The Regulation of NFE2L2 (NRF2) Signalling and Epithelial-to-Mesenchymal Transition in Age-Related Macular Degeneration Pathology



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
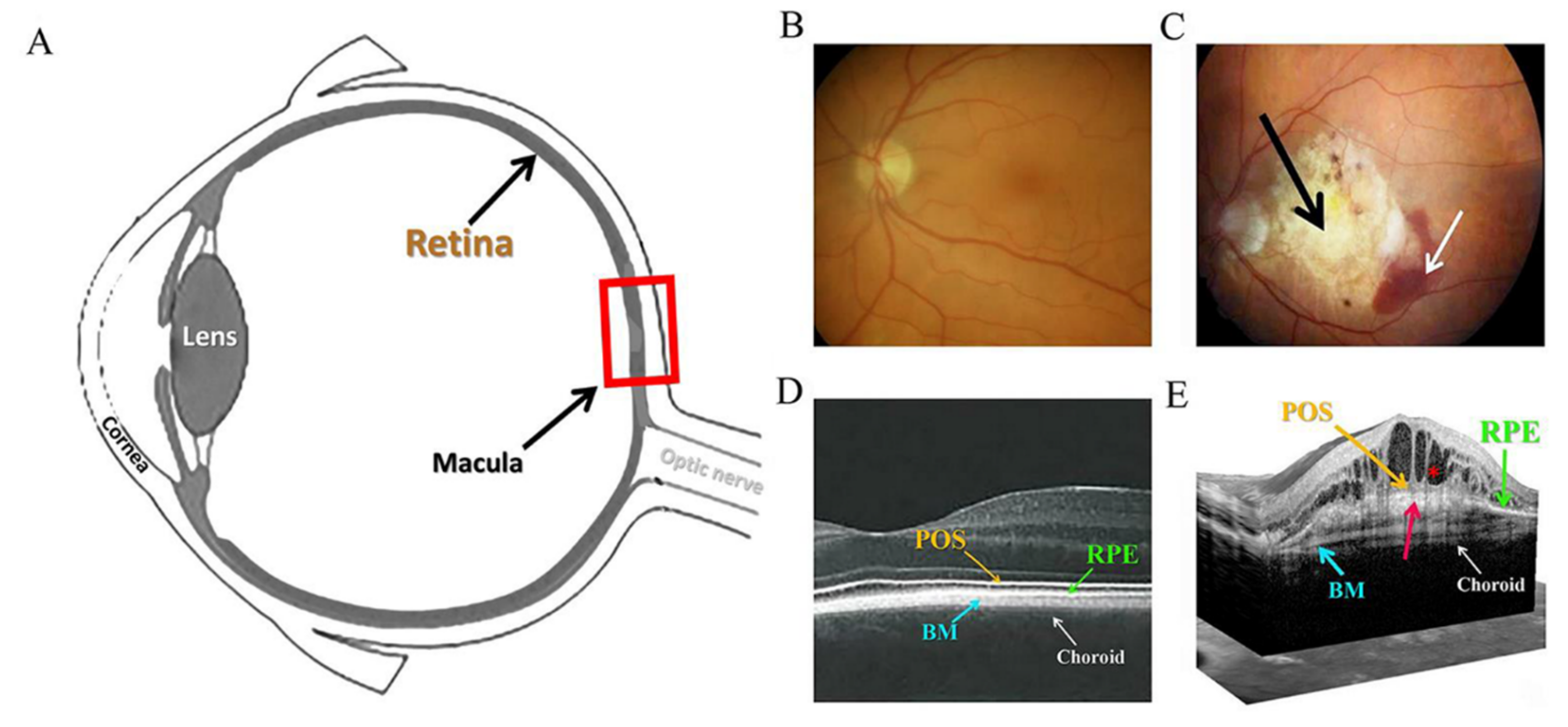
1.1. Age-Related Macular Degeneration (AMD)
1.2. Retinal Pigment Epithelium (RPE) and Oxidative Stress
2. The Kelch-Like ECH-Associated Protein 1 (KEAP1)-Nuclear Factor Erythroid 2-Rlated Factor (NFE2L2) Pathway
2.1. Overview
2.2. NFE2L2, the Master Regulator of Oxidative Stress Response
2.3. Regulation of NFE2L2 by KEAP1, the Sensor of Oxidative Stress
2.4. The Transmission and the Action of Oxidative Stress Response in the Nucleus
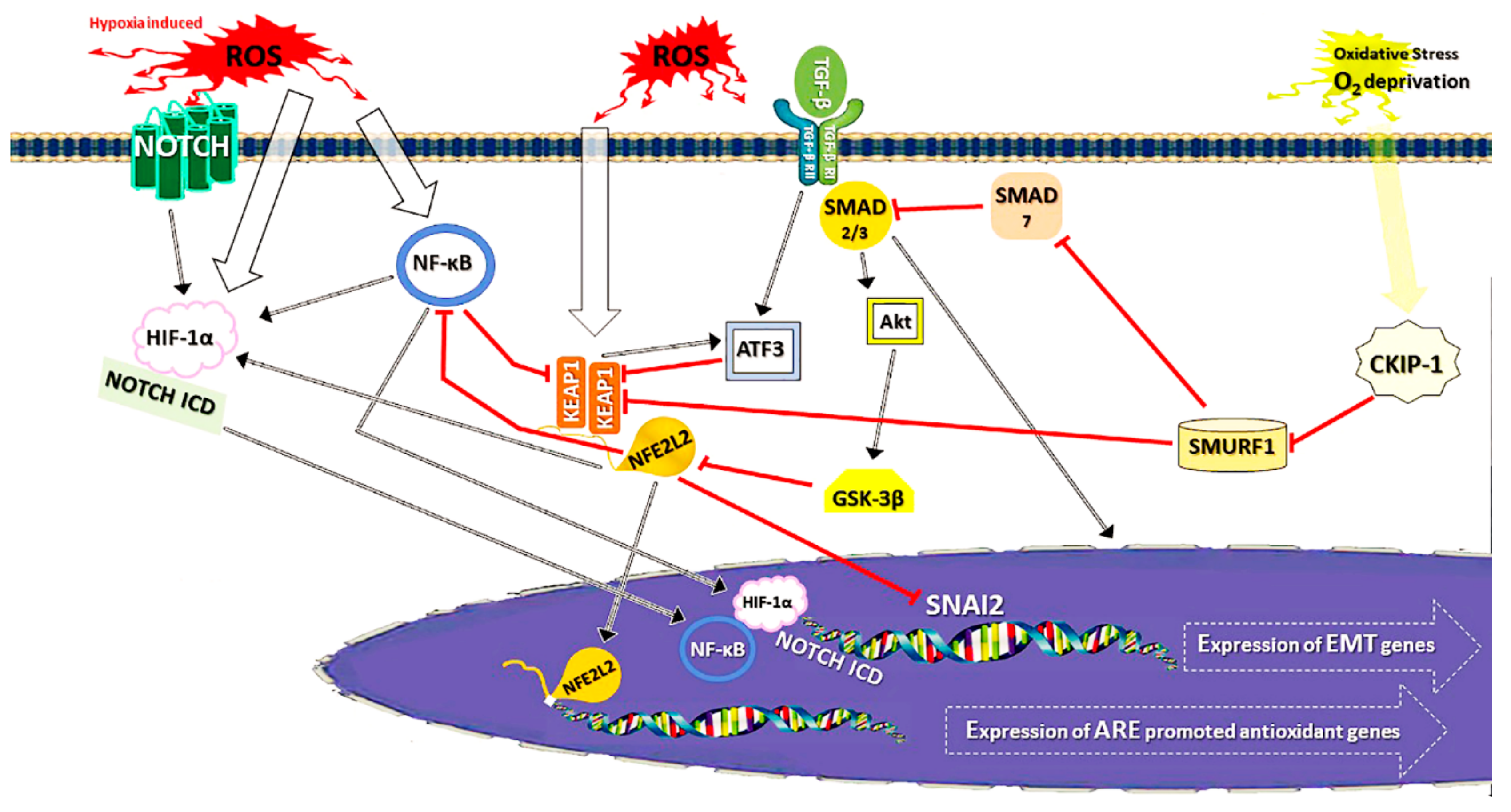
2.4.1. Non-Canonical KEAP1-NFE2L2 Regulation
2.4.2. KEAP1-Independent Control of NFE2L2
2.5. NFE2L2 Signalling and Autophagy
2.6. NFE2L2 and Connections to RPE Functioning and AMD
3. Epithelial to Mesenchymal Transition (EMT)
3.1. General
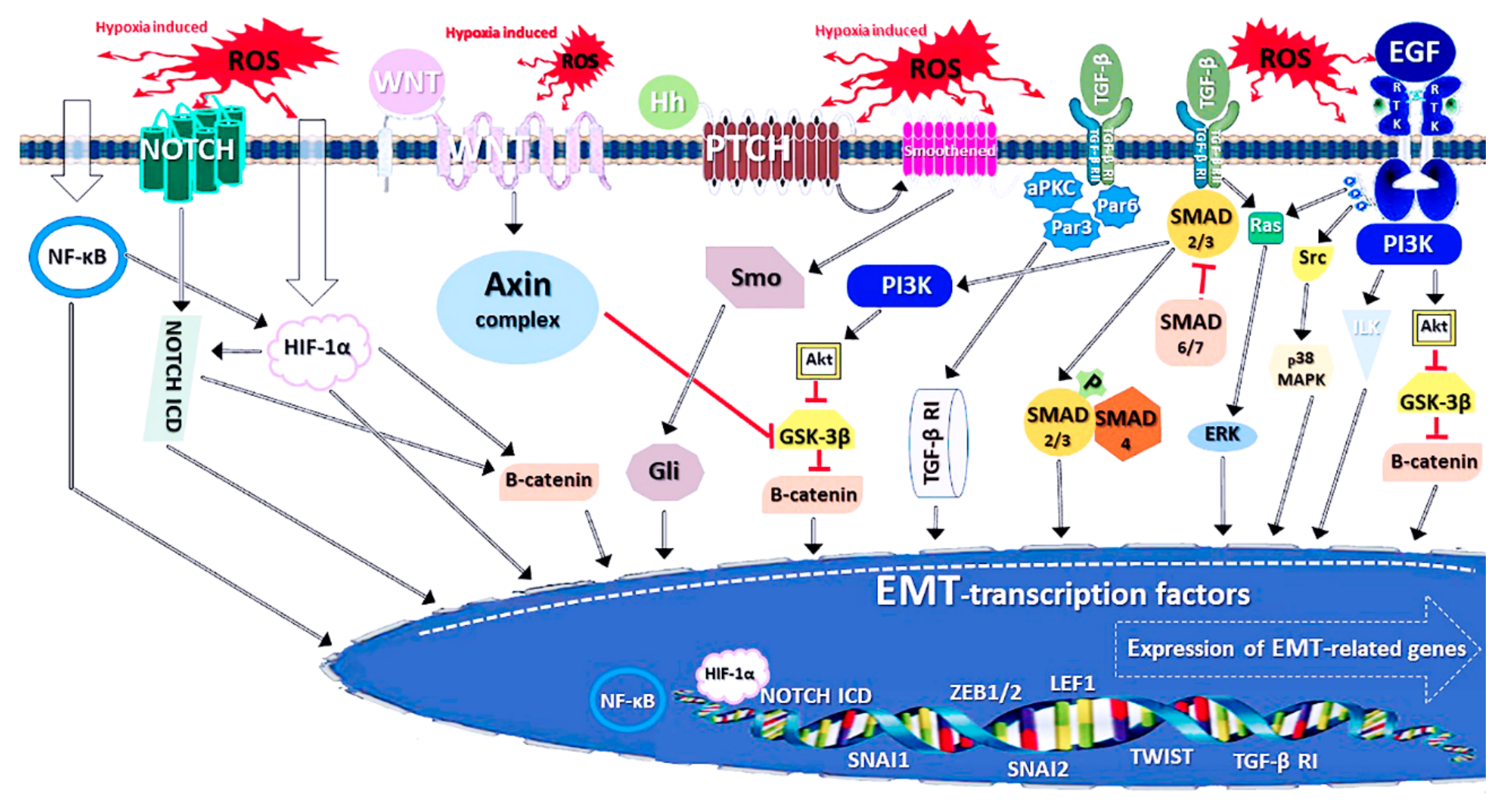
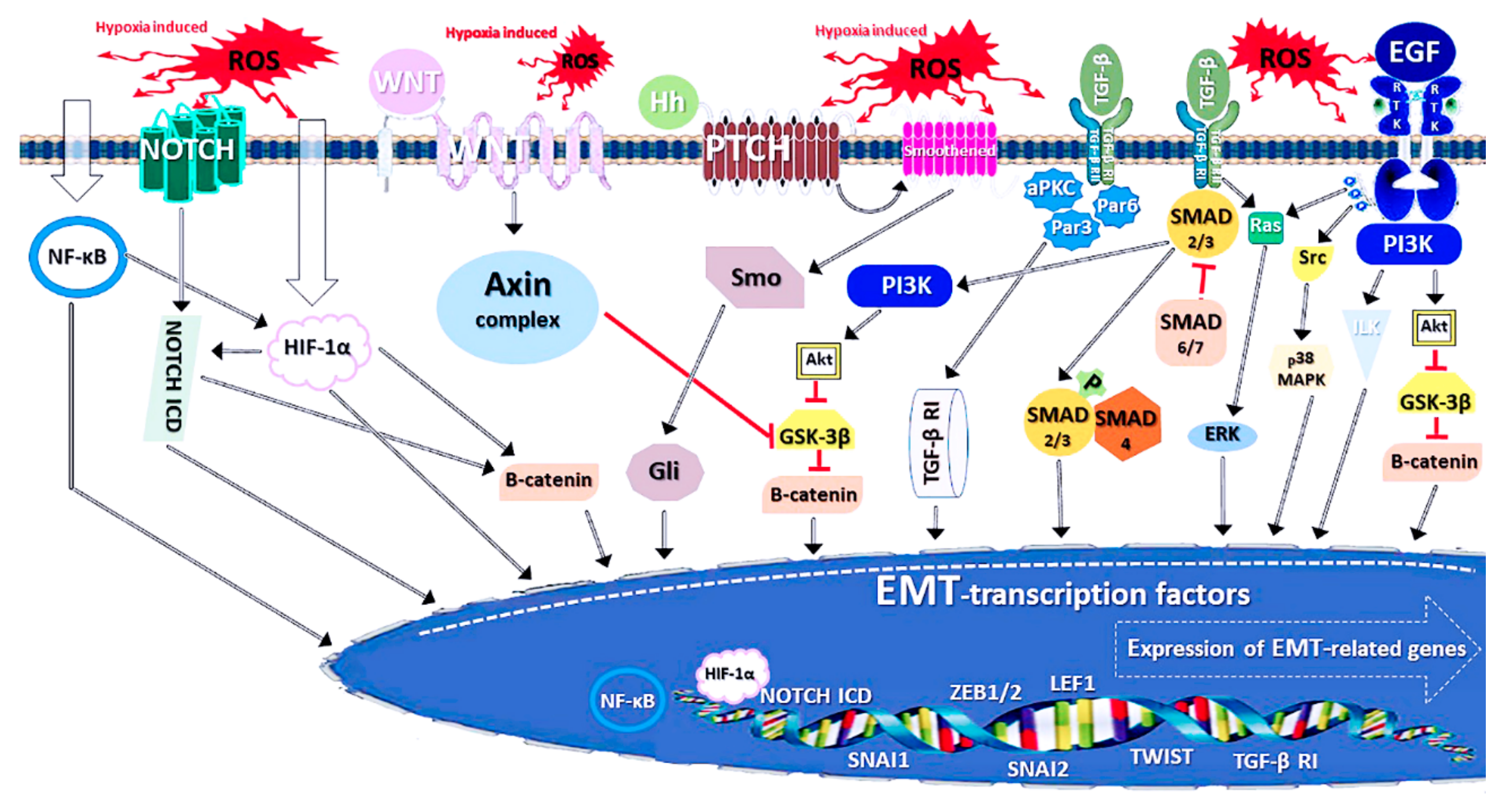
3.2. The Molecular Indications and Mechanisms of EMT
3.3. Connections Between EMT and NFE2L2 Signalling
3.4. EMT and Autophagy
3.5. EMT and AMD
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Akt | Protein kinase B |
| α-SMA | α-Smooth muscle actin |
| AMD | Age-related macular degeneration |
| AMPK | AMP-activated protein kinase |
| aPKC | Atypical protein kinase C |
| ATF4 | Activating transcription factor 4 |
| ARE | Antioxidant response element |
| Atg | Autophagy-related protein |
| BACH | BTB and CNC homologue protein |
| BM | Bruch’s membrane |
| BTB | Broad-complex, Tramtrac and Bric-à-brac |
| CNC | Cap’n’collar protein |
| CREB | cAMP response element-binding protein |
| GSH | Reduced glutathione |
| GSK-3β | Glycogen synthase kinase 3β |
| ECM | Extracellular matrix |
| EMT | Epithelial-to-mesenchymal transition |
| ERK | Extracellular signal-regulated kinase |
| HIF-1α | Hypoxia-induced factor 1, α subunit |
| HO-1 | Heme oxygenase 1 |
| KEAP1 | Kelch-like ECH-associated protein 1 |
| MAPK | p38 mitogen-activated protein kinase |
| MAPK1-LC3 | Microtubule-associated protein 1 light chain 3 |
| MET | Mesenchymal-to-epithelial transition |
| mtDNA | Mitochondrial DNA |
| NF-κB | Nuclear factor kappa-light chain-enhancer of activated B cells |
| NQO-1 | NAD(P)H:quinone-oxidoreductase 1 |
| NFE2L2 | Nuclear factor erythroid 2-related factor 2 |
| OS | Oxidative stress |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1- α |
| PI3K | Phosphoinositide-3-kinase (phosphatidylinositol-3-kinase) |
| POS | Photoreceptor outer segments |
| RAC3 | Co-regulatory receptor-associated cofactor 3 |
| RAR | Retinoic acid receptor |
| ROS | Reactive oxygen species |
| RPE | Retinal pigment epithelium |
| sMAF | Small musculoaponeurotic fibrosarcoma protein |
| SMURF1 | SMAD ubiquitination regulatory factor 1 |
| SOD1 | Cu/Zn superoxide dismutase 1 |
| SQSTM1/p62 | Sequestosome 1/p62 |
| TGF-β | Transforming growth factor β |
| Ub | Ubiquitin |
| UVA | Long ultraviolet radiation |
| VEGF | Vascular endothelial growth factor |
References
- Ambati, J.; Fowler, B.J. Mechanisms of age-related macular degeneration. Neuron 2012, 75, 26–39. [Google Scholar] [CrossRef]
- Hamdi, H.K.; Kenney, C. Age-related macular degeneration: A new viewpoint. Front. Biosci. 2003, 8, e305–e314. [Google Scholar] [CrossRef]
- United Nations, Department of Economic and Social Affairs, Population Division. World Population Prospects: The 2012 Revision, Highlights and Advance Tables; Working paper no. ESA/P/WP.228; United Nations, Department of Economic and Social Affairs, Population: New York, NY, USA, 2013. [Google Scholar]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef]
- National Guideline Alliance. Age-Related Macular Degeneration: Diagnosis and Management; National Institute for Health and Care Excellence: Clinical Guidelines; National Guideline Alliance: London, UK, 2018. [Google Scholar]
- Winkler, T.W.; Brandl, C.; Grassmann, F.; Gorski, M.; Stark, K.; Loss, J.; Weber, B.H.F.; Heid, I.M. International Age-related Macular Degeneration Genomics Consortium (IAMDGC). Investigating the modulation of genetic effects on late AMD by age and sex: Lessons learned and two additional loci. PLoS ONE 2018, 13, e0194321. [Google Scholar] [CrossRef]
- Arunkumar, R.; Calvo, C.M.; Conrady, C.D.; Bernstein, P.S. What do we know about the macular pigment in AMD: The past, the present, and the future. Eye 2018, 32, 992–1004. [Google Scholar] [CrossRef]
- Delcourt, C.; Cougnard-Grégoire, A.; Boniol, M.; Carrière, I.; Doré, J.F.; Delyfer, M.N.; Rougier, M.B.; Le Goff, M.; Dartigues, J.F.; Barberger-Gateau, P.; et al. Lifetime exposure to ambient ultraviolet radiation and the risk for cataract extraction and age-related macular degeneration: The Alienor Study. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7619–7627. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, H.; Yu, A.; Xie, J. Association between sunlight exposure and risk of age-related macular degeneration: A meta-analysis. BMC Ophthalmol. 2018, 18, 331. [Google Scholar] [CrossRef]
- Bowes Rickman, C.; Farsiu, S.; Toth, C.A.; Klingeborn, M. Dry age-related macular degeneration: Mechanisms, therapeutic targets, and imaging. Investig. Ophthalmol. Vis. Sci. 2013, 54, ORSF68–ORSF80. [Google Scholar] [CrossRef] [PubMed]
- Jobling, A.I.; Guymer, R.H.; Vessey, K.A.; Greferath, U.; Mills, S.A.; Brassington, K.H.; Luu, C.D.; Aung, K.Z.; Trogrlic, L.; Plunkett, M.; et al. Nanosecond laser therapy reverses pathologic and molecular changes in age-related macular degeneration without retinal damage. FASEB J. 2015, 29, 696–710. [Google Scholar] [CrossRef] [PubMed]
- Gillies, M.C.; Walton, R.; Simpson, J.M.; Arnold, J.J.; Guymer, R.H.; McAllister, I.L.; Hunyor, A.P.; Essex, R.W.; Morlet, N.; Barthelmes, D. Fight Retinal Blindness! Project Investigators. Prospective audit of exudative age-related macular degeneration: 12-month outcomes in treatment-naive eyes. Investig. Ophthalmol. Vis. Sci. 2013, 54, 5754–5760. [Google Scholar] [CrossRef] [PubMed]
- Kiser, P.D.; Palczewski, K. Retinoids and retinal diseases. Annu. Rev. Vis. Sci. 2016, 2, 197–234. [Google Scholar] [CrossRef] [PubMed]
- Plafker, S.M.; O’Mealey, G.B.; Szweda, L.I. Mechanisms for countering oxidative stress and damage in retinal pigment epithelium. Int. Rev. Cell Mol. Biol. 2012, 298, 135–177. [Google Scholar] [CrossRef] [PubMed]
- Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Ben, T.A.; Hong, S.; Heintzmann, R.; Gerig, G.; Ablonczy, Z.; Curcio, C.A.; Ach, T.; Smith, R.T. Hyperspectral autofluorescence imaging of drusen and retinal pigment epithelium in donor eyes with age-related macular degeneration. Retina 2016, 36 (Suppl. 1), S127–S136. [Google Scholar] [CrossRef]
- Rudolf, M.; Clark, M.E.; Chimento, M.F.; Li, C.M.; Medeiros, N.E.; Curcio, C.A. Prevalence and morphology of druse types in the macula and periphery of eyes with age-related maculopathy. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1200–1209. [Google Scholar] [CrossRef]
- Algvere, P.V.; Kvanta, A.; Seregard, S. Drusen maculopathy: A risk factor for visual deterioration. Acta Ophthalmol. 2016, 94, 427–433. [Google Scholar] [CrossRef]
- Bandello, F.; Sacconi, R.; Querques, L.; Corbelli, E.; Cicinelli, M.V.; Querques, G. Recent advances in the management of dry age-related macular degeneration: A review. F1000Research 2017, 6, 245. [Google Scholar] [CrossRef]
- Bhutto, I.; Lutty, G. Understanding age-related macular degeneration (AMD): Relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Mol. Aspects Med. 2012, 33, 295–317. [Google Scholar] [CrossRef]
- Hussain, A.A.; Starita, C.; Hodgetts, A.; Marshall, J. Macromolecular diffusion characteristics of ageing human Bruch’s membrane: Implications for age-related macular degeneration (AMD). Exp. Eye Res. 2010, 90, 703–710. [Google Scholar] [CrossRef]
- Moreira-Neto, C.A.; Moult, E.M.; Fujimoto, J.G.; Waheed, N.K.; Ferrara, D. Choriocapillaris loss in advanced age-related macular degeneration. J. Ophthalmol. 2018, 2018, 8125267. [Google Scholar] [CrossRef]
- Dysli, C.; Wolf, S.; Berezin, M.Y.; Sauer, L.; Hammer, M.; Zinkernagel, M.S. Fluorescence lifetime imaging ophthalmoscopy. Prog. Retin. Eye Res. 2017, 60, 120–143. [Google Scholar] [CrossRef] [PubMed]
- Ach, T.; Huisingh, C.; McGwin, G., Jr.; Messinger, J.D.; Zhang, T.; Bentley, M.J.; Gutierrez, D.B.; Ablonczy, Z.; Smith, R.T.; Sloan, K.R.; et al. Quantitative autofluorescence and cell density maps of the human retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4832–4841. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Hyttinen, J.; Ryhänen, T.; Viiri, J.; Paimela, T.; Toropainen, E.; Sorri, I.; Salminen, A. Mechanisms of protein aggregation in the retinal pigment epithelial cells. Front. Biosci. 2010, 2, 1374–1384. [Google Scholar] [CrossRef] [PubMed]
- Handa, J.T. How does the macula protect itself from oxidative stress? Mol. Asp. Med. 2012, 33, 418–435. [Google Scholar] [CrossRef]
- Roehlecke, C.; Schumann, U.; Ader, M.; Brunssen, C.; Bramke, S.; Morawietz, H.; Funk, R.H. Stress reaction in outer segments of photoreceptors after blue light irradiation. PLoS ONE 2013, 8, e71570. [Google Scholar] [CrossRef]
- Hyttinen, J.M.T.; Błasiak, J.; Niittykoski, M.; Kinnunen, K.; Kauppinen, A.; Salminen, A.; Kaarniranta, K. DNA damage response and autophagy in the degeneration of retinal pigment epithelial cells—Implications for age-related macular degeneration (AMD). Ageing Res. Rev. 2017, 36, 64–77. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Hyttinen, J.M.T.; Toropainen, E.; Kaarniranta, K. Endoplasmic reticulum stress in age-related macular degeneration: Trigger for neovascularization. Mol. Med. 2010, 16, 535–542. [Google Scholar] [CrossRef]
- Boulton, M.E. Studying melanin and lipofuscin in RPE cell culture models. Exp. Eye Res. 2014, 126, 61–67. [Google Scholar] [CrossRef]
- Fernandez-Godino, R.; Pierce, E.A.; Garland, D.L. Extracellular matrix alterations and deposit formation in AMD. Adv. Exp. Med. Biol. 2016, 854, 53–58. [Google Scholar] [CrossRef]
- Hyttinen, J.M.T.; Amadio, M.; Viiri, J.; Pascale, A.; Salminen, A.; Kaarniranta, K. Clearance of misfolded and aggregated proteins by aggrephagy and implications for aggregation diseases. Ageing Res. Rev. 2014, 18, 16–28. [Google Scholar] [CrossRef]
- Kauppinen, A.; Paterno, J.J.; Błasiak, J.; Salminen, A.; Kaarniranta, K. Inflammation and its role in age-related macular degeneration. Cell. Mol. Life Sci. 2016, 73, 1765–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maresca, V.; Flori, E.; Briganti, S.; Mastrofrancesco, A.; Fabbri, C.; Mileo, A.M.; Paggi, M.G.; Picardo, M. Correlation between melanogenic and catalase activity in in vitro human melanocytes: A synergic strategy against oxidative stress. Pigment Cell Melanoma Res. 2008, 21, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Cunha, E.S.; Kawahara, R.; Kadowaki, M.K.; Amstalden, H.G.; Noleto, G.R.; Cadena, S.M.; Winnischofer, S.M.; Martinez, G.R. Melanogenesis stimulation in B16-F10 melanoma cells induces cell cycle alterations, increased ROS levels and a differential expression of proteins as revealed by proteomic analysis. Exp. Cell Res. 2012, 318, 1913–1925. [Google Scholar] [CrossRef] [PubMed]
- Korytowski, W.; Pilas, B.; Sarna, T.; Kalyanaraman, B. Photoinduced generation of hydrogen peroxide and hydroxyl radicals in melanins. Photochem. Photobiol. 1987, 45, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Li, J.; Lin, X.; Wu, W.; Kang, K.; Fu, W. Oxidation levels differentially impact melanocytes: Low versus high concentration of hydrogen peroxide promotes melanin synthesis and melanosome transfer. Dermatology 2012, 224, 145–153. [Google Scholar] [CrossRef]
- Nag, T.C. Ultrastructural changes in the melanocytes of aging human choroid. Micron 2015, 79, 16–23. [Google Scholar] [CrossRef]
- Błasiak, J.; Petrovski, G.; Veréb, Z.; Facskó, A.; Kaarniranta, K. Oxidative stress, hypoxia, and autophagy in the neovascular processes of age-related macular degeneration. Biomed. Res. Int. 2014, 2014, 768026. [Google Scholar] [CrossRef]
- Zhao, Z.; Chen, Y.; Wang, J.; Sternberg, P.; Freeman, M.L.; Grossniklaus, H.E.; Cai, J. Age-related retinopathy in NRF2-deficient mice. PLoS ONE 2011, 6, e19456. [Google Scholar] [CrossRef] [Green Version]
- Felszeghy, S.; Viiri, J.; Paterno, J.J.; Hyttinen, J.M.T.; Koskela, A.; Chen, M.; Leinonen, H.O.; Tanila, H.; Kivinen, N.; Koistinen, A.; et al. Loss of NRF-2- and PGC-1α genes leads to retinal pigment epithelium damage resembling dry age-related macular degeneration. Redox Biol. 2018, 20, 1–12. [Google Scholar] [CrossRef]
- Ishikawa, K.; Kannan, R.; Hinton, D.R. Molecular mechanisms of subretinal fibrosis in age-related macular degeneration. Exp. Eye Res. 2014, 142, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Baldelli, S.; Aquilano, K.; Ciriolo, M.R. Punctum on two different transcription factors regulated by PGC-1α: Nuclear factor erythroid-derived 2-like 2 and nuclear respiratory factor 2. Biochim. Biophys. Acta 2013, 1830, 4137–4146. [Google Scholar] [CrossRef] [PubMed]
- Silva-Palacios, A.; Ostolga-Chavarría, M.; Zazueta, C.; Königsberg, M. Nrf2: Molecular and epigenetic regulation during aging. Ageing Res. Rev. 2018, 47, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Chui, D.H.; Tang, W.; Orkin, S.H. cDNA cloning of murine Nrf 2 gene, coding for a p45 NF-E2 related transcription factor. Biochem. Biophys. Res. Commun. 1995, 209, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Sihvola, V.; Levonen, A.L. Keap1 as the redox sensor of the antioxidant response. Arch. Biochem. Biophys. 2017, 617, 94–100. [Google Scholar] [CrossRef]
- Suzuki, T.; Yamamoto, M. Molecular basis of the Keap1-Nrf2 system. Free Radic. Biol. Med. 2015, 88 Pt B, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.; Doan, A.; Hybertson, B.M. The clinical potential of influencing Nrf2 signaling in degenerative and immunological disorders. Clin. Pharmacol. 2014, 6, 19–34. [Google Scholar] [CrossRef] [Green Version]
- Larabee, C.M.; Desai, S.; Agasing, A.; Georgescu, C.; Wren, J.D.; Axtell, R.C.; Plafker, S.M. Loss of Nrf2 exacerbates the visual deficits and optic neuritis elicited by experimental autoimmune encephalomyelitis. Mol. Vis. 2016, 22, 1503–1513. [Google Scholar]
- Lewis, K.N.; Mele, J.; Hayes, J.D.; Buffenstein, R. Nrf2, a guardian of healthspan and gatekeeper of species longevity. Integr. Comp. Biol. 2010, 50, 829–843. [Google Scholar] [CrossRef] [Green Version]
- Nakagami, Y. Nrf2 is an attractive therapeutic target for retinal diseases. Oxid. Med. Cell. Longev. 2016, 2016, 7469326. [Google Scholar] [CrossRef] [Green Version]
- Chaharbakhshi, E.; Jemc, J.C. Broad-complex, tramtrack, and bric-à-brac (BTB) proteins: Critical regulators of development. Genesis 2016, 54, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Numazawa, S.; Yoshida, T. Redox regulation of the transcriptional repressor Bach1. Free Radic. Biol. Med. 2005, 38, 1344–1352. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; O’Connor, T.; Katsuoka, F.; Engel, J.D.; Yamamoto, M. Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene 2002, 294, 1–12. [Google Scholar] [CrossRef]
- Tian, W.; de la Vega, M.R.; Schmidlin, C.J.; Ooi, A.; Zhang, D.D. Kelch-like ECH-associated protein 1 (KEAP1) differentially regulates nuclear factor erythroid-2-related factors 1 and 2 (NRF1 and NRF2). J. Biol. Chem. 2018, 293, 2029–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Yu, S.; Chen, J.D.; Kong, A.N. The nuclear cofactor RAC3/AIB1/SRC-3 enhances Nrf2 signaling by interacting with transactivation domains. Oncogene 2013, 32, 514–527. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Chen, Y.; Hou, X.; Huang, M.; Jin, J. Emerging role of NRF2 in chemoresistance by regulating drug-metabolizing enzymes and efflux transporters. Drug Metab. Rev. 2016, 48, 541–567. [Google Scholar] [CrossRef]
- Dodson, M.; Redmann, M.; Rajasekaran, N.S.; Darley-Usmar, V.; Zhang, J. KEAP1-NRF2 signalling and autophagy in protection against oxidative and reductive proteotoxicity. Biochem. J. 2015, 469, 347–355. [Google Scholar] [CrossRef]
- Kawai, Y.; Garduño, L.; Theodore, M.; Yang, J.; Arinze, I.J. Acetylation-deacetylation of the transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2) regulates its transcriptional activity and nucleocytoplasmic localization. J. Biol. Chem. 2011, 286, 7629–7640. [Google Scholar] [CrossRef] [Green Version]
- Zenkov, N.K.; Kozhin, P.M.; Chechushkov, A.V.; Martinovich, G.G.; Kandalintseva, N.V.; Menshchikova, E.B. Mazes of Nrf2 regulation. Biochemistry 2017, 82, 556–564. [Google Scholar] [CrossRef]
- Choudhary, S.; Xiao, T.; Srivastava, S.; Zhang, W.; Chan, L.L.; Vergara, L.A.; Van Kuijk, F.J.; Ansari, N.H. Toxicity and detoxification of lipid-derived aldehydes in cultured retinal pigmented epithelial cells. Toxicol. Appl. Pharmacol. 2005, 204, 122–134. [Google Scholar] [CrossRef]
- Woodell, A.; Rohrer, B. A mechanistic review of cigarette smoke and age-related macular degeneration. Adv. Exp. Med. Biol. 2014, 801, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Bertram, K.M.; Baglole, C.J.; Phipps, R.P.; Libby, R.T. Molecular regulation of cigarette smoke induced-oxidative stress in human retinal pigment epithelial cells: Implications for age-related macular degeneration. Am. J. Physiol. Cell Physiol. 2009, 297, C1200–C1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connell, M.A.; Hayes, J.D. The Keap1/Nrf2 pathway in health and disease: From the bench to the clinic. Biochem. Soc. Trans. 2015, 43, 687–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frede, K.; Ebert, F.; Kipp, A.P.; Schwerdtle, T.; Baldermann, S. Lutein activates the transcription factor Nrf2 in human retinal pigment epithelial cells. J. Agric. Food Chem. 2017, 65, 5944–5952. [Google Scholar] [CrossRef] [Green Version]
- Koskela, A.; Reinisalo, M.; Hyttinen, J.M.T.; Kaarniranta, K.; Karjalainen, R.O. Pinosylvin-mediated protection against oxidative stress in human retinal pigment epithelial cells. Mol. Vis. 2014, 20, 760–769. [Google Scholar]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [Green Version]
- Perepechaeva, M.L.; Kolosova, N.G.; Stefanova, N.A.; Fursova, A.Z.; Grishanova, A.Y. The influence of changes in expression of redox-sensitive genes on the development of retinopathy in rats. Exp. Mol. Pathol. 2016, 101, 124–132. [Google Scholar] [CrossRef]
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol. 2018, 17, 297–314. [Google Scholar] [CrossRef]
- Zhu, M.; Fahl, W.E. Functional characterization of transcription regulators that interact with the electrophile response element. Biochem. Biophys. Res. Commun. 2001, 289, 212–219. [Google Scholar] [CrossRef]
- De los Fayos Alonso, I.G.; Liang, H.C.; Turner, S.D.; Lagger, S.; Merkel, O.; Kenner, L. The role of activator protein-1 (AP-1) family members in CD30-positive lymphomas. Cancers 2018, 10, 93. [Google Scholar] [CrossRef] [Green Version]
- Rössler, O.G.; Thiel, G. Specificity of stress-responsive transcription factors Nrf2, ATF4, and AP-1. J. Cell. Biochem. 2017, 118, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, K.I.; Katoh, Y.; Kusunoki, H.; Itoh, K.; Tanaka, T.; Yamamoto, Y. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: Characterization of the two-site molecular recognition model. Mol. Cell. Biol. 2006, 26, 2887–2900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, S.C.; Hannink, M. CAND1-mediated substrate adaptor recycling is required for efficient repression of Nrf2 by Keap1. Mol. Cell. Biol. 2006, 26, 1235–1244. [Google Scholar] [CrossRef] [Green Version]
- Iso, T.; Suzuki, T.; Baird, L.; Yamamoto, M. Absolute amounts and status of the Nrf2-Keap1-Cul3 complex within cells. Mol. Cell. Biol. 2016, 36, 3100–3112. [Google Scholar] [CrossRef] [Green Version]
- Hong, F.; Freeman, M.L.; Liebler, D.C. Identification of sensor cysteines in human Keap1 modified by the cancer chemopreventive agent sulforaphane. Chem. Res. Toxicol. 2005, 18, 1917–1926. [Google Scholar] [CrossRef]
- Furukawa, M.; Xiong, Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol. Cell. Biol. 2005, 25, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.D.; Lo, S.C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [Green Version]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef] [Green Version]
- Tao, S.; Liu, P.; Luo, G.; de la Vega, M.R.; Chen, H.; Wu, T.; Tillotson, J.; Chapman, E.; Zhang, D.D. p97 negatively regulates NRF2 by extracting ubiquitylated NRF2 from the KEAP1-CUL3 E3 complex. Mol. Cell. Biol. 2017, 37, e00660-16. [Google Scholar] [CrossRef] [Green Version]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, R.; Suzuki, T.; Hiramoto, K.; Asami, S.; Naganuma, E.; Suda, H.; Iso, T.; Yamamoto, H.; Morita, M.; Baird, L.; et al. Characterizations of three major cysteine sensors of Keap1 in stress response. Mol. Cell. Biol. 2016, 36, 271–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rachakonda, G.; Xiong, Y.; Sekhar, K.R.; Stamer, S.L.; Liebler, D.C.; Freeman, M.L. Covalent modification at Cys151 dissociates the electrophile sensor Keap1 from the ubiquitin ligase CUL3. Chem. Res. Toxicol. 2008, 21, 705–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, B.S.; Boulton, M.E.; Gottsch, J.D.; Sternberg, P. Oxidative damage and age-related macular degeneration. Mol. Vis. 1999, 5, 32. [Google Scholar]
- Takaya, K.; Suzuki, T.; Motohashi, H.; Onodera, K.; Satomi, S.; Kensler, T.W.; Yamamoto, M. Validation of the multiple sensor mechanism of the Keap1-Nrf2 system. Free Radic. Biol. Med. 2012, 53, 817–827. [Google Scholar] [CrossRef] [Green Version]
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; Mack, N.J.; Ahmad, N. The role of sirtuins in antioxidant and redox signaling. Antioxid. Redox Signal. 2018, 28, 643–661. [Google Scholar] [CrossRef]
- Huang, K.; Gao, X.; Wei, W. The crosstalk between Sirt1 and Keap1/Nrf2/ARE anti-oxidative pathway forms a positive feedback loop to inhibit FN and TGF-β1 expressions in rat glomerular mesangial cells. Exp. Cell Res. 2017, 361, 63–72. [Google Scholar] [CrossRef]
- Huang, H.C.; Nguyen, T.; Pickett, C.B. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 2002, 277, 42769–42774. [Google Scholar] [CrossRef] [Green Version]
- Joo, M.S.; Kim, W.D.; Lee, K.Y.; Kim, J.H.; Koo, J.H.; Kim, S.G. AMPK facilitates nuclear accumulation of Nrf2 by phosphorylating at Serine 550. Mol. Cell. Biol. 2016, 36, 1931–1942. [Google Scholar] [CrossRef] [Green Version]
- Numazawa, S.; Ishikawa, M.; Yoshida, A.; Tanaka, S.; Yoshida, T. Atypical protein kinase C mediates activation of NF-E2-related factor 2 in response to oxidative stress. Am. J. Physiol. Cell. Physiol. 2003, 285, C334–C342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva-Islas, C.A.; Maldonado, P.D. Canonical and non-canonical mechanisms of Nrf2 activation. Pharmacol. Res. 2018, 134, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Theodore, M.; Kawai, Y.; Yang, J.; Kleshchenko, Y.; Reddy, S.P.; Villalta, F.; Arinze, I.J. Multiple nuclear localization signals function in the nuclear import of the transcription factor Nrf2. J. Biol. Chem. 2008, 283, 8984–8994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Yu, S.W.; Kong, A.N.T. Nrf2 possesses a redox-sensitive nuclear exporting signal in the Neh5 transactivation domain. J. Biol. Chem. 2006, 281, 27251–27263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Yu, S.; Liu, T.; Kim, J.H.; Blank, V.; Li, H.; Kong, A.N. Heterodimerization with small Maf proteins enhances nuclear retention of Nrf2 via masking the NESzip motif. Biochim. Biophys. Acta 2008, 1783, 1847–1856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Lai, Q.; Chen, C.; Li, N.; Sun, F.; Huang, W.; Zhang, S.; Yu, Q.; Yang, P.; Xiong, F.; et al. Both conditional ablation and overexpression of E2 SUMO-conjugating enzyme (UBC9) in mouse pancreatic beta cells result in impaired beta cell function. Diabetologia 2018, 61, 881–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Chin, E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol. Cell. Biol. 2009, 29, 2659–2672. [Google Scholar] [CrossRef] [Green Version]
- Su, C.; Liu, Z.; Wang, Y.; Wang, Y.; Song, E.; Song, Y. The electrophilic character of quinones is essential for the suppression of Bach1. Toxicology 2017, 387, 17–26. [Google Scholar] [CrossRef]
- Levy, S.; Forman, H.J. C-Myc is a Nrf2-interacting protein that negatively regulates phase II genes through their electrophile responsive elements. IUBMB Life 2010, 62, 237–246. [Google Scholar] [CrossRef]
- Davies, K.J.A.; Forman, H.J. Does Bach1 & c-Myc dependent redox dysregulation of Nrf2 & adaptive homeostasis decrease cancer risk in ageing? Free Radic. Biol. Med. 2019, 134, 708–714. [Google Scholar] [CrossRef]
- Rushmore, T.H.; Morton, M.R.; Pickett, C.B. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J. Biol. Chem. 1991, 266, 11632–11639. [Google Scholar] [PubMed]
- Wasserman, W.W.; Fahl, W.E. Functional antioxidant responsive elements. Proc. Natl. Acad. Sci. USA 1997, 94, 5361–5366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, C.Y.; Hsiao, H.Y.; Wu, W.Y.; Liu, C.A.; Tsai, Y.C.; Chao, Y.J.; Wang, D.L.; Hsieh, H.J. Regulation of shear-induced nuclear translocation of the Nrf2 transcription factor in endothelial cells. J. Biomed. Sci. 2009, 16, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaekashiwa, M.; Wang, L.H. Nrf2 regulates thromboxane synthase gene expression in human lung cells. DNA Cell Biol. 2003, 22, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Stalder, R.; McKercher, S.R.; Williamson, R.E.; Roth, G.P.; Lipton, S.A. Nrf2 and HSF-1 pathway activation via hydroquinone-based proelectrophilic small molecules is regulated by electrochemical oxidation potential. ASN Neuro 2015, 7, 1759091415593294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Ma, Q. NRF2 cysteine residues are critical for oxidant/electrophile-sensing, Kelch-like ECH-associated protein-1-dependent ubiquitination-proteasomal degradation, and transcription activation. Mol. Pharmacol. 2009, 76, 1265–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alarcón-Aguilar, A.; Luna-López, A.; Ventura-Gallegos, J.L.; Lazzarini, R.; Galván-Arzate, S.; González-Puertos, V.Y.; Morán, J.; Santamaría, A.; Königsberg, M. Primary cultured astrocytes from old rats are capable to activate the Nrf2 response against MPP+ toxicity after tBHQ pretreatment. Neurobiol. Aging 2014, 35, 1901–1912. [Google Scholar] [CrossRef]
- Sekine, H.; Okazaki, K.; Ota, N.; Shima, H.; Katoh, Y.; Suzuki, N.; Igarashi, K.; Ito, M.; Motohashi, H.; Yamamoto, M. The mediator subunit MED16 transduces NRF2-activating signals into antioxidant gene expression. Mol. Cell Biol. 2015, 36, 407–420. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Ye, X.; Tang, N.; Shen, S.; Li, Z.; Niu, X.; Lu, S.; Xu, L. The histone acetyltranseferase hMOF acetylates Nrf2 and regulates anti-drug responses in human non-small cell lung cancer. Br. J. Pharmacol. 2014, 171, 3196–3211. [Google Scholar] [CrossRef]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell. Biol. 2005, 25, 10895–10906. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Zhang, S.; Chan, J.Y.; Zhang, D.D. Keap1 controls postinduction repression of the Nrf2-mediated antioxidant response by escorting nuclear export of Nrf2. Mol. Cell. Biol. 2007, 27, 6334–6349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sykiotis, G.P.; Habeos, I.G.; Samuelson, A.V.; Bohmann, D. The role of the antioxidant and longevity-promoting Nrf2 pathway in metabolic regulation. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namani, A.; Li, Y.; Wang, X.J.; Tang, X. Modulation of NRF2 signaling pathway by nuclear receptors: Implications for cancer. Biochim. Biophys. Acta 2014, 1843, 1875–1885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chio, I.I.C.; Jafarnejad, S.M.; Ponz-Sarvise, M.; Park, Y.; Rivera, K.; Palm, W.; Wilson, J.; Sangar, V.; Hao, Y.; Ohlund, D.; et al. NRF2 promotes tumor maintenance by modulating mRNA translation in pancreatic cancer. Cell 2016, 166, 963–976. [Google Scholar] [CrossRef] [Green Version]
- Jeong, Y.; Hoang, N.T.; Lovejoy, A.; Stehr, H.; Newman, A.M.; Gentles, A.J.; Kong, W.; Truong, D.; Martin, S.; Chaudhuri, A.; et al. Role of KEAP1/NRF2 and TP53 mutations in lung squamous cell carcinoma development and radiation resistance. Cancer Discov. 2017, 7, 86–101. [Google Scholar] [CrossRef] [Green Version]
- Zavattari, P.; Perra, A.; Menegon, S.; Kowalik, M.A.; Petrelli, A.; Angioni, M.M.; Follenzi, A.; Quagliata, L.; Ledda-Columbano, G.M.; Terracciano, L.; et al. Nrf2, but not β-catenin, mutation represents an early event in rat hepatocarcinogenesis. Hepatology 2015, 62, 851–862. [Google Scholar] [CrossRef] [Green Version]
- Stępkowski, T.M.; Kruszewski, M.K. Molecular cross-talk between the NRF2/KEAP1 signaling pathway, autophagy, and apoptosis. Free Radic. Biol. Med. 2011, 50, 1186–1195. [Google Scholar] [CrossRef]
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88 Pt B, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Copple, I.M.; Lister, A.; Obeng, A.D.; Kitteringham, N.R.; Jenkins, R.E.; Layfield, R.; Foster, B.J.; Goldring, C.E.; Park, B.K. Physical and functional interaction of sequestosome 1 with Keap1 regulates the Keap1-Nrf2 cell defense pathway. J. Biol. Chem. 2010, 285, 16782–16788. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Wang, X.J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.S.; Kang, D.H.; Lee, D.H.; Bae, S.H. PF-4708671, a specific inhibitor of p70 ribosomal S6 kinase 1, activates Nrf2 by promoting p62-dependent autophagic degradation of Keap1. Biochem. Biophys. Res. Commun. 2015, 466, 499–504. [Google Scholar] [CrossRef]
- Dotto, G.P. p21(WAF1/Cip1): More than a break to the cell cycle? Biochim. Biophys. Acta 2000, 1471, M43–M56. [Google Scholar] [CrossRef]
- Marazita, M.C.; Dugour, A.; Marquioni-Ramella, M.D.; Figueroa, J.M.; Suburo, A.M. Oxidative stress-induced premature senescence dysregulates VEGF and CFH expression in retinal pigment epithelial cells: Implications for age-related macular degeneration. Redox Biol. 2016, 7, 78–87. [Google Scholar] [CrossRef] [Green Version]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi, K.B.; Cano, M.; Rhee, J.; Datta, S.; Wang, L.; Handa, J.T. Oxidative Stress induces an interactive decline in Wnt and Nrf2 signaling in degenerating retinal pigment epithelium. Antioxid. Redox Signal. 2018, 29, 389–407. [Google Scholar] [CrossRef]
- Brown, S.L.; Sekhar, K.R.; Rachakonda, G.; Sasi, S.; Freeman, M.L. Activating transcription factor 3 is a novel repressor of the nuclear factor erythroid-derived 2-related factor 2 (Nrf2)-regulated stress pathway. Cancer Res. 2008, 68, 364–368. [Google Scholar] [CrossRef] [Green Version]
- Faraonio, R.; Vergara, P.; Di Marzo, D.; Pierantoni, M.G.; Napolitano, M.; Russo, T.; Cimino, F. p53 suppresses the Nrf2-dependent transcription of antioxidant response genes. J. Biol. Chem. 2006, 281, 39776–39784. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Lo, S.C.; Liu, J.H.; Hannink, M.; Lubahn, D.B. ERRβ: A potent inhibitor of Nrf2 transcriptional activity. Mol. Cell. Endocrinol. 2007, 278, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Ki, S.H.; Cho, I.J.; Choi, D.W.; Kim, S.G. Glucocorticoid receptor (GR)-associated SMRT binding to C/EBPβ TAD and Nrf2 Neh4/5: Role of SMRT recruited to GR in GSTA2 gene repression. Mol. Cell. Biol. 2005, 25, 4150–4165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta 2018, 1865, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Cuadrado, A.; Martín-Moldes, Z.; Ye, J.; Lastres-Becker, I. Transcription factors NRF2 and NF-κB are coordinated effectors of the Rho family, GTP-binding protein RAC1 during inflammation. J. Biol. Chem. 2014, 289, 15244–15258. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Zhu, X.; Kim, Y.; Li, J.; Huang, S.; Saleem, S.; Li, R.C.; Xu, Y.; Dore, S.; Cao, W. Histone deacetylase inhibition activates transcription factor Nrf2 and protects against cerebral ischemic damage. Free Radic. Biol. Med. 2012, 52, 928–936. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Salminen, A. NF-kappaB signaling as a putative target for omega-3 metabolites in the prevention of age-related macular degeneration (AMD). Exp. Gerontol. 2009, 44, 685–688. [Google Scholar] [CrossRef]
- Paimela, T.; Hyttinen, J.M.T.; Viiri, J.; Ryhänen, T.; Karjalainen, R.O.; Salminen, A.; Kaarniranta, K. Celastrol regulates innate immunity response via NF-κB and Hsp70 in human retinal pigment epithelial cells. Pharmacol. Res. 2011, 64, 501–508. [Google Scholar] [CrossRef]
- Boya, P.; Esteban-Martínez, L.; Serrano-Puebla, A.; Gómez-Sintes, R.; Villarejo-Zori, B. Autophagy in the eye: Development, degeneration, and aging. Prog. Retin. Eye Res. 2016, 55, 206–245. [Google Scholar] [CrossRef]
- Eskelinen, E.L.; Saftig, P. Autophagy: A lysosomal degradation pathway with a central role in health and disease. Biochim. Biophys. Acta 2009, 1793, 664–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ktistakis, N.T.; Tooze, S.A. Digesting the expanding mechanisms of autophagy. Trends Cell Biol. 2016, 26, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bales, K.L.; Gross, A.K. Aberrant protein trafficking in retinal degenerations: The initial phase of retinal remodeling. Exp. Eye Res. 2016, 150, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Kaarniranta, K.; Tokarz, P.; Koskela, A.; Paterno, J.; Błasiak, J. Autophagy regulates death of retinal pigment epithelium cells in age-related macular degeneration. Cell. Biol. Toxicol. 2017, 33, 113–128. [Google Scholar] [CrossRef] [Green Version]
- Keeling, E.; Lotery, A.J.; Tumbarello, D.A.; Ratnayaka, J.A. Impaired cargo clearance in the retinal pigment epithelium (RPE) underlies irreversible blinding diseases. Cells 2018, 7, E16. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.M.; Maltagliati, A.J. Nrf2 at the heart of oxidative stress and cardiac protection. Physiol. Genom. 2018, 50, 77–97. [Google Scholar] [CrossRef]
- Johansson, I.; Monsen, V.T.; Pettersen, K.; Mildenberger, J.; Misund, K.; Kaarniranta, K.; Schønberg, S.; Bjørkøy, G. The marine n-3 PUFA DHA evokes cytoprotection against oxidative stress and protein misfolding by inducing autophagy and NFE2L2 in human retinal pigment epithelial cells. Autophagy 2015, 11, 1636–1651. [Google Scholar] [CrossRef]
- Darvekar, S.R.; Elvenes, J.; Brenne, H.B.; Johansen, T.; Sjøttem, E. SPBP is a sulforaphane induced transcriptional coactivator of NRF2 regulating expression of the autophagy receptor p62/SQSTM1. PLoS ONE 2014, 9, e85262. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; García-Yagüe, A.J.; Scannevin, R.H.; Casarejos, M.J.; Kügler, S.; Rábano, A.; Cuadrado, A. Repurposing the NRF2 activator dimethyl fumarate as therapy against synucleinopathy in Parkinson’s disease. Antioxid. Redox Signal. 2016, 25, 61–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Wu, W.; Song, H.; Wang, F.; Li, H.; Chen, L.; Lai, Y.; Janicki, J.S.; Ward, K.W.; Meyer, C.J.; et al. Targeting Nrf2 by dihydro-CDDO-trifluoroethyl amide enhances autophagic clearance and viability of β-cells in a setting of oxidative stress. FEBS Lett. 2014, 588, 2115–2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Wang, H.; Fan, Y.; Gao, Y.; Li, X.; Hu, Z.; Ding, K.; Wang, Y.; Wang, X. Fucoxanthin provides neuroprotection in models of traumatic brain injury via the Nrf2-ARE and Nrf2-autophagy pathways. Sci. Rep. 2017, 7, 46763. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.; Kuse, Y.; Inoue, Y.; Nakamura, S.; Hara, H.; Shimazawa, M. Transient acceleration of autophagic degradation by pharmacological Nrf2 activation is important for retinal pigment epithelium cell survival. Redox Biol. 2018, 19, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Choi, K.J.; Cho, S.J.; Yun, S.M.; Jeon, J.P.; Koh, Y.H.; Song, J.; Johnson, G.V.; Jo, C. Fisetin stimulates autophagic degradation of phosphorylated tau via the activation of TFEB and Nrf2 transcription factors. Sci. Rep. 2016, 6, 24933. [Google Scholar] [CrossRef] [PubMed]
- Dayalan Naidu, S.; Dikovskaya, D.; Gaurilcikaite, E.; Knatko, E.V.; Healy, Z.R.; Mohan, H.; Koh, G.; Laurell, A.; Ball, G.; Olagnier, D.; et al. Transcription factors NRF2 and HSF1 have opposing functions in autophagy. Sci. Rep. 2017, 7, 11023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitto, A.; Lerner, C.A.; Nacarelli, T.; Crowe, E.; Torres, C.; Sell, C. p62/SQSTM1 at the interface of aging, autophagy, and disease. Age 2014, 36, 1123–1137. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Martín, P.; Komatsu, M. p62/SQSTM1—steering the cell through health and disease. J. Cell Sci. 2018, 131, jcs222836. [Google Scholar] [CrossRef] [Green Version]
- Viiri, J.; Hyttinen, J.M.T.; Ryhänen, T.; Rilla, K.; Paimela, T.; Kuusisto, E.; Siitonen, A.; Urtti, A.; Salminen, A.; Kaarniranta, K. p62/sequestosome 1 as a regulator of proteasome inhibitor-induced autophagy in human retinal pigment epithelial cells. Mol. Vis. 2010, 16, 1399–1414. [Google Scholar]
- Ichimura, Y.; Komatsu, M. Activation of p62/SQSTM1-Keap1-nuclear factor erythroid 2-related factor 2 pathway in cancer. Front. Oncol. 2018, 8, 210. [Google Scholar] [CrossRef]
- Shah, S.Z.A.; Zhao, D.; Hussain, T.; Sabir, N.; Mangi, M.H.; Yang, L. p62-Keap1-NRF2-ARE pathway: A contentious player for selective targeting of autophagy, oxidative stress and mitochondrial dysfunction in prion diseases. Front. Mol. Neurosci. 2018, 11, 310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, K.; Fujikawa, N.; Komatsu, M.; Ishii, T.; Unno, M.; Akaike, T.; Motohashi, H.; Yamamoto, M. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 13561–13566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Cano, M.; Handa, J.T. p62 provides dual cytoprotection against oxidative stress in the retinal pigment epithelium. Biochim. Biophys. Acta 2014, 1843, 1248–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, C.; Gundemir, S.; Pritchard, S.; Jin, Y.N.; Rahman, I.; Johnson, G.V. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat. Commun. 2014, 5, 3496. [Google Scholar] [CrossRef]
- Pajares, M.; Jiménez-Moreno, N.; García-Yagüe, Á.J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rábano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef] [Green Version]
- Hyttinen, J.M.T.; Petrovski, G.; Salminen, A.; Kaarniranta, K. 5’-Adenosine monophosphate-activated protein kinase-mammalian target of rapamycin axis as therapeutic target for age-related macular degeneration. Rejuv. Res. 2011, 14, 651–660. [Google Scholar] [CrossRef]
- O’Mealey, G.B.; Plafker, K.S.; Berry, W.L.; Janknecht, R.; Chan, J.Y.; Plafker, S.M. A PGAM5-KEAP1-Nrf2 complex is required for stress-induced mitochondrial retrograde trafficking. J. Cell Sci. 2017, 130, 3467–3480. [Google Scholar] [CrossRef] [Green Version]
- Sachdeva, M.M.; Cano, M.; Handa, J.T. Nrf2 signaling is impaired in the aging RPE given an oxidative insult. Exp. Eye Res. 2013, 119, 111–114. [Google Scholar] [CrossRef] [Green Version]
- Synowiec, E.; Sliwinski, T.; Danisz, K.; Błasiak, J.; Sklodowska, A.; Romaniuk, D.; Watala, C.; Szaflik, J.; Szaflik, J.P. Association between polymorphism of the NQO1, NOS3 and NFE2L2 genes and AMD. Front. Biosci. 2013, 18, 80–90. [Google Scholar]
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D.; et al. RXRα inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013, 73, 3097–3108. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.J.; Hayes, J.D.; Henderson, C.J.; Wolf, C.R. Identification of retinoic acid as an inhibitor of transcription factor Nrf2 through activation of retinoic acid receptor alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 19589–19594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Xue, J.; Liu, Y.; Gu, H.; Wei, X.; Ma, W.; Luo, W.; Ma, L.; Jia, S.; Dong, N.; et al. Inhibition of NRF2 signaling and increased reactive oxygen species during embryogenesis in a rat model of retinoic acid-induced neural tube defects. Neurotoxicology 2018, 69, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Golestaneh, N.; Chu, Y.; Cheng, S.K.; Cao, H.; Poliakov, E.; Berinstein, D.M. Repressed SIRT1/PGC-1α pathway and mitochondrial disintegration in iPSC-derived RPE disease model of age-related macular degeneration. J. Transl. Med. 2016, 14, 344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maugeri, A.; Barchitta, M.; Mazzone, M.G.; Giuliano, F.; Basile, G.; Agodi, A. Resveratrol modulates SIRT1 and DNMT functions and restores LINE-1 methylation levels in ARPE-19 cells under oxidative stress and inflammation. Int. J. Mol. Sci. 2018, 19, E2118. [Google Scholar] [CrossRef] [Green Version]
- Zhuge, C.C.; Xu, J.Y.; Zhang, J.; Li, W.; Li, P.; Li, Z.; Chen, L.; Liu, X.; Shang, P.; Xu, H.; et al. Fullerenol protects retinal pigment epithelial cells from oxidative stress-induced premature senescence via activating SIRT1. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4628–4638. [Google Scholar] [CrossRef] [Green Version]
- Kaarniranta, K.; Kajdanek, J.; Morawiec, J.; Pawlowska, E.; Błasiak, J. PGC-1α protects RPE cells of the aging retina against oxidative stress-induced degeneration through the regulation of senescence and mitochondrial quality control. The significance for AMD pathogenesis. Int. J. Mol. Sci. 2018, 19, E2317. [Google Scholar] [CrossRef] [Green Version]
- Aquilano, K.; Baldelli, S.; Pagliei, B.; Cannata, S.M.; Rotilio, G.; Ciriolo, M.R. p53 orchestrates the PGC-1α-mediated antioxidant response upon mild redox and metabolic imbalance. Antioxid. Redox Signal. 2013, 18, 386–399. [Google Scholar] [CrossRef] [Green Version]
- Cherry, A.D.; Suliman, H.B.; Bartz, R.R.; Piantadosi, C.A. Peroxisome proliferator-activated receptor γ co-activator 1-α as a critical co-activator of the murine hepatic oxidative stress response and mitochondrial biogenesis in Staphylococcus aureus sepsis. J. Biol. Chem. 2014, 289, 41–52. [Google Scholar] [CrossRef] [Green Version]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Chu, Y.; Mowery, J.; Konkel, B.; Galli, S.; Theos, A.C.; Golestaneh, N. Pgc-1α repression and high-fat diet induce age-related macular degeneration-like phenotypes in mice. Dis. Model Mech. 2018, 11, dmm032698. [Google Scholar] [CrossRef] [Green Version]
- Ferrington, D.A.; Ebeling, M.C.; Kapphahn, R.J.; Terluk, M.R.; Fisher, C.R.; Polanco, J.R.; Roehrich, H.; Leary, M.M.; Geng, Z.; Dutton, J.R.; et al. Altered bioenergetics and enhanced resistance to oxidative stress in human retinal pigment epithelial cells from donors with age-related macular degeneration. Redox Biol. 2017, 13, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Jian, Z.; Li, K.; Liu, L.; Zhang, Y.; Zhou, Z.; Li, C.; Gao, T. Heme oxygenase-1 protects human melanocytes from H2O2-induced oxidative stress via the Nrf2-ARE pathway. J. Investig. Dermatol. 2011, 131, 1420–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrot, L.; Jones, C.; Perez, P.; Meunier, J.R. The significance of Nrf2 pathway in (photo)-oxidative stress response in melanocytes and keratinocytes of the human epidermis. Pigment Cell Melanoma Res. 2008, 21, 79–88. [Google Scholar] [CrossRef]
- Shin, J.M.; Kim, M.Y.; Sohn, K.C.; Jung, S.Y.; Lee, H.E.; Lim, J.W.; Kim, S.; Lee, Y.H.; Im, M.; Seo, Y.J.; et al. Nrf2 negatively regulates melanogenesis by modulating PI3K/Akt signaling. PLoS ONE 2014, 9, e96035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannito, S.; Novo, E.; di Bonzo, L.V.; Busletta, C.; Colombatto, S.; Parola, M. Epithelial-mesenchymal transition: From molecular mechanisms, redox regulation to implications in human health and disease. Antioxid. Redox Signal. 2010, 12, 1383–1430. [Google Scholar] [CrossRef]
- Giannoni, E.; Parri, M.; Chiarugi, P. EMT and oxidative stress: A bidirectional interplay affecting tumor malignancy. Antioxid. Redox Signal. 2012, 16, 1248–1263. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, K.; Chen, Y.; Chen, H.; Nice, E.C.; Huang, C. Redox regulation in tumor cell epithelial-mesenchymal transition: Molecular basis and therapeutic strategy. Signal Transduct. Target. Ther. 2017, 2, 17036. [Google Scholar] [CrossRef] [Green Version]
- Piera-Velazquez, S.; Jimenez, S.A. Endothelial to mesenchymal transition: Role in physiology and in the pathogenesis of human diseases. Physiol. Rev. 2019, 99, 1281–1324. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Cubillo, E.; Diaz-Lopez, A.; Cuevas, E.P.; Moreno-Bueno, G.; Peinado, H.; Montes, A.; Santos, V.; Portillo, F.; Cano, A. E47 and Id1 interplay in epithelial-mesenchymal transition. PLoS ONE 2013, 8, e59948. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitoh, M. Epithelial-mesenchymal transition is regulated at post-transcriptional levels by transforming growth factor-β signaling during tumor progression. Cancer Sci. 2015, 106, 481–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, C.; Eddy, S.F.; Sherr, D.H.; Sonenshein, G.E. NF-kappaB and epithelial to mesenchymal transition of cancer. J. Cell. Biochem. 2008, 104, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Cieply, B.; Riley, P.; Pifer, P.M.; Widmeyer, J.; Addison, J.B.; Ivanov, A.V.; Denvir, J.; Frisch, S.M. Suppression of the epithelial-mesenchymal transition by Grainyhead-like-2. Cancer Res. 2012, 72, 2440–2453. [Google Scholar] [CrossRef]
- Deakin, N.O.; Pignatelli, J.; Turner, C.E. Diverse roles for the paxillin family of proteins in cancer. Genes Cancer 2012, 3, 362–370. [Google Scholar] [CrossRef] [Green Version]
- Scanlon, C.S.; Van Tubergen, E.A.; Inglehart, R.C.; D’Silva, N.J. Biomarkers of epithelial-mesenchymal transition in squamous cell carcinoma. J. Dent. Res. 2013, 92, 114–121. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Sancho, E.; Francí, C.; Domínguez, D.; Monfar, M.; Baulida, J.; García De Herreros, A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef]
- Jobling, M.F.; Mott, J.D.; Finnegan, M.T.; Jurukovski, V.; Erickson, A.C.; Walian, P.J.; Taylor, S.E.; Ledbetter, S.; Lawrence, C.M.; Rifkin, D.B.; et al. Isoform-specific activation of latent transforming growth factor β (LTGF-β) by reactive oxygen species. Radiat. Res. 2006, 166, 839–848. [Google Scholar] [CrossRef]
- Gonzalez, D.M.; Medici, D. Signaling mechanism of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Song, K.; Sponseller, T.L.; Danielpour, D. Novel function of androgen receptor-associated protein 55/Hic-5 as a negative regulator of Smad3 signaling. J. Biol. Chem. 2005, 280, 5154–5162. [Google Scholar] [CrossRef] [Green Version]
- Thuan, D.T.B.; Zayed, H.; Eid, A.H.; Abou-Saleh, H.; Nasrallah, G.K.; Mangoni, A.A.; Pintus, G. A potential link between oxidative stress and endothelial-to-mesenchymal transition in systemic sclerosis. Front. Immunol. 2018, 9, 1985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.M.; Gaston Pravia, K.A. Oxidative stress and glutathione in TGF-β-mediated fibrogenesis. Free Radic. Biol. Med. 2010, 48, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overstreet, J.M.; Samarakoon, R.; Meldrum, K.K.; Higgins, P.J. Redox control of p53 in the transcriptional regulation of TGF-β1 target genes through SMAD cooperativity. Cell. Signal. 2014, 26, 1427–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaffer, O.A.; Carter, A.B.; Sanders, P.N.; Dibbern, M.E.; Winters, C.J.; Murthy, S.; Ryan, A.J.; Rokita, A.G.; Prasad, A.M.; Zabner, J.; et al. Mitochondrial-targeted antioxidant therapy decreases transforming growth factor-β-mediated collagen production in a murine asthma model. Am. J. Respir. Cell Mol. Biol. 2015, 52, 106–115. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, W.; Zhang, T.; Zhou, Q.; Liu, J.; Liu, Y.; Kong, D.; Yu, W.; Liu, R.; Hai, C. TGF-β1 induces epithelial-to-mesenchymal transition via inhibiting mitochondrial functions in A549 cells. Free Radic. Res. 2018, 52, 1432–1444. [Google Scholar] [CrossRef]
- De Paepe, B.; Van Coster, R. A critical assessment of the therapeutic potential of resveratrol supplements for treating mitochondrial disorders. Nutrients 2017, 9, E1017. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Jeong, E.K.; Ju, M.K.; Jeon, H.M.; Kim, M.Y.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation. Mol. Cancer 2017, 16, 10. [Google Scholar] [CrossRef] [Green Version]
- Gunaratne, A.; Di Guglielmo, G.M. Par6 is phosphorylated by aPKC to facilitate EMT. Cell Adhes. Migr. 2013, 7, 357–361. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Ning, X.; Li, R.; Yang, Z.; Yang, X.; Sun, S.; Qian, Q. Signalling pathways involved in hypoxia-induced renal fibrosis. J. Cell. Mol. Med. 2017, 21, 1248–1259. [Google Scholar] [CrossRef]
- Movafagh, S.; Crook, S.; Vo, K. Regulation of hypoxia-inducible factor-1a by reactive oxygen species: New developments in an old debate. J. Cell. Biochem. 2015, 116, 696–703. [Google Scholar] [CrossRef]
- Liu, K.H.; Tsai, Y.T.; Chin, S.Y.; Lee, W.R.; Chen, Y.C.; Shen, S.C. Hypoxia stimulates the epithelial-to-mesenchymal transition in lung cancer cells through accumulation of nuclear β-catenin. Anticancer Res. 2018, 38, 6299–6308. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Maggiolini, M.; Musti, A.M. Crosstalk between Notch, HIF-1α and GPER in breast cancer EMT. Int. J. Mol. Sci. 2018, 19, 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahlgren, C.; Gustafsson, M.V.; Jin, S.; Poellinger, L.; Lendahl, U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci. USA 2008, 105, 6392–6397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arjamaa, O.; Nikinmaa, M.; Salminen, A.; Kaarniranta, K. Regulatory role of HIF-1α in the pathogenesis of age-related macular degeneration (AMD). Ageing Res. Rev. 2009, 8, 349–358. [Google Scholar] [CrossRef]
- Cheng, Z.X.; Wang, D.W.; Liu, T.; Liu, W.X.; Xia, W.B.; Xu, J.; Zhang, Y.H.; Qu, Y.K.; Guo, L.Q.; Ding, L.; et al. Effects of the HIF-1α and NF-κB loop on epithelial-mesenchymal transition and chemoresistance induced by hypoxia in pancreatic cancer cells. Oncol. Rep. 2014, 31, 1891–1898. [Google Scholar] [CrossRef] [PubMed]
- Cannito, S.; Novo, E.; Compagnone, A.; Valfrè di Bonzo, L.; Busletta, C.; Zamara, E.; Paternostro, C.; Povero, D.; Bandino, A.; Bozzo, F.; et al. Redox mechanisms switch on hypoxia-dependent epithelial-mesenchymal transition in cancer cells. Carcinogenesis 2008, 29, 2267–2278. [Google Scholar] [CrossRef] [PubMed]
- Gammons, M.; Bienz, M. Multiprotein complexes governing Wnt signal transduction. Curr. Opin. Cell Biol. 2018, 51, 42–49. [Google Scholar] [CrossRef]
- Li, W.; Cao, L.; Chen, X.; Lei, J.; Ma, Q. Resveratrol inhibits hypoxia-driven ROS-induced invasive and migratory ability of pancreatic cancer cells via suppression of the Hedgehog signaling pathway. Oncol. Rep. 2016, 35, 1718–1726. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.H.; Suh, Y.; Lee, H.J.; Yoo, K.C.; Uddin, N.; Jeong, Y.J.; Lee, J.S.; Hwang, S.G.; Nam, S.Y.; Kim, M.J.; et al. Radiation promotes invasiveness of non-small-cell lung cancer cells through granulocyte-colony-stimulating factor. Oncogene 2015, 34, 5372–5382. [Google Scholar] [CrossRef]
- Abba, M.L.; Patil, N.; Leupold, J.H.; Allgayer, H. MicroRNA regulation of epithelial to mesenchymal transition. J. Clin. Med. 2016, 5, E8. [Google Scholar] [CrossRef]
- Zou, X.Z.; Liu, T.; Gong, Z.C.; Hu, C.P.; Zhang, Z. MicroRNAs-mediated epithelial-mesenchymal transition in fibrotic diseases. Eur. J. Pharmacol. 2017, 796, 190–206. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Mo, X.; Cui, W.; Zhang, Z.; Li, D.; Li, L.; Xu, L.; Yao, H.; Gao, J. Nrf2 inhibits epithelial-mesenchymal transition by suppressing snail expression during pulmonary fibrosis. Sci. Rep. 2016, 16, 38646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Jiang, Z.; Zhou, C.; Chen, K.; Li, X.; Wang, Z.; Wu, Z.; Ma, J.; Ma, Q.; Duan, W. Activation of Nrf2 by sulforaphane inhibits high glucose-induced progression of pancreatic cancer via AMPK dependent signaling. Cell. Physiol. Biochem. 2018, 50, 1201–1215. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Tai, W.; Qu, X.; Wu, W.; Li, Z.; Deng, S.; Vongphouttha, C.; Dong, Z. Rapamycin protects against paraquat-induced pulmonary fibrosis: Activation of Nrf2 signaling pathway. Biochem. Biophys. Res. Commun. 2017, 490, 535–540. [Google Scholar] [CrossRef]
- Qu, J.; Zhang, Z.; Zhang, P.; Zheng, C.; Zhou, W.; Cui, W.; Xu, L.; Gao, J. Downregulation of HMGB1 is required for the protective role of Nrf2 in EMT-mediated PF. J. Cell. Physiol. 2018, 2018, 8862–8872. [Google Scholar] [CrossRef]
- Lee, D.F.; Kuo, H.P.; Liu, M.; Chou, C.K.; Xia, W.; Du, Y.; Shen, J.; Chen, C.T.; Huo, L.; Hsu, M.C.; et al. KEAP1 E3 ligase-mediated downregulation of NF-kappaB signaling by targeting IKKβ. Mol. Cell 2009, 3, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Mallano, T.; Palumbo-Zerr, K.; Zerr, P.; Ramming, A.; Zeller, B.; Beyer, C.; Dees, C.; Huang, J.; Hai, T.; Distler, O.; et al. Activating transcription factor 3 regulates canonical TGFβ signalling in systemic sclerosis. Ann. Rheum. Dis. 2016, 75, 586–592. [Google Scholar] [CrossRef] [Green Version]
- Ryoo, I.G.; Ha, H.; Kwak, M.K. Inhibitory role of the KEAP1-NRF2 pathway in TGFβ1-stimulated renal epithelial transition to fibroblastic cells: A modulatory effect on SMAD signaling. PLoS ONE 2014, 9, e93265. [Google Scholar] [CrossRef]
- Kim, K.H.; Jeong, J.Y.; Surh, Y.J.; Kim, K.W. Expression of stress-response ATF3 is mediated by Nrf2 in astrocytes. Nucleic Acids Res. 2010, 38, 48–59. [Google Scholar] [CrossRef] [Green Version]
- Salazar, M.; Rojo, A.I.; Velasco, D.; de Sagarra, R.M.; Cuadrado, A. Glycogen synthase kinase-3β inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Zhang, L. A Smurf1 tale: Function and regulation of an ubiquitin ligase in multiple cellular networks. Cell. Mol. Life Sci. 2013, 70, 2305–2317. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Chen, Z.; Zou, Y.; Zhang, L.; Huang, J.; Liu, P.; Huang, H. CKIP-1 affects the polyubiquitination of Nrf2 and Keap1 via mediating Smurf1 to resist HG-induced renal fibrosis in GMCs and diabetic mice kidneys. Free Radic. Biol. Med. 2018, 115, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Fan, X.; Zhao, M.; Guo, Q.; Guo, S. CKIP-1 alleviates oxygen-glucose deprivation/reoxygenation-induced apoptosis and oxidative stress in cultured hippocampal neurons by downregulating Keap1 and activating Nrf2/ARE signaling. Eur. J. Pharmacol. 2019, 848, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.S.; Zhang, Z.G.; Du, G.Y.; Sun, H.L.; Liu, H.Y.; Zhou, Z.; Gou, X.M.; Wu, X.H.; Yu, X.Y.; Huang, Y.H. Nrf2 promotes breast cancer cell migration via up-regulation of G6PD/HIF-1α/Notch1 axis. J. Cell. Mol. Med. 2019, 23, 3451–3463. [Google Scholar] [CrossRef] [Green Version]
- Ghavami, S.; Cunnington, R.H.; Gupta, S.; Yeganeh, B.; Filomeno, K.L.; Freed, D.H.; Chen, S.; Klonisch, T.; Halayko, A.J.; Ambrose, E.; et al. Autophagy is a regulator of TGF-β1-induced fibrogenesis in primary human atrial myofibroblasts. Cell Death Dis. 2015, 6, e1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, B.B.; Li, B.; Yang, Y.H.; Chen, J.W.; Chen, K.; Jiang, S.D.; Jiang, L.S. The effect of transforming growth factor β1 on the crosstalk between autophagy and apoptosis in the annulus fibrosus cells under serum deprivation. Cytokine 2014, 70, 87–96. [Google Scholar] [CrossRef]
- Zou, M.; Zhu, W.; Wang, L.; Shi, L.; Gao, R.; Ou, Y.; Chen, X.; Wang, Z.; Jiang, A.; Liu, K.; et al. AEG-1/MTDH-activated autophagy enhances human malignant glioma susceptibility to TGF-β1-triggered epithelial-mesenchymal transition. Oncotarget 2016, 7, 13122–13138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertrand, M.; Petit, V.; Jain, A.; Amsellem, R.; Johansen, T.; Larue, L.; Codogno, P.; Beau, I. SQSTM1/p62 regulates the expression of junctional proteins through epithelial-mesenchymal transition factors. Cell Cycle 2015, 14, 364–374. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Chen, X.; Liu, X.; Huang, S.; He, C.; Chen, B.; Liu, Y. Autophagy regulates TGF-β2-induced epithelial-mesenchymal transition in human retinal pigment epithelium cells. Mol. Med. Rep. 2018, 17, 3607–3614. [Google Scholar] [CrossRef] [Green Version]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [Green Version]
- Tracy, K.; Dibling, B.C.; Spike, B.T.; Knabb, J.R.; Schumacker, P.; Macleod, K.F. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol. Cell. Biol. 2007, 27, 6229–6242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassi, G.; Di Caprio, G.; Santangelo, L.; Fimia, G.M.; Cozzolino, A.M.; Komatsu, M.; Ippolito, G.; Tripodi, M.; Alonzi, T. Autophagy regulates hepatocyte identity and epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions promoting Snail degradation. Cell Death Dis. 2015, 6, e1880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, H.; Zhao, X.; Guo, Q.; Feng, Y.; Ma, M.; Guo, W.; Dong, X.; Deng, C.; Li, C.; Song, X.; et al. Autophagy resists EMT process to maintain retinal pigment epithelium homeostasis. Int. J. Biol. Sci. 2019, 15, 507–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniel, E.; Toth, C.A.; Grunwald, J.E.; Jaffe, G.J.; Martin, D.F.; Fine, S.L.; Huang, J.; Ying, G.S.; Hagstrom, S.A.; Winter, K.; et al. Risk of scar in the comparison of age-related macular degeneration treatments trials. Ophthalmology 2014, 121, 656–666. [Google Scholar] [CrossRef] [Green Version]
- Kent, D.; Sheridan, C. Choroidal neovascularization: A wound healing perspective. Mol. Vis. 2003, 9, 747–755. [Google Scholar]
- Priglinger, C.S.; Obermann, J.; Szober, C.M.; Merl-Pham, J.; Ohmayer, U.; Behler, J.; Gruhn, F.; Kreutzer, T.C.; Wertheimer, C.; Geerlof, A.; et al. Epithelial-to-mesenchymal transition of RPE cells in vitro confers increased β1,6-N-glycosylation and increased susceptibility to galectin-3 binding. PLoS ONE 2016, 11, e0146887. [Google Scholar] [CrossRef]
- Wu, D.; Kanda, A.; Liu, Y.; Kase, S.; Noda, K.; Ishida, S. Galectin-1 promotes choroidal neovascularization and subretinal fibrosis mediated via epithelial-mesenchymal transition. FASEB J. 2019, 33, 2498–2513. [Google Scholar] [CrossRef] [Green Version]
- Rosales, M.A.B.; Shu, D.Y.; Iacovelli, J.; Saint-Geniez, M. Loss of PGC-1α in RPE induces mesenchymal transition and promotes retinal degeneration. Life Sci. Alliance 2019, 2, e201800212. [Google Scholar] [CrossRef]
- Ghosh, S.; Shang, P.; Terasaki, H.; Stepicheva, N.; Hose, S.; Yazdankhah, M.; Weiss, J.; Sakamoto, T.; Bhutto, I.A.; Xia, S.; et al. A role for βA3/A1-crystallin in type 2 EMT of RPE cells occurring in dry age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2018, 9, AMD104–AMD113. [Google Scholar] [CrossRef] [Green Version]
- Valapala, M.; Wilson, C.; Hose, S.; Bhutto, I.A.; Grebe, R.; Dong, A.; Greenbaum, S.; Gu, L.; Sengupta, S.; Cano, M.; et al. Lysosomal-mediated waste clearance in retinal pigment epithelial cells is regulated by CRYBA1/βA3/A1-crystallin via V-ATPase-MTORC1 signaling. Autophagy 2014, 10, 480–496. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, K.; Sreekumar, P.G.; Spee, C.; Nazari, H.; Zhu, D.; Kannan, R.; Hinton, D.R. αB-crystallin regulates subretinal fibrosis by modulation of epithelial-mesenchymal transition. Am. J. Pathol. 2016, 186, 859–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pursiheimo, J.P.; Rantanen, K.; Heikkinen, P.T.; Johansen, T.; Jaakkola, P.M. Hypoxia-activated autophagy accelerates degradation of SQSTM1/p62. Oncogene 2009, 28, 334–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, A.; Yoon, S.; Kim, J.; Baek, Y.M.; Park, H.; Lim, D.; Chung, H.; Kim, D.E. Autophagy and KRT8/keratin 8 protect degeneration of retinal pigment epithelium under oxidative stress. Autophagy 2017, 13, 248–263. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Tokuda, K.; Kobayashi, Y.; Yamashiro, C.; Uchi, S.H.; Hatano, M.; Kimura, K. Suppression of epithelial-mesenchymal transition in retinal pigment epithelial cells by an MRTF-A inhibitor. Investig. Ophthalmol. Vis. Sci. 2019, 60, 528–537. [Google Scholar] [CrossRef] [Green Version]
- Kimura, K.; Orita, T.; Liu, Y.; Yang, Y.; Tokuda, K.; Kurakazu, T.; Noda, T.; Yanai, R.; Morishige, N.; Takeda, A.; et al. Attenuation of EMT in RPE cells and subretinal fibrosis by an RAR-γ agonist. J. Mol. Med. 2015, 93, 749–758. [Google Scholar] [CrossRef]
- Kim, S.M.; Hwang, K.A.; Choi, D.W.; Choi, K.C. The cigarette smoke components induced the cell proliferation and epithelial to mesenchymal transition via production of reactive oxygen species in endometrial adenocarcinoma cells. Food Chem. Toxicol. 2018, 121, 657–665. [Google Scholar] [CrossRef]
- Yan, G.; Zhang, L.; Feng, C.; Gong, R.; Idiiatullina, E.; Huang, Q.; He, M.; Guo, S.; Yang, F.; Li, Y.; et al. Blue light emitting diodes irradiation causes cell death in colorectal cancer by inducing ROS production and DNA damage. Int. J. Biochem. Cell Biol. 2018, 103, 81–88. [Google Scholar] [CrossRef]
- Terluk, M.R.; Kapphahn, R.J.; Soukup, L.M.; Gong, H.; Gallardo, C.; Montezuma, S.R.; Ferrington, D.A. Investigating mitochondria as a target for treating age-related macular degeneration. J. Neurosci. 2015, 35, 7304–7311. [Google Scholar] [CrossRef] [Green Version]
- Hyttinen, J.M.T.; Viiri, J.; Kaarniranta, K.; Błasiak, J. Mitochondrial quality control in AMD: Does mitophagy play a pivotal role? Mol. Cell. Life Sci. 2018, 75, 2991–3008. [Google Scholar] [CrossRef] [Green Version]
- Riazi-Esfahani, M.; Kuppermann, B.D.; Kenney, M.C. The role of mitochondria in AMD: Current knowledge and future applications. J. Ophthalmic Vis. Res. 2017, 12, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Favre, C.; Zhdanov, A.; Leahy, M.; Papkovsky, D.; O’Connor, R. Mitochondrial pyrimidine nucleotide carrier (PNC1) regulates mitochondrial biogenesis and the invasive phenotype of cancer cells. Oncogene 2010, 29, 3964–3976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, K.; Guo, X.; Liu, Y.; Li, J.; Hu, Y.; Wang, D.; Song, J. TUFM downregulation induces epithelial-mesenchymal transition and invasion in lung cancer cells via a mechanism involving AMPK-GSK3β signaling. Cell. Mol. Life Sci. 2016, 73, 2105–2121. [Google Scholar] [CrossRef] [PubMed]
- Shagieva, G.; Domnina, L.; Makarevich, O.; Chernyak, B.; Skulachev, V.; Dugina, V. Depletion of mitochondrial reactive oxygen species downregulates epithelial-to-mesenchymal transition in cervical cancer cells. Oncotarget 2017, 8, 4901–4913. [Google Scholar] [CrossRef] [Green Version]
- Stone, R.C.; Pastar, I.; Ojeh, N.; Chen, V.; Liu, S.; Garzon, K.I.; Tomic-Canic, M. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 2016, 365, 495–506. [Google Scholar] [CrossRef]
- Vu, K.T.; Hulleman, J.D. An inducible form of Nrf2 confers enhanced protection against acute oxidative stresses in RPE cells. Exp. Eye Res. 2017, 164, 31–36. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hyttinen, J.M.T.; Kannan, R.; Felszeghy, S.; Niittykoski, M.; Salminen, A.; Kaarniranta, K. The Regulation of NFE2L2 (NRF2) Signalling and Epithelial-to-Mesenchymal Transition in Age-Related Macular Degeneration Pathology. Int. J. Mol. Sci. 2019, 20, 5800. https://doi.org/10.3390/ijms20225800
Hyttinen JMT, Kannan R, Felszeghy S, Niittykoski M, Salminen A, Kaarniranta K. The Regulation of NFE2L2 (NRF2) Signalling and Epithelial-to-Mesenchymal Transition in Age-Related Macular Degeneration Pathology. International Journal of Molecular Sciences. 2019; 20(22):5800. https://doi.org/10.3390/ijms20225800
Chicago/Turabian StyleHyttinen, Juha M. T., Ram Kannan, Szabolcs Felszeghy, Minna Niittykoski, Antero Salminen, and Kai Kaarniranta. 2019. "The Regulation of NFE2L2 (NRF2) Signalling and Epithelial-to-Mesenchymal Transition in Age-Related Macular Degeneration Pathology" International Journal of Molecular Sciences 20, no. 22: 5800. https://doi.org/10.3390/ijms20225800
APA StyleHyttinen, J. M. T., Kannan, R., Felszeghy, S., Niittykoski, M., Salminen, A., & Kaarniranta, K. (2019). The Regulation of NFE2L2 (NRF2) Signalling and Epithelial-to-Mesenchymal Transition in Age-Related Macular Degeneration Pathology. International Journal of Molecular Sciences, 20(22), 5800. https://doi.org/10.3390/ijms20225800