Molecular Signatures for Combined Targeted Treatments in Diffuse Malignant Peritoneal Mesothelioma

 , ,
, ,  , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
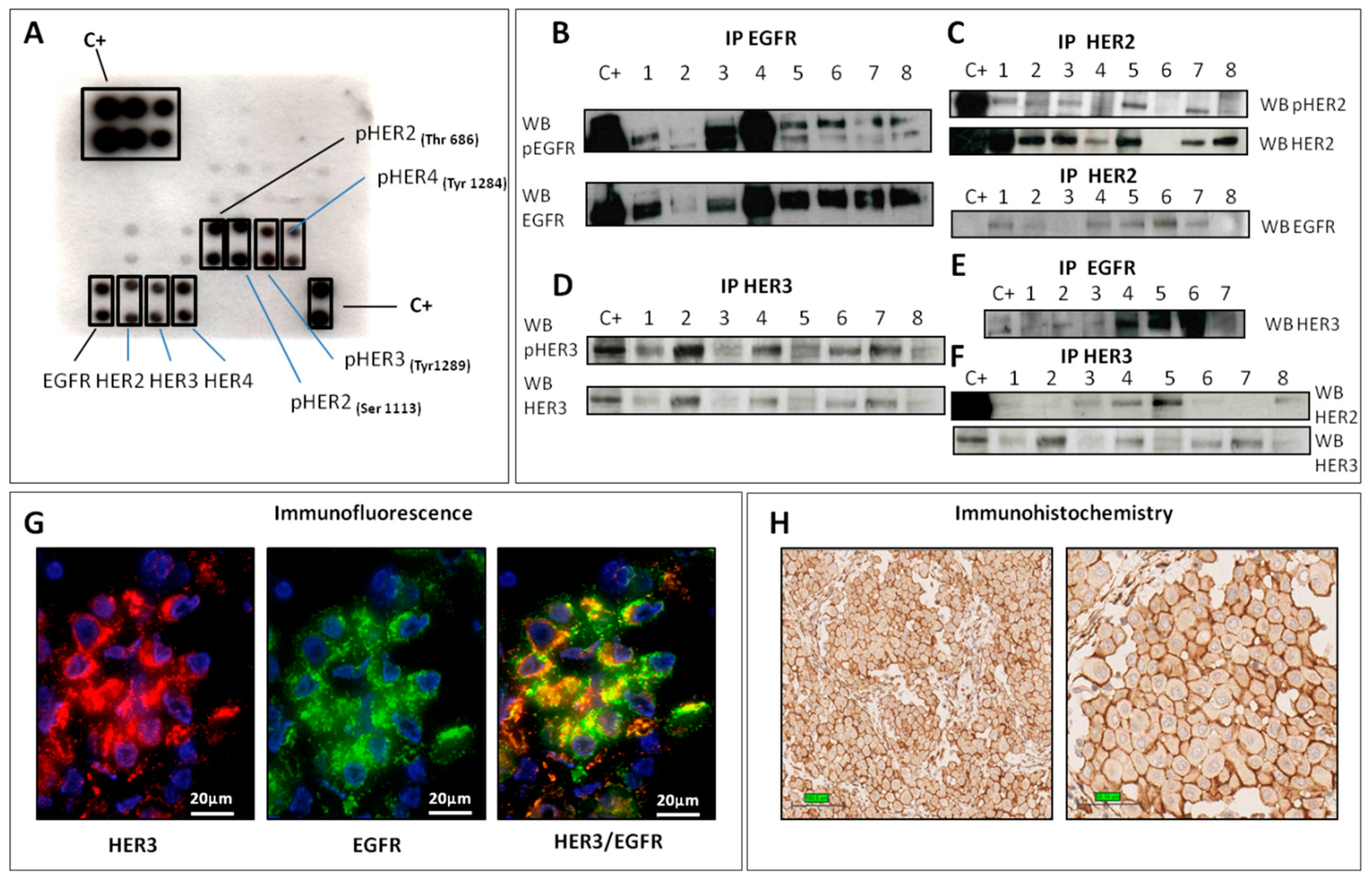
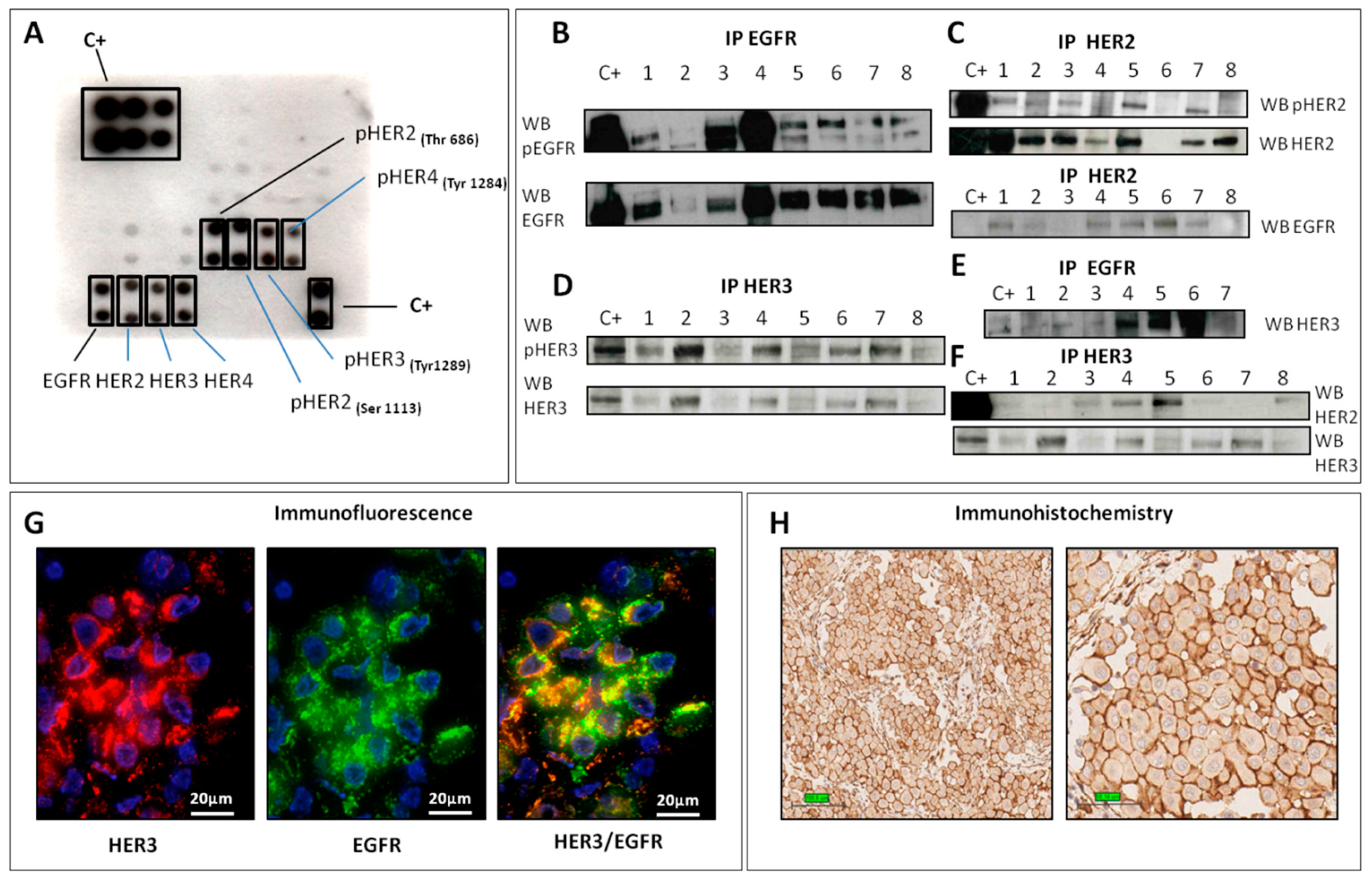
2.1. HER Family Analysis
2.1.1. EGFR
2.1.2. HER2
2.1.3. HER3
2.2. Phosphorylation Antibody Array
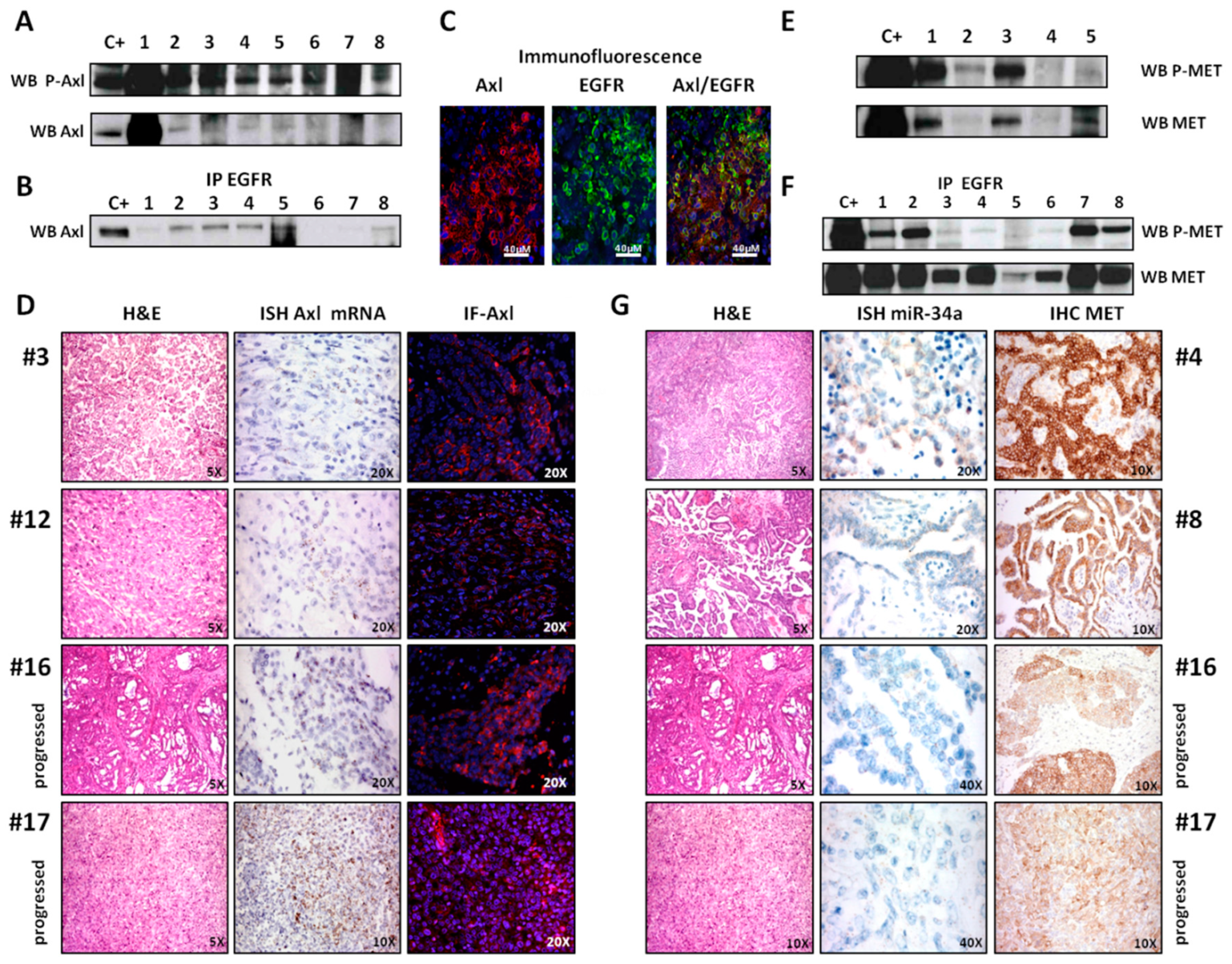
2.3. Axl Analysis
2.4. MET Analysis
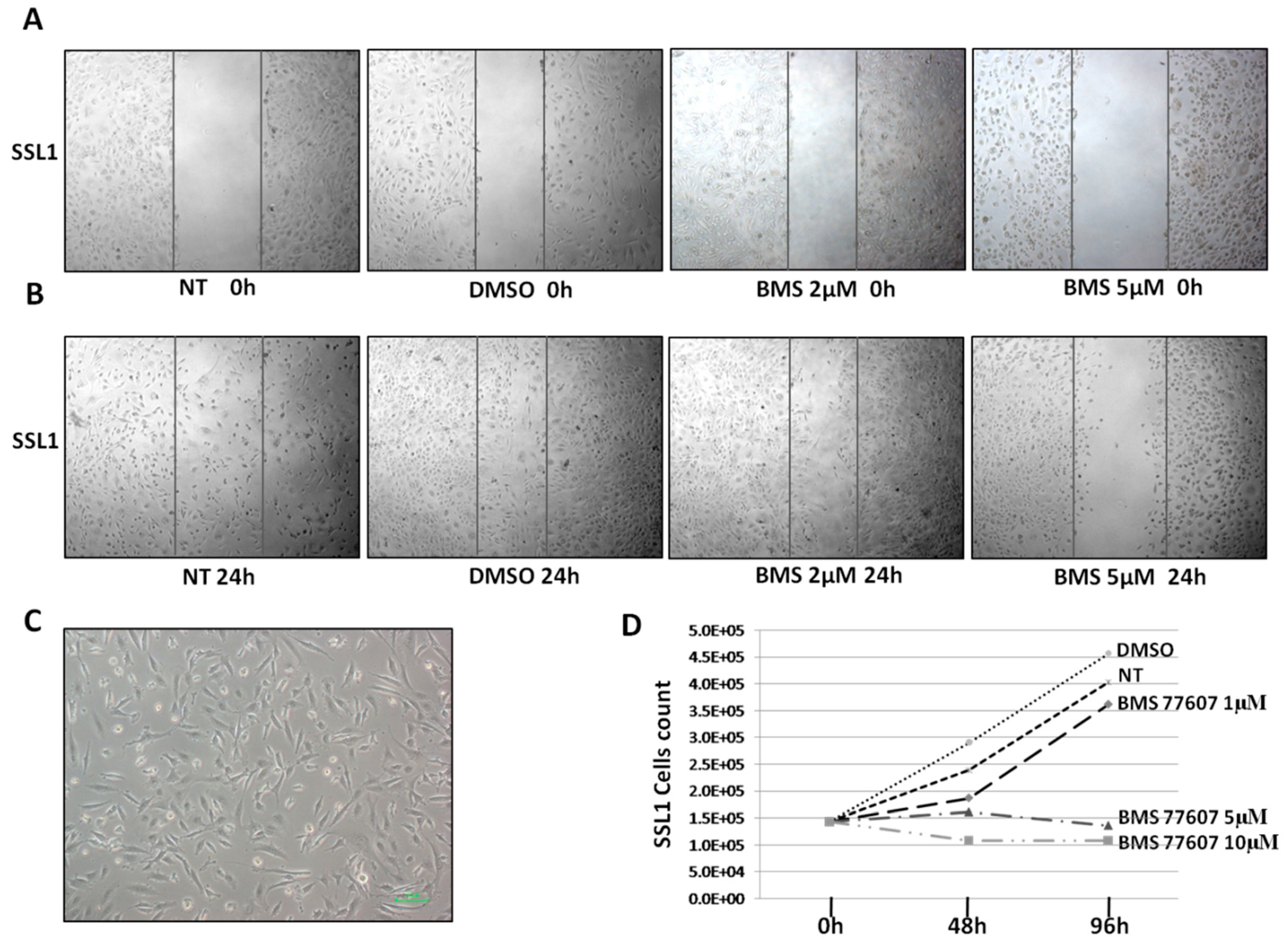
2.5. Cell Motility and Proliferation Assay
2.6. miRNA 34a Analysis
3. Discussion
4. Material and Method
4.1. Patients and Samples
4.2. Biochemical Analysis
4.3. Real-Time PCR
4.4. Immunofluorescence (IF)
4.5. Immunohistochemistry (IHC)
4.6. Next-Generation Sequencing (NGS)
4.7. miRNA in Situ Hybridization (ISH)
4.8. mRNA in Situ Hybridization (ISH)
4.9. In Vitro Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Helm, J.H.; Miura, J.T.; Glenn, J.A.; Marcus, R.K.; Larrieux, G.; Jayakrishnan, T.T.; Donahue, A.E.; Gamblin, T.C.; Turaga, K.K.; Johnston, F.M. Cytoreductive surgery and hyperthermic intraperitoneal chemotherapy for malignant peritonealmesothelioma: A systematic review and meta-analysis. Ann. Surg. Oncol. 2015, 22, 1686–1693. [Google Scholar] [CrossRef]
- Alexander, H.R., Jr.; Li, C.Y.; Kennedy, T.J. Current Management and Future Opportunities for Peritoneal Metastases: Peritoneal Mesothelioma. Ann. Surg. Oncol. 2018, 25, 2159–2164. [Google Scholar] [CrossRef] [PubMed]
- Baratti, D.; Kusamura, S.; Cabras, A.D.; Bertulli, R.; Hutanu, I.; Deraco, M. Diffuse malignant peritoneal mesothelioma: Long-term survival with complete cytoreductive surgery followed by hyperthermic intraperitoneal chemotherapy (HIPEC). Eur. J. Cancer 2013, 49, 3140–3148. [Google Scholar] [CrossRef] [PubMed]
- Cartenì, G.; Manegold, C.; Garcia, G.M.; Siena, S.; Zielinski, C.; Amadori, D.; Liu, Y.; Blatter, J.; Visseren-Grul, C.; Stahel, R. Malignant peritoneal mesothelioma-Results from the International Expanded Access Program using pemetrexed alone or in combination with a platinum agent. Lung Cancer 2009, 64, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Manzini, V.D.P.; Recchia, L.; Cafferata, M.; Porta, C.; Siena, S.; Giannetta, L.; Morelli, F.; Oniga, F.; Bearz, A.; Torri, V.; et al. Malignant peritoneal mesothelioma: A multicenter study on 81 cases. Ann. Oncol. 2010, 21, 348–353. [Google Scholar] [CrossRef]
- Shrestha, R.; Nabavi, N.; Lin, Y.-Y.; Mo, F.; Anderson, S.; Volik, S.; Adomat, H.H.; Lin, D.; Xue, H.; Dong, X.; et al. BAP1 haploinsufficiency predicts a distinct immunogenic class of malignant peritoneal mesothelioma. Genome Med. 2019, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Joseph, N.M.; Chen, Y.Y.; Nasr, A.; Yeh, I.; Talevich, E.; Onodera, C.; Bastian, B.C.; Rabban, J.T.; Garg, K.; Zaloudek, C.; et al. Genomic profiling of malignant peritoneal mesothelioma reveals recurrent alterations in epigenetic regulatory genes BAP1, SETD2, and DDX3X. Mod. Pathol. 2017, 30, 246–254. [Google Scholar] [CrossRef]
- Hung, Y.P.; Dong, F.; Watkins, J.C.; Nardi, V.; Bueno, R.; Cin, P.D.; Godleski, J.J.; Crum, C.P.; Chirieac, L.R. Identification of ALK Rearrangements in Malignant Peritoneal Mesothelioma. JAMA Oncol. 2018, 4, 235–238. [Google Scholar] [CrossRef]
- Perrone, F.; Jocollè, G.; Pennati, M.; Deraco, M.; Baratti, D.; Brich, S.; Orsenigo, M.; Tarantino, E.; De Marco, C.; Bertan, C.; et al. Receptor tyrosine kinase and downstream signalling analysis in diffuse malignant peritoneal mesothelioma. Eur. J. Cancer 2010, 46, 2837–2848. [Google Scholar] [CrossRef]
- Varghese, S.; Chen, Z.; Bartlett, D.L.; Pingpank, J.F.; Libutti, S.K.; Steinberg, S.M.; Wunderlich, J.; Alexander, H.R., Jr. Activation of the phosphoinositide-3-kinase and mammalian target of rapamycin signaling pathways are associated with shortened survival in patients with malignant peritoneal mesothelioma. Cancer 2011, 117, 361–371. [Google Scholar] [CrossRef]
- Dolly, S.O.; Migali, C.; Tunariu, N.; Della-Pepa, C.; Khakoo, S.; Hazell, S.; de Bono, J.S.; Kaye, S.B.; Banerjee, S. Indolent peritoneal mesothelioma: PI3K-mTOR inhibitors as a novel therapeutic strategy. ESMO Open 2017, 2, e000101. [Google Scholar] [CrossRef] [PubMed]
- Bozzi, F.; Brich, S.; Dagrada, G.P.; Negri, T.; Conca, E.; Cortelazzi, B.; Belfiore, A.; Perrone, F.; Gualeni, A.V.; Gloghini, A.; et al. Epithelioid peritoneal mesothelioma: A hybrid phenotype within a mesenchymal-epithelial/epithelial-mesenchymal transition framework. Oncotarget 2016, 7, 75503–75517. [Google Scholar] [CrossRef] [PubMed]
- Kalra, N.; Ashai, A.; Xi, L.; Zhang, J.; Avital, I.; Raffeld, M.; Hassan, R. Patients with peritoneal mesothelioma lack epidermal growth factor receptor tyrosine kinase mutations that would make them sensitive to tyrosine kinase inhibitors. Oncol. Rep. 2012, 27, 1794–1800. [Google Scholar] [PubMed]
- Sponziello, M.; Benvenuti, S.; Gentile, A.; Pecce, V.; Rosignolo, F.; Virzì, A.R.; Milan, M.; Comoglio, P.M.; Londin, E.; Fortina, P.; et al. Whole exome sequencing identifies a germline MET mutation in two siblings with hereditary wild-type RET medullary thyroid cancer. Hum. Mutat. 2018, 39, 371–377. [Google Scholar] [CrossRef]
- Mudduluru, G.; Ceppi, P.; Kumarswamy, R.; Scagliotti, G.V.; Papotti, M.; Allgayer, H. Regulation of Axl receptor tyrosine kinase expression by miR-34a and miR-199a/b in solid cancer. Oncogene 2011, 30, 2888–2899. [Google Scholar] [CrossRef]
- El Bezawy, R.; De Cesare, M.; Pennati, M.; Deraco, M.; Gandellini, P.; Zuco, V.; Zaffaroni, N. Antitumor activity of miR-34a in peritoneal mesothelioma relies on c-MET and AXL inhibition: Persistent activation of ERK and AKT signaling as a possible cytoprotective mechanism. J. Hematol. Oncol. 2017, 10, 19. [Google Scholar] [CrossRef]
- Govindan, R.; Kratzke, R.A.; Herndon, J.E.; Niehans, G.A.; Vollmer, R.; Watson, D.; Green, M.R.; Kindler, H.L. Gefitinib in patients with malignant mesothelioma: A phase II study by the Cancer and Leukemia Group B. Clin. Cancer Res. 2005, 11, 2300–2304. [Google Scholar] [CrossRef]
- Garland, L.L.; Rankin, C.; Gandara, D.R.; Rivkin, S.E.; Scott, K.M.; Nagle, R.B.; Klein-Szanto, A.J.; Testa, J.R.; Altomare, D.A.; Borden, E.C. Phase II study of erlotinib in patients with malignant pleural mesothelioma: A Southwest Oncology Group Study. J. Clin. Oncol. 2007, 25, 2406–2413. [Google Scholar] [CrossRef]
- Yonesaka, K.; Hirotani, K.; Kawakami, H.; Takeda, M.; Kaneda, H.; Sakai, K.; Okamoto, I.; Nishio, K.; Jänne, P.A.; Nakagawa, K. Anti-HER3 monoclonal antibody patritumab sensitizes refractory non-small cell lung cancer to the epidermal growth factor receptor inhibitor erlotinib. Oncogene 2016, 35, 878–886. [Google Scholar] [CrossRef]
- Ye, X.; Li, Y.; Stawicki, S.; Couto, S.; Eastham-Anderson, J.; Kallop, D.; Weimer, R.; Wu, Y.; Pei, L. An anti-Axl monoclonal antibody attenuates xenograft tumor growth and enhances the effect of multiple anticancer therapies. Oncogene 2010, 29, 5254–5264. [Google Scholar] [CrossRef]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Greger, J.; Shi, H.; Liu, Y.; Greshock, J.; Annan, R.; Halsey, W.; Sathe, G.M.; Martin, A.M.; Gilmer, T.M. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: Activation of AXL. Cancer Res. 2009, 69, 6871–6878. [Google Scholar] [CrossRef] [PubMed]
- Brand, T.M.; Iida, M.; Corrigan, K.L.; Braverman, C.M.; Coan, J.P.; Flanigan, B.G.; Stein, A.P.; Salgia, R.; Rolff, J.; Kimple, R.J.; et al. The receptor tyrosine kinase AXL mediates nuclear translocation of the epidermal growth factor receptor. Sci. Signal. 2017, 10, eaag1064. [Google Scholar] [CrossRef] [PubMed]
- Brevet, M.; Shimizu, S.; Bott, M.J.; Shukla, N.; Zhou, Q.; Olshen, A.B.; Rusch, V.; Ladanyi, M. Coactivation of receptor tyrosine kinases in malignant mesothelioma as a rationale for combination targeted therapy. J. Thorac. Oncol. 2011, 6, 864–874. [Google Scholar] [CrossRef] [PubMed]
- Ou, W.-B.; Hubert, C.; Fletcher, J.A.; Corson, J.M.; Bueno, R.; Flynn, D.L.; Sugarbaker, D.J. Targeted inhibition of multiple receptor tyrosine kinases in mesothelioma. Neoplasia 2011, 13, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Kim, J.H.; Rho, J.K.; Lee, J.C.; Son, J. AXL and MET receptor tyrosine kinases are essential for lung cancer metastasis. Oncol. Rep. 2017, 37, 2201–2208. [Google Scholar] [CrossRef]
- Nie, D.; Fu, J.; Chen, H.; Cheng, J.; Fu, J. Roles of MicroRNA-34a in Epithelial to Mesenchymal Transition, Competing Endogenous RNA Sponging and Its Therapeutic Potential. Int. J. Mol. Sci. 2019, 20, 861. [Google Scholar] [CrossRef]
- Asiedu, M.K.; Beauchamp-Perez, F.D.; Ingle, J.N.; Behrens, M.D.; Radisky, D.C.; Knutson, K.L. AXL induces epithelial-to-mesenchymal transition and regulates the function of breast cancer stem cells. Oncogene 2014, 33, 1316–1324. [Google Scholar] [CrossRef]
- Cichoń, M.A.; Szentpetery, Z.; Caley, M.; Papadakis, E.; Mackenzie, I.; Brennan, C.; O’toole, E.A. The receptor tyrosine kinase Axl regulates cell-cell adhesion and stemness in cutaneous squamous cell carcinoma. Oncogene 2014, 33, 4185–4192. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Gupta, P.B.; Chaffer, C.L.; Weinberg, R.A. Cancer stem cells: Mirage or reality? Nat. Med. 2009, 15, 1010–1012. [Google Scholar] [CrossRef] [PubMed]
- Biddle, A.; Mackenzie, I.C. Cancer stem cells and EMT in carcinoma. Cancer Metastasis Rev. 2012, 31, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.H.I.; Moon, J.; Garland, L.L.; Mack, P.C.; Testa, J.R.; Tsao, A.S.; Wozniak, A.J.; Gandara, D.R. SWOG S0722: Phase II study of mTOR inhibitor everolimus (RAD001) in advanced malignant pleural mesothelioma (MPM). J. Thorac. Oncol. 2015, 10, 387–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Liu, L.; Li, H.; Eilers, G.; Kuang, Y.; Shi, S.; Yan, Z.; Li, X.; Corson, J.M.; Meng, F.; et al. Multipoint targeting of the PI3K/mTOR pathway in mesothelioma. Br. J. Cancer 2014, 110, 2479–2488. [Google Scholar] [CrossRef] [Green Version]
- Dolly, S.O.; Wagner, A.J.; Bendell, J.C.; Kindler, H.L.; Krug, L.M.; Seiwert, T.Y.; Zauderer, M.G.; Lolkema, M.P.; Apt, D.; Yeh, R.F.; et al. Phase I Study of Apitolisib (GDC-0980), Dual Phosphatidylinositol-3-Kinase and Mammalian Target of Rapamycin Kinase Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 2874–2884. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Gualeni, A.V.; Volpi, C.C.; Carbone, A.; Gloghini, A. A novel semi-automated in situ hybridization protocol for microRNA detection in paraffin embedded tissue sections. J. Clin. Pathol. 2015, 68, 661–664. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belfiore, A.; Busico, A.; Bozzi, F.; Brich, S.; Dallera, E.; Conca, E.; Capone, I.; Gloghini, A.; Volpi, C.C.; Cabras, A.D.; et al. Molecular Signatures for Combined Targeted Treatments in Diffuse Malignant Peritoneal Mesothelioma. Int. J. Mol. Sci. 2019, 20, 5817. https://doi.org/10.3390/ijms20225817
Belfiore A, Busico A, Bozzi F, Brich S, Dallera E, Conca E, Capone I, Gloghini A, Volpi CC, Cabras AD, et al. Molecular Signatures for Combined Targeted Treatments in Diffuse Malignant Peritoneal Mesothelioma. International Journal of Molecular Sciences. 2019; 20(22):5817. https://doi.org/10.3390/ijms20225817
Chicago/Turabian StyleBelfiore, Antonino, Adele Busico, Fabio Bozzi, Silvia Brich, Elena Dallera, Elena Conca, Iolanda Capone, Annunziata Gloghini, Chiara C. Volpi, Antonello D. Cabras, and et al. 2019. "Molecular Signatures for Combined Targeted Treatments in Diffuse Malignant Peritoneal Mesothelioma" International Journal of Molecular Sciences 20, no. 22: 5817. https://doi.org/10.3390/ijms20225817
APA StyleBelfiore, A., Busico, A., Bozzi, F., Brich, S., Dallera, E., Conca, E., Capone, I., Gloghini, A., Volpi, C. C., Cabras, A. D., Pilotti, S., Baratti, D., Guaglio, M., Deraco, M., Kusamura, S., & Perrone, F. (2019). Molecular Signatures for Combined Targeted Treatments in Diffuse Malignant Peritoneal Mesothelioma. International Journal of Molecular Sciences, 20(22), 5817. https://doi.org/10.3390/ijms20225817