biomarker of TBI-induced hyper- and hypo-glycaemia and neuronal vulnerability. Neurosci. Lett. 2012, 525, 140–145. [Google Scholar]
- Carpenter, K.L.; Jalloh, I.; Hutchinson, P.J. Glycolysis and the significance of lactate in traumatic brain injury. Front. Neurosci. 2015, 9. [Google Scholar] [CrossRef]
- Reinert, M.; Schaller, B.; Widmer, H.R.; Seiler, R.; Bullock, R. Influence of oxygen therapy on glucose-lactate metabolism after diffuse brain injury. J. Neurosurg. 2004, 101, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Lama, S.; Auer, R.N.; Tyson, R.; Gallagher, C.N.; Tomanek, B.; Sutherland, G.R. Lactate storm marks cerebral metabolism following brain trauma. J. Biol. Chem. 2014, 289, 20200–20208. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Gurdziel, K.; Liu, J.; Qu, W.; Nuga, O.O.; Burl, R.B.; Hüttemann, M.; Pique-Regi, R.; Ruden, D.M. Smooth, an hnRNP-L homolog, might decrease mitochondrial metabolism by post-transcriptional regulation of isocitrate dehydrogenase (Idh) and other metabolic genes in the sub-acute phase of traumatic brain injury. Front. Genet. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, V.; Amorini, A.M.; Tavazzi, B.; Hovda, D.A.; Signoretti, S.; Giza, C.C.; Lazzarino, G.; Vagnozzi, R.; Lazzarino, G.; Belli, A. Potentially neuroprotective gene modulation in an in vitro model of mild traumatic brain injury. Mol. Cell Biochem. 2013, 375, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Kilbaugh, T.J.; Karlsson, M.; Byro, M.; Bebee, A.; Ralston, J.; Sullivan, S.; Duhaime, A.C.; Hansson, M.J.; Elmér, E.; Margulies, S.S. Mitochondrial bioenergetic alterations after focal traumatic brain injury in the immature brain. Exp. Neurol. 2015, 271, 136–144. [Google Scholar] [CrossRef]
- Jiang, X.B.; Kuroiwa, T.; Ohno, K.; Duan, L.; Aoyagi, M.; Hirakawa, K. Local mitochondrial function following traumatic brain injury in rats. Neurol. Med. Chir. 1999, 39, 649–656. [Google Scholar] [CrossRef][Green Version]
- Jiang, X.B.; Ohno, K.; Qian, L.; Tominaga, B.; Kuroiwa, T.; Nariai, T.; Hirakawa, K. Changes in local cerebral blood flow, glucose utilization, and mitochondrial function following traumatic brain injury in rats. Neurol. Med. Chir. 2000, 40, 16–29. [Google Scholar] [CrossRef]
- Gerbatin, R.D.R.; Cassol, G.; Dobrachinski, F.; Ferreira, A.P.O.; Quines, C.B.; Pace, I.D.D.; Busanello, G.L.; Gutierres, J.M.; Nogueira, C.W.; Oliveira, M.S.; et al. Guanosine protects against traumatic brain injury-induced functional impairments and neuronal loss by modulating excitotoxicity, mitochondrial dysfunction, and inflammation. Mol. Neurobiol. 2017, 54, 7585–7596. [Google Scholar] [CrossRef]
- Singh, K.; Trivedi, R.; Verma, A.; D’souza, M.M.; Koundal, S.; Rana, P.; Baishya, B.; Khushu, S. Altered metabolites of the rat hippocampus after mild and moderate traumatic brain injury—A combined in vivo and in vitro 1 H-MRS study. NMR Biomed. 2017, 30, e3764. [Google Scholar] [CrossRef]
- Bartnik-Olson, B.L.; Oyoyo, U.; Hovda, D.A.; Sutton, R.L. Astrocyte oxidative metabolism and metabolite trafficking after fluid percussion brain injury in adult rats. J. Neurotrauma 2010, 27, 2191–2202. [Google Scholar] [CrossRef]
- Sibon, O.C.; Strauss, E. Coenzyme A: To make it or uptake it? Nat. Rev. Mol. Cell Biol. 2016, 17, 605–606. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, I.; Carecchio, M.; Tiranti, V. Inborn errors of coenzyme A metabolism and neurodegeneration. J. Inherit. Metab. Dis. 2019, 42, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Shumar, S.A.; Fagone, P.; Alfonso-Pecchio, A.; Gray, J.T.; Rehg, J.E.; Jackowski, S.; Leonardi, R. Induction of neuron-specific degradation of coenzyme A models pantothenate kinase-associated neurodegeneration by reducing motor coordination in mice. PLoS ONE 2015. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Di Pietro, V.; Lazzarino, G.; Amorini, A.M.; Tavazzi, B.; D’Urso, S.; Longo, S.; Vagnozzi, R.; Signoretti, S.; Clementi, E.; Giardina, B.; et al. Neuroglobin expression and oxidant/antioxidant balance after graded traumatic brain injury in the rat. Free Radic. Biol. Med. 2014, 69, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Marmarou, A.; Foda, M.A.; van den Brink, W.; Campbell, J.; Kita, H.; Demetriadou, K. A new model of diffuse brain injury in rats. J. Neurosurg. 1994, 80, 291–300. [Google Scholar] [CrossRef]
- Lazzarino, G.; Amorini, A.M.; Fazzina, G.; Vagnozzi, R.; Signoretti, S.; Donzelli, S.; Di Stasio, E.; Giardina, B.; Tavazzi, B. Single-sample preparation for simultaneous cellular redox and energy state determination. Anal. Biochem. 2003, 322, 51–59. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Tavazzi, B.; Lazzarino, G.; Leone, P.; Amorini, A.M.; Bellia, F.; Janson, C.G.; Di Pietro, V.; Ceccarelli, L.; Donzelli, S.; Francis, J.S.; et al. Simultaneous high performance liquid chromatographic separation of purines, pyrimidines, N-acetylated amino acids, and dicarboxylic acids for the chemical diagnosis of inborn errors of metabolism. Clin. Biochem. 2005, 38, 997–1008. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Species | Gene Sequence | Forward | Reverse |
|---|---|---|---|---|
| CS | Rattus norvegicus | NM_130755.1 | TATGGCATGACGGAGATGAA | CATGAACTTGGGCCTTTCTA |
| DLAT | Rattus norvegicus | NM_031025.1 | AGACATCCCCATCAGCAACA | CAACGCTGACATCAACCACA |
| DLD | Rattus norvegicus | NM_199385.2 | TGGTTGTTATTGGTGCAGGA | CCCACATGACCCAAAAATTC |
| DLST | Rattus norvegicus | NM_001006981.2 | GAGCAGCCTGTAGTAAACGC | TTCATTCTTCCGGGCCTTCT |
| IDH2 | Rattus norvegicus | NM_001014161.1 | TGGGCCTGCAAGAACTATGA | ACCTTCTCCAGAGTCTGTGC |
| IDH3A | Rattus norvegicus | NM_053638.1 | ACACAAATCAGGCCAAGCTG | TGGCCAGGGTACTTATGCAA |
| IDH3B | Rattus norvegicus | NM_053581.1 | GATGTGCTTGTGATGCCCAA | ATGGCTGTGGGATTGGCTAT |
| IDH3G | Rattus norvegicus | NM_031551.1 | TTGCTAACCCTACTGCCACA | CCACAGCCCGTCCATTAATG |
| MDH1 | Rattus norvegicus | NM_001316877.1 | GATGGAGCTGCAAGACTGTG | GGTTCCCCACAACAATGACC |
| MDH2 | Rattus norvegicus | NM_031151.2 | GTTGACTTTCCCCAAGACCA | GTCCACCAGGGAGAAGACAA |
| OGDH | Rattus norvegicus | NM_001017461.1 | AACCCTTCCCACTTAGAGGC | TCAGTGGGGTAGGGAGAAGA |
| PDHA1 | Rattus norvegicus | NM_001004072.2 | TGGAGTTGCAGACATACCGT | AGCTGCTTCCTCGACTTCTT |
| PDHB | Rattus norvegicus | NM_001007620.1 | CCCGGTTTGAAAGTGGTCAG | ACTACAGTGATGTGGGTCCC |
| PDK1 | Rattus norvegicus | NM_053826.2 | TGCACAGTACTTCCAGGGAG | GTCGTCATGTCTTTCGGCTC |
| PDK2 | Rattus norvegicus | NM_030872.1 | ACCTCAGCCGCATCTCTATC | ACACAGGAGCTTAGCCATGT |
| PDK3 | Rattus norvegicus | NM_001106581.1 | CTGTGGCATCATTAGCACCC | GGAAGAACCCTGGGACTGAA |
| PDK4 | Rattus norvegicus | NM_053551.1 | TTGGCTGGTTTTGGTTACGG | CACCAGTCATCAGCCTCAGA |
| PDP1 | Rattus norvegicus | NM_001271108.2 | GTTCTCTGATGCCATGCCAG | CAGTTCTCCCTTGGCCTACA |
| PDP2 | Rattus norvegicus | NM_145091.4 | TGGAAACGGGACTAAGCACT | TATGTGCAGGTGAACTCCGT |
| SDHA | Rattus norvegicus | NM_130428.1 | GGGGAACATGGAAGAGGACA | AGGAGCTTGCTCTGTCATGT |
| SDHB | Rattus norvegicus | NM_001100539.1 | AGACCTTGGCAAAGTCTCGA | ATACTGTTGCTTGCCCTCCT |
| SDHC | Rattus norvegicus | NM_001005534.1 | ATTGAATGGGGTCCGACACT | GACAGCACTGCAAGAACCAA |
| SDHD | Rattus norvegicus | NM_198788.2 | TTGAATCCCTGCTCTGTGGT | AAGCCCAGCAAAGGTCAAAG |
| SUCLG2 | Rattus norvegicus | NM_001100750.1 | GGAGTGAGAGTGCAGAGGTT | AACACCTCCTTTCAAACCGC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazzarino, G.; Amorini, A.M.; Signoretti, S.; Musumeci, G.; Lazzarino, G.; Caruso, G.; Pastore, F.S.; Di Pietro, V.; Tavazzi, B.; Belli, A. Pyruvate Dehydrogenase and Tricarboxylic Acid Cycle Enzymes Are Sensitive Targets of Traumatic Brain Injury Induced Metabolic Derangement. Int. J. Mol. Sci. 2019, 20, 5774. https://doi.org/10.3390/ijms20225774
Lazzarino G, Amorini AM, Signoretti S, Musumeci G, Lazzarino G, Caruso G, Pastore FS, Di Pietro V, Tavazzi B, Belli A. Pyruvate Dehydrogenase and Tricarboxylic Acid Cycle Enzymes Are Sensitive Targets of Traumatic Brain Injury Induced Metabolic Derangement. International Journal of Molecular Sciences. 2019; 20(22):5774. https://doi.org/10.3390/ijms20225774
Chicago/Turabian StyleLazzarino, Giacomo, Angela Maria Amorini, Stefano Signoretti, Giuseppe Musumeci, Giuseppe Lazzarino, Giuseppe Caruso, Francesco Saverio Pastore, Valentina Di Pietro, Barbara Tavazzi, and Antonio Belli. 2019. "Pyruvate Dehydrogenase and Tricarboxylic Acid Cycle Enzymes Are Sensitive Targets of Traumatic Brain Injury Induced Metabolic Derangement" International Journal of Molecular Sciences 20, no. 22: 5774. https://doi.org/10.3390/ijms20225774
APA StyleLazzarino, G., Amorini, A. M., Signoretti, S., Musumeci, G., Lazzarino, G., Caruso, G., Pastore, F. S., Di Pietro, V., Tavazzi, B., & Belli, A. (2019). Pyruvate Dehydrogenase and Tricarboxylic Acid Cycle Enzymes Are Sensitive Targets of Traumatic Brain Injury Induced Metabolic Derangement. International Journal of Molecular Sciences, 20(22), 5774. https://doi.org/10.3390/ijms20225774