Advances in Sphingolipidoses: CRISPR-Cas9 Editing as an Option for Modelling and Therapy


Abstract
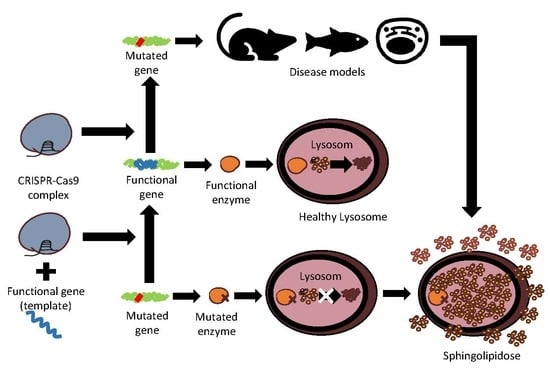
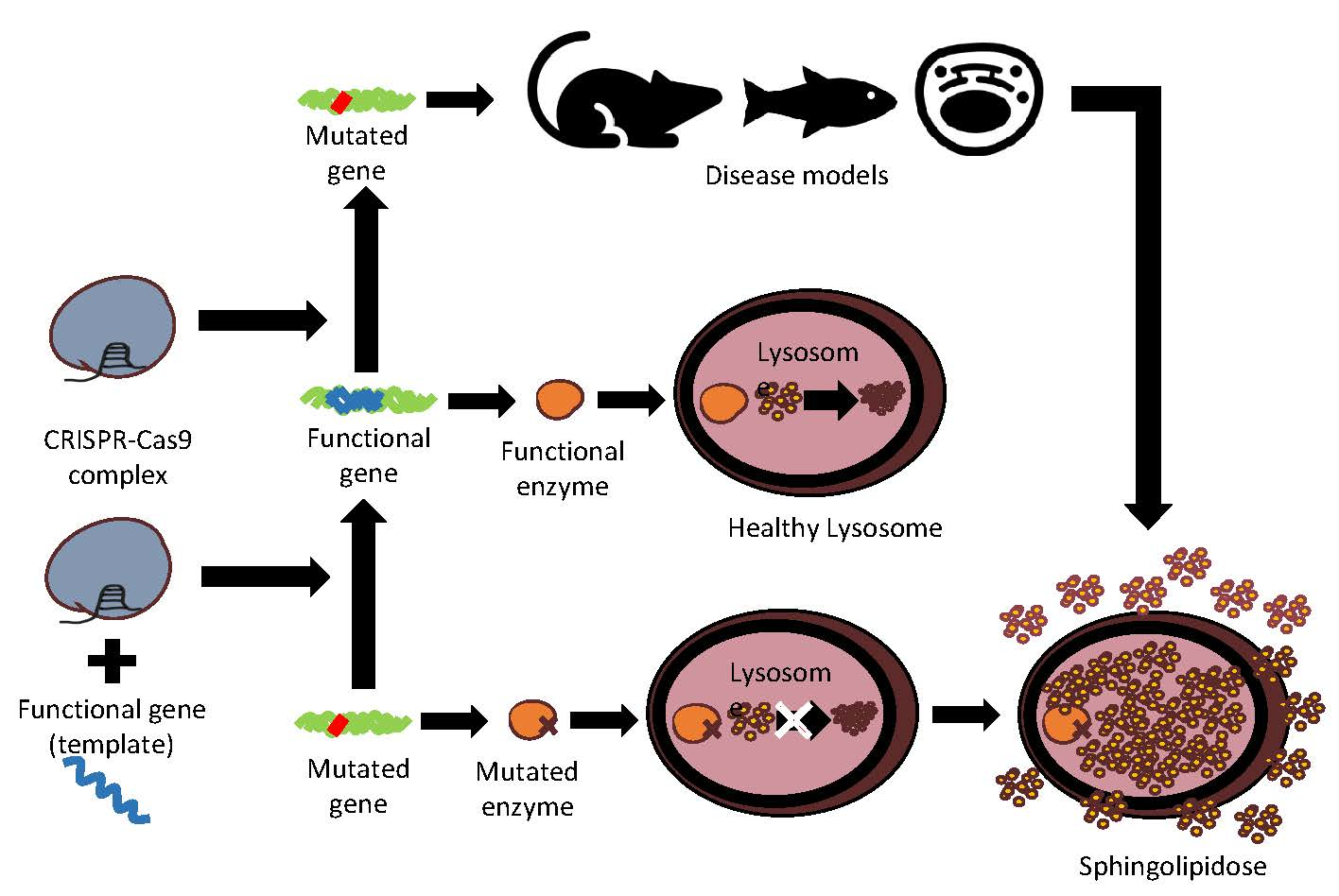
:
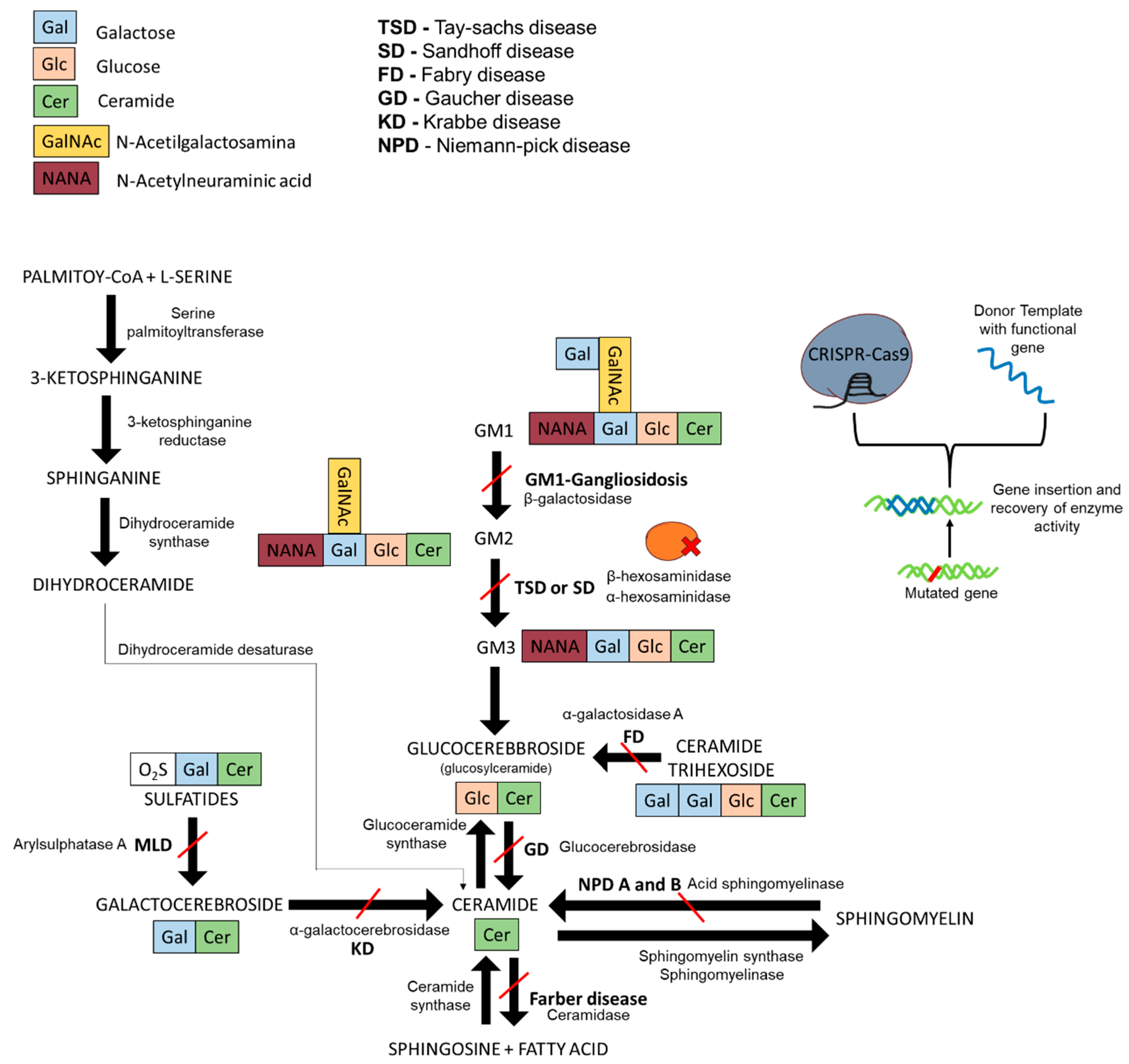
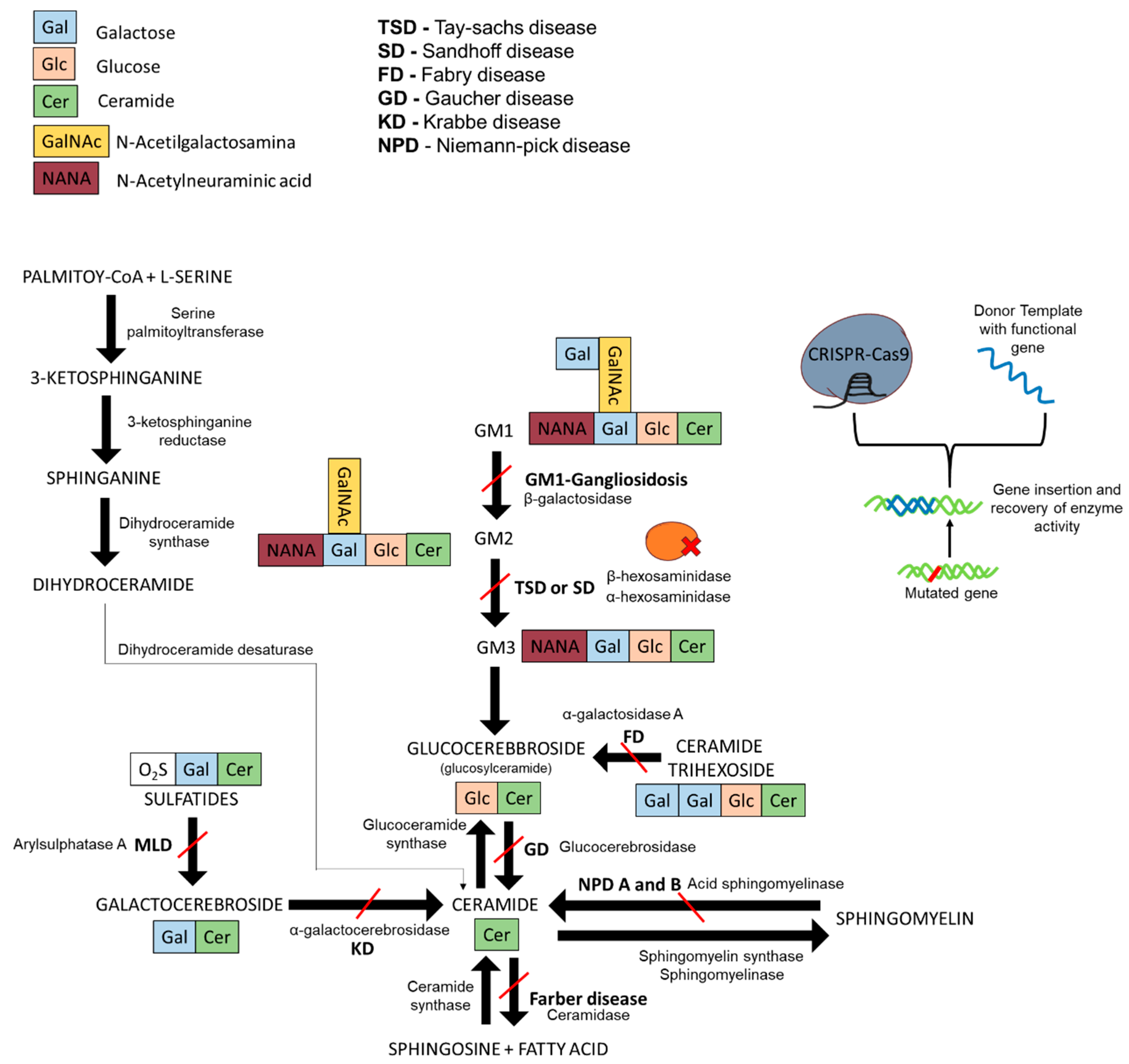
1. Lysosomal Storage Diseases and Sphingolipidoses
1.1. Gaucher Disease
1.2. Fabry Disease
1.3. GM2-Gangliosidoses
1.4. Niemann–Pick Disease
1.5. Krabbe Disease
1.6. GM1-Gangliosidosis and Morquio B Syndrome
1.7. Current Treatments for Sphingolipidoses
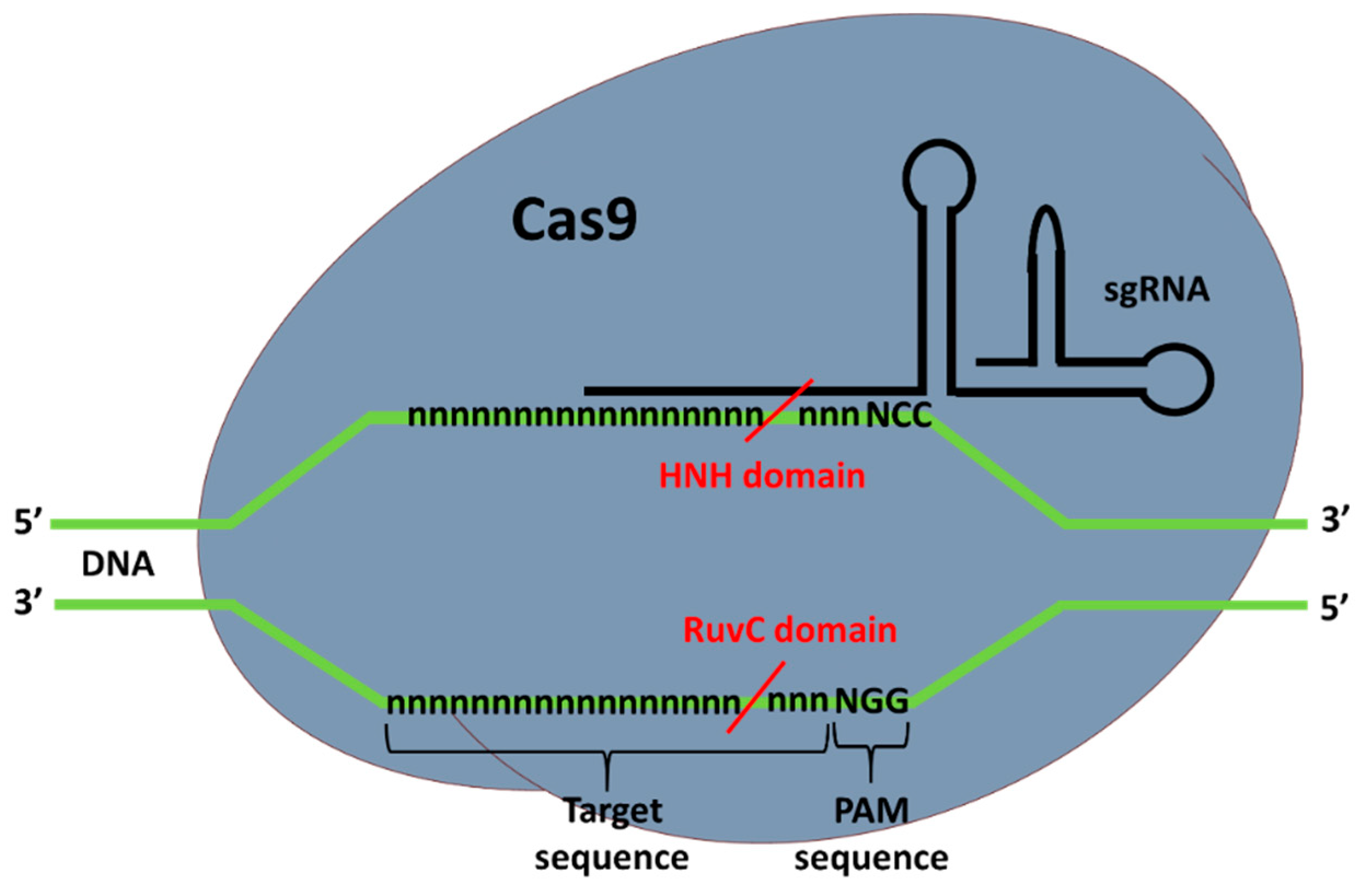
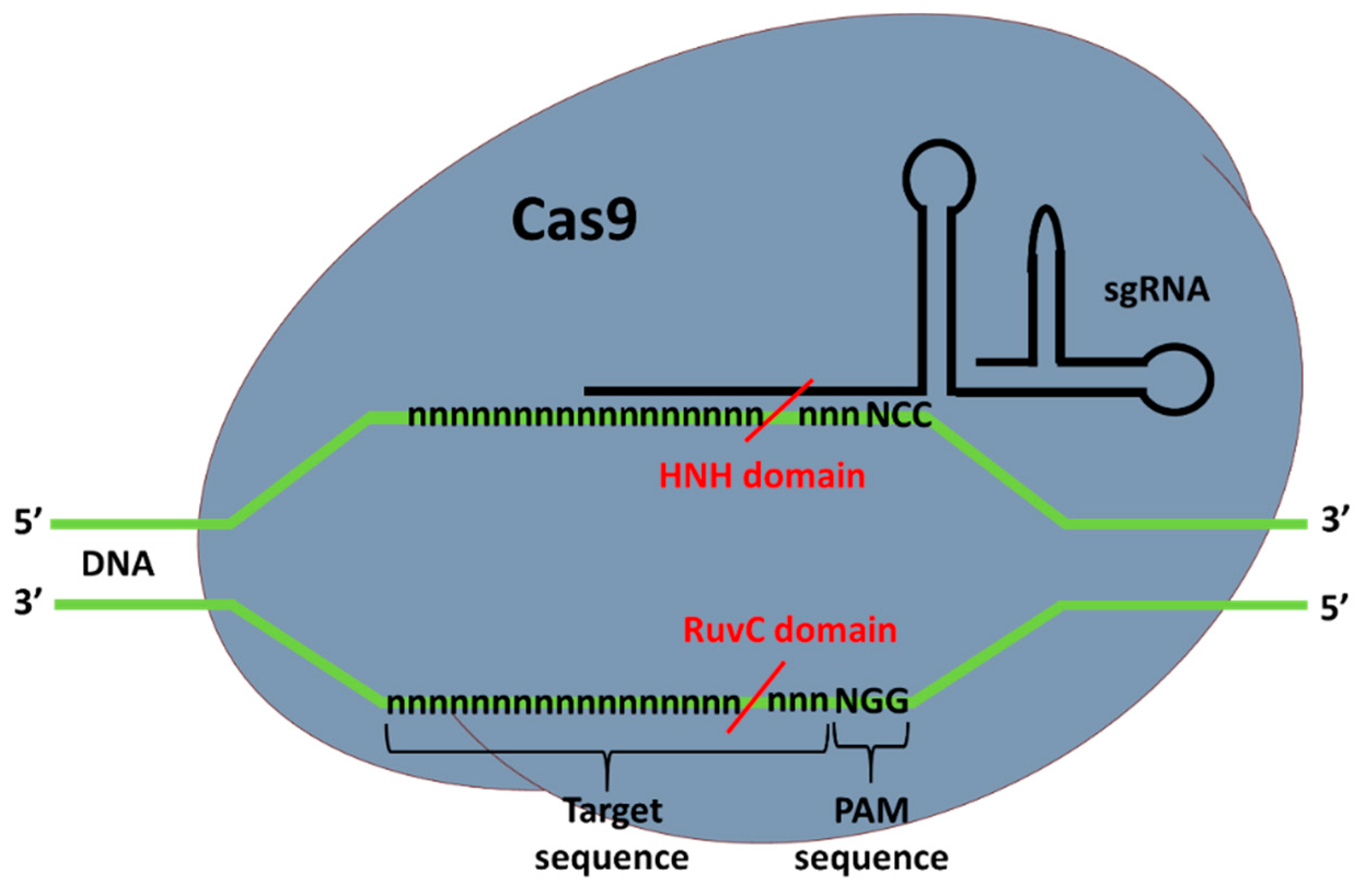
2. CRISPR-Cas9 and Prime Editing Background
3. CRISPR-Cas9 Edition for Disease Models and Therapeutic Approaches of Sphingolipidoses
3.1. Gaucher Disease
3.2. Fabry Disease
3.3. Tay–Sachs and Sandhoff Diseases
3.4. Niemann–Pick Disease
3.5. Krabbe Disease
3.6. GM1-Gangliosidosis and Morquio Syndrome
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Cox, T.M.; Cachon-Gonzalez, M.B. The cellular pathology of lysosomal diseases. J. Pathol. 2012, 226, 241–254. [Google Scholar] [CrossRef]
- Platt, F.M.; D’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primers 2018, 4, 27. [Google Scholar] [CrossRef]
- Sandhoff, K. Sphingolipidoses. J. Clin. Pathol. Suppl. 1974, 8, 94–105. [Google Scholar] [CrossRef]
- Platt, F.M. Sphingolipid lysosomal storage disorders. Nature 2014, 510, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Arenz, C. Recent advances and novel treatments for sphingolipidoses. Future Med. Chem. 2017, 9, 1685–1698. [Google Scholar] [CrossRef] [PubMed]
- Do, J.; McKinney, C.; Sharma, P.; Sidransky, E. Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener. 2019, 14, 36. [Google Scholar] [CrossRef] [PubMed]
- Ysselstein, D.; Shulman, J.M.; Krainc, D. Emerging links between pediatric lysosomal storage diseases and adult parkinsonism. Mov Disord. 2019, 34, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Melum, E.; Jiang, X.; Baker, K.D.; Macedo, M.F.; Fritsch, J.; Dowds, C.M.; Wang, J.; Pharo, A.; Kaser, A.; Tan, C.; et al. Control of CD1d-restricted antigen presentation and inflammation by sphingomyelin. Nat. Immunol. 2019, 20, 1644–1655. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef]
- Palmieri Impey, S.; Kang, H.; di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef]
- Farfel-Becker, T.; Do, J.; Tayebi, N.; Sidransky, E. Can GBA1-Associated Parkinson Disease Be Modeled in the Mouse? Trends Neurosci. 2019, 42, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, M.; Elstein, D.; Zimran, A.; Goker-Alpan, O. New Directions in Gaucher Disease. Hum. Mutat. 2016, 37, 1121–1136. [Google Scholar] [CrossRef] [PubMed]
- Bennett, L.L.; Mohan, D. Gaucher Disease and Its Treatment Options. Ann. Pharmacother. 2013, 47, 1182–1193. [Google Scholar] [CrossRef] [PubMed]
- Zimran, A.; Belmatoug, N.; Bembi, B.; Deegan, P.; Elstein, D.; Fernandez-Sasso, D.; Giraldo, P.; Goker-Alpan, O.; Lau, H.; Lukina, E.; et al. GOS Study group.Demographics and patient characteristics of 1209 patients with Gaucher disease: Descriptive analysis from the Gaucher Outcome Survey (GOS). Am. J. Hematol. 2018, 93, 205–212. [Google Scholar] [CrossRef]
- Bendikov-Bar, I.; Horowitz, M.; Bendikov-Bar, I. Gaucher disease paradigm: From ERAD to comorbidity. Hum. Mutat. 2012, 33, 1398–1407. [Google Scholar] [CrossRef]
- Maor, G.; Rapaport, D.; Horowitz, M. The effect of mutant GBA1 on accumulation and aggregation of α-synuclein. Hum. Mol. Genet. 2019, 28, 1768–1781. [Google Scholar] [CrossRef]
- Migdalska-Richards, A.; Wegrzynowicz, M.; Rusconi, R.; Deangeli, G.; Di Monte, D.A.; Spillantini, M.G.; Schapira, A.H.V. The L444P Gba1 mutation enhances alpha-synuclein induced loss of nigral dopaminergic neurons in mice. Brain 2017, 140, 2706–2721. [Google Scholar] [CrossRef]
- Duro, G.; Zizzo, C.; Cammarata, G.; Burlina, A.; Burlina, A.; Polo, G.; Scalia, S.; Oliveri, R.; Sciarrino, S.; Francofonte, D.; et al. Mutations in the GLA Gene and LysoGb3: Is It Really Anderson-Fabry Disease? Int. J. Mol. Sci. 2018, 19, 3726. [Google Scholar] [CrossRef]
- Germain, D.P.; Elliott, P.M.; Falissard, B.; Fomin, V.V.; Hilz, M.J.; Jovanovic, A.; Kantola, I.; Linhart, A.; Mignani, R.; Namdar, M.; et al. The effect of enzyme replacement therapy on clinical outcomes in male patients with Fabry disease: A systematic literature review by a European panel of experts. Mol. Genet. Metab. Rep. 2019, 19, 100454. [Google Scholar] [CrossRef]
- Mehta, A.; Widmer, U. Chapter 19 Natural history of Fabry disease. In Fabry Dis Ease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006. [Google Scholar]
- Waldek, S.; Feriozzi, S. Fabry nephropathy: A review—How can we optimize the management of Fabry nephropathy? BMC Nephrol. 2014, 15, 72. [Google Scholar] [CrossRef]
- Vu, M.; Li, R.; Baskfield, A.; Lu, B.; Farkhondeh, A.; Gorshkov, K.; Motabar, O.; Beers, J.; Chen, G.; Zou, J.; et al. Neural stem cells for disease modeling and evaluation of therapeutics for Tay-Sachs disease. Orphanet J. Rare Dis. 2018, 13, 152. [Google Scholar] [CrossRef] [PubMed]
- Hall, P.; Minnich, S.; Teigen, C.; Raymond, K. Diagnosing Lysosomal Storage Disorders: The GM2 Gangliosidoses. Curr. Protoc. Hum. Genet. 2014, 83, 16.1–16.8. [Google Scholar] [PubMed]
- Karimzadeh, P.; Jafari, N.; Biglari, H.N.; Dari, S.J.; Abadi, F.A.; Alaee, M.-R.; Nemati, H.; Saket, S.; Tonekaboni, S.H.; Taghdiri, M.-M.; et al. GM2-Gangliosidosis (Sandhoff and Tay Sachs disease): Diagnosis and Neuroimaging Findings (An Iranian Pediatric Case Series). Iran. J. Child Neurol. 2014, 8, 55–60. [Google Scholar] [PubMed]
- Solovyeva, V.V.; Shaimardanova, A.A.; Chulpanova, D.S.; Kitaeva, K.V.; Chakrabarti, L.; Rizvanov, A.A. New Approaches to Tay-Sachs Disease Therapy. Front. Physiol. 2018, 9, 1663. [Google Scholar] [CrossRef]
- Duarte, A.J.; Ribeiro, D.; Oliveira, P.; Amaral, O. Mutation Frequency of Three Neurodegenerative Lysosomal Storage Diseases: From Screening to Treatment? Arch. Med. Res. 2017, 48, 263–269. [Google Scholar] [CrossRef]
- Kaback, M.M.; Desnick, R.J. Hexosaminidase A Deficiency. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Vanier, M.T. Niemann-Pick diseases. Handb. Clin. Neurol. 2013, 113, 1717–1721. [Google Scholar]
- Pineda, M.; Juríčková, K.; Karimzadeh, P.; Kolnikova, M.; Malinova, V.; Insua, J.L.; Velten, C.; Kolb, S.A. Disease characteristics, prognosis and miglustat treatment effects on disease progression in patients with Niemann-Pick disease Type C: An international, multicenter, retrospective chart review. Orphanet J. Rare Dis. 2019, 14, 32. [Google Scholar] [CrossRef]
- Simonaro, C.M.; Park, J.-H.; Eliyahu, E.; Shtraizent, N.; McGovern, M.M.; Schuchman, E.H. Imprinting at the SMPD1 Locus: Implications for Acid Sphingomyelinase—Deficient Niemann-Pick Disease. Am. J. Hum. Genet. 2006, 78, 865–870. [Google Scholar] [CrossRef]
- Schuchman, E.H.; Desnick, R.J. Types A and B Niemann-Pick disease. Mol. Genet. Metab. 2017, 120, 27–33. [Google Scholar] [CrossRef]
- Graziano, A.C.E.; Cardile, V. History, genetic, and recent advances on Krabbe disease. Gene 2015, 555, 2–13. [Google Scholar] [CrossRef]
- Spratley, S.J.; Hill, C.H.; Viuff, A.H.; Edgar, J.R.; Skjødt, K.; Deane, J.E. Molecular Mechanisms of Disease Pathogenesis Differ in Krabbe Disease Variants. Traffic 2016, 17, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Bascou, N.; Derenzo, A.; Poe, M.D.; Escolar, M.L. A prospective natural history study of Krabbe disease in a patient cohort with onset between 6 months and 3 years of life. Orphanet J. Rare Dis. 2018, 13, 126. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Oshima, A.; Namba, E. β-Galactosidase deficiency (β-galactosidosis) GM1 gangliosidosis and Morquio B disease. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3775–3809. [Google Scholar]
- Morita, M.; Saito, S.; Ikeda, K.; Ohno, K.; Sugawara, K.; Suzuki, T.; Togawa, T.; Sakuraba, H. Structural bases of GM1 gangliosidosis and Morquio B disease. J. Hum. Genet. 2009, 54, 510–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caciotti, A.; Garman, S.C.; Rivera-Colón, Y.; Procopio, E.; Catarzi, S.; Ferri, L.; Guido, C.; Martelli, P.; Parini, R.; Antuzzi, D.; et al. GM1 gangliosidosis and Morquio B disease: An update on genetic alterations and clinical findings. Biochim. Biophys. Acta 2011, 1812, 782–790. [Google Scholar] [CrossRef]
- Barton, N.W.; Brady, R.O.; Dambrosia, J.M.; Di Bisceglie, A.M.; Doppelt, S.H.; Hill, S.C.; Mankin, H.J.; Murray, G.J.; Parker, R.I.; Argoff, C.E.; et al. Replacement therapy for inherited enzyme deficiency—Macrophage-targeted glucocerebrosidase for Gaucher’s disease. N. Engl. J. Med. 1991, 324, 1464–1470. [Google Scholar] [CrossRef]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F. ZFN, TALEN and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Doudna, J.A. CRISPR—Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [Green Version]
- Trevino, A.E.; Zhang, F. Genome editing using Cas9 nickases. Methods Enzymol. 2014, 546, 161–174. [Google Scholar]
- Jensen, M.K. Design principles for nuclease-deficient CRISPR-based transcriptional regulators. FEMS Yeast Res. 2018, 18, foy039. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Oberdoerffer, P. Chromatin dynamics in DNA double-strand break repair. Biochim. Biophys. Acta 2012, 1819, 811–819. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Rehman, S.; Tang, X.; Gu, K.; Fan, Q.; Chen, D.; Ma, W. Methodologies for Improving HDR Efficiency. Front. Genet. 2019, 9, 691. [Google Scholar] [CrossRef]
- Han, X.; Liu, Z.; Jo, M.C.; Zhang, K.; Li, Y.; Zeng, Z.; Li, N.; Zu, Y.; Qin, L. CRISPR-Cas9 delivery to hard-to-transfect cells via membrane deformation. Sci. Adv. 2015, 1, e1500454. [Google Scholar] [CrossRef] [Green Version]
- Kouranova, E.; Forbes, K.; Zhao, G.; Warren, J.; Bartels, A.; Wu, Y.; Cui, X. CRISPRs for Optimal Targeting: Delivery of CRISPR Components as DNA, RNA, and Protein into Cultured Cells and Single-Cell Embryos. Hum. Gene Ther. 2016, 27, 464–475. [Google Scholar] [CrossRef]
- Zhang, Y.; Long, C.; Li, H.; McAnally, J.R.; Baskin, K.K.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Sci. Adv. 2017, 3, e1602814. [Google Scholar] [CrossRef] [Green Version]
- Sanz, D.J.; Hollywood, J.A.; Scallan, M.F.; Harrison, P.T. Cas9/gRNA targeted excision of cystic fibrosis-causing deep-intronic splicing mutations restores normal splicing of CFTR mRNA. PLoS ONE 2017, 12, e0184009. [Google Scholar] [CrossRef] [Green Version]
- García-Tuñón, I.; Hernández-Sánchez, M.; Ordoñez, J.L.; Alonso-Pérez, V.; Álamo-Quijada, M.; Benito, R.; Guerrero, C.; Hernández-Rivas, J.M.; Sánchez-Martín, M. The CRISPR/Cas9 system efficiently reverts the tumorigenic ability of BCR/ABL in vitro and in a xenograft model of chronic myeloid leukemia. Oncotarget 2017, 8, 26027–26040. [Google Scholar] [CrossRef] [Green Version]
- György, B.; Nist-Lund, C.; Pan, B.; Asai, Y.; Karavitaki, K.D.; Kleinstiver, B.P.; Garcia, S.P.; Zaborowski, M.P.; Solanes, P.; Spataro, S.; et al. Allele-specific gene editing prevents deafness in a model of dominant progressive hearing loss. Nat. Med. 2019, 25, 1123–1130. [Google Scholar] [CrossRef]
- Diakatou, M.; Manes, G.; Bocquet, B.; Meunier, I.; Kalatzis, V. Genome Editing as a Treatment for the Most Prevalent Causative Genes of Autosomal Dominant Retinitis Pigmentosa. Int. J. Mol. Sci. 2019, 20, 2542. [Google Scholar] [CrossRef] [Green Version]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019. [Google Scholar] [CrossRef]
- Drews, K.; Calgi, M.P.; Harrison, W.C.; Drews, C.M.; Costa-Pinheiro, P.; Shaw, J.J.P.; Jobe, K.A.; Nelson, E.A.; Han, J.D.; Fox, T.; et al. Glucosylceramidase Maintains Influenza Virus Infection by Regulating Endocytosis. J. Virol. 2019, 93, e00017–e00019. [Google Scholar] [CrossRef] [Green Version]
- Lelieveld, L.T.; Mirzaian, M.; Kuo, C.-L.; Artola, M.; Ferraz, M.J.; Peter, R.E.A.; Akiyama, H.; Greimel, P.; Berg, R.J.B.H.N.V.D.; Overkleeft, H.S.; et al. Role of β-glucosidase 2 in aberrant glycosphingolipid metabolism: Model of glucocerebrosidase deficiency in zebrafish. J. Lipid Res. 2019, 60, 1851–1867. [Google Scholar] [CrossRef] [PubMed]
- Song, H.-Y.; Chiang, H.-C.; Tseng, W.-L.; Wu, P.; Chien, C.-S.; Leu, H.-B.; Yang, Y.-P.; Wang, M.-L.; Jong, Y.-J.; Chen, C.-H.; et al. Using CRISPR/Cas9-Mediated GLA Gene Knockout as an In Vitro Drug Screening Model for Fabry Disease. Int. J. Mol. Sci. 2016, 17, 2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenders, M.; Stappers, F.; Niemietz, C.; Schmitz, B.; Boutin, M.; Ballmaier, P.J.; Zibert, A.; Schmidt, H.; Brand, S.-M.; Auray-Blais, C.; et al. Mutation-specific Fabry disease patient-derived cell model to evaluate the amenability to chaperone therapy. J. Med. Genet. 2019, 56, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Song, H.Y.; Chien, C.S.; Yarmishyn, AA.; Chou, S.J.; Yang, Y.P.; Wang, M.L.; Wang, C.Y.; Leu, H.B.; Yu, W.C.; Chang, Y.L.; et al. Generation of GLA-Knockout Human Embryonic Stem Cell Lines to Model Autophagic Dysfunction and Exosome Secretion in Fabry Disease-Associated Hypertrophic Cardiomyopathy. Cells 2019, 8, 327. [Google Scholar] [CrossRef] [Green Version]
- Pereira, E.M.; Labilloy, A.; Eshbach, M.L.; Roy, A.; Subramanya, A.R.; Monte, S.; Labilloy, G.; Weisz, O.A. Characterization and phosphoproteomic analysis of a human immortalized podocyte model of Fabry disease generated using CRISPR/Cas9 technology. Am. J. Physiol. Physiol. 2016, 311, F1015–F1024. [Google Scholar] [CrossRef]
- Kang, J.J.; Kaissarian, N.M.; Desch, K.C.; Kelly, R.J.; Shu, L.; Bodary, P.F.; Shayman, J.A. α-galactosidase A deficiency promotes von Willebrand factor secretion in models of Fabry disease. Kidney Int. 2019, 95, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.J.; Aoki, K.; Moehring, F.; Murphy, C.A.; O’Hara, C.L.; Tiemeyer, M.; Stucky, C.L.; Dahms, N.M. Neuropathic pain in a Fabry disease rat model. JCI Insight 2018, 3, e99171. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.K.; Lu, Y.H.; Chen, Y.R.; Hsieh, Y.P.; Lin, W.J.; Hsu, T.; Niu, D.M. AB043. Correction of the GLA IVS4 + 919 G > A mutation with CRISPR/Cas9 deletion strategy in fibroblasts of Fabry disease. Ann. Transl. Med 2017, 5 (Suppl. 2), AB043. [Google Scholar] [CrossRef] [Green Version]
- Tropak, M.B.; Yonekawa, S.; Karumuthil-Melethil, S.; Thompson, P.; Wakarchuk, W.; Gray, S.J.; Walia, J.S.; Mark, B.L.; Mahuran, D. Construction of a hybrid β-hexosaminidase subunit capable of forming stable homodimers that hydrolyze GM2 ganglioside in vivo. Mol. Ther. Methods Clin. Dev. 2016, 3, 15057. [Google Scholar] [CrossRef]
- Allende, M.L.; Cook, E.K.; Larman, B.C.; Nugent, A.; Brady, J.M.; Golebiowski, D.; Sena-Esteves, M.; Tifft, C.J.; Proia, R.L. Cerebral organoids derived from Sandhoff disease-induced pluripotent stem cells exhibit impaired neurodifferentiation. J. Lipid Res. 2018, 59, 550–563. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Lukmantara, I.; Yang, H. CRISPR/Cas9-Mediated Generation of Niemann–Pick C1 Knockout Cell Line. Methods Mol. Biol. 2017, 1583, 73–83. [Google Scholar] [PubMed]
- Tseng, W.-C.; Loeb, H.E.; Pei, W.; Tsai-Morris, C.-H.; Xu, L.; Cluzeau, C.V.; Wassif, C.A.; Feldman, B.; Burgess, S.M.; Pavan, W.J.; et al. Modeling Niemann-Pick disease type C1 in zebrafish: A robust platform for in vivo screening of candidate therapeutic compounds. Dis. Model. Mech. 2018, 11, dmm034165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Cai, X.; Wang, G.; Ouyang, G.; Cao, H.; Caia, X. Model construction of Niemann-Pick type C disease in zebrafish. Biol. Chem. 2018, 399, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Dever, D.P.; Scharenberg, S.G.; Camarena, J.; Kildebeck, E.J.; Clark, J.T.; Martin, R.M.; Bak, R.O.; Tang, Y.; Dohse, M.; Birgmeier, J.A.; et al. CRISPR/Cas9 Genome Engineering in Engraftable Human Brain-Derived Neural Stem Cells. iScience 2019, 15, 524–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zizioli, D.; Guarienti, M.; Tobia, C.; Gariano, G.; Borsani, G.; Bresciani, R.; Ronca, R.; Giacopuzzi, E.; Preti, A.; Gaudenzi, G.; et al. Molecular cloning and knockdown of galactocerebrosidase in zebrafish: New insights into the pathogenesis of Krabbe’s disease. Biochim. Biophys. Acta 2014, 1842, 665–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latour, Y.L.; Yoon, R.; Thomas, S.E.; Grant, C.; Li, C.; Sena-Esteves, M.; Allende, M.L.; Proia, R.L.; Tifft, C.J.; et al. Human GLB1 knockout cerebral organoids: A model system for testing AAV9-mediated GLB1 gene therapy for reducing GM1 ganglioside storage in GM1 gangliosidosis. Mol. Genet. Metab. Rep. 2019, 21, 100513. [Google Scholar] [CrossRef] [PubMed]
- Przybilla, M.J.; Ou, L.; Tăbăran, A.-F.; Jiang, X.; Sidhu, R.; Kell, P.J.; Ory, D.S.; O’Sullivan, M.G.; Whitley, C.B. Comprehensive behavioral and biochemical outcomes of novel murine models of GM1-gangliosidosis and Morquio syndrome type B. Mol. Genet. Metab. 2019, 126, 139–150. [Google Scholar] [CrossRef]
- Martinez-Lage, M.; Puig-Serra, P.; Menendez, P.; Torres-Ruiz, R.; Rodriguez-Perales, S. CRISPR/Cas9 for Cancer Therapy: Hopes and Challenges. Biomedicines 2018, 6, 105. [Google Scholar] [CrossRef] [Green Version]
- Dai, W.-J.; Zhu, L.-Y.; Yan, Z.-Y.; Xu, Y.; Wang, Q.-L.; Lu, X.-J. CRISPR-Cas9 for in vivo Gene Therapy: Promise and Hurdles. Mol. Ther. Nucleic Acids 2016, 5, e349. [Google Scholar] [CrossRef] [Green Version]
- Mollanoori, H.; Teimourian, S. Therapeutic applications of CRISPR/Cas9 system in gene therapy. Biotechnol. Lett. 2018, 40, 907–914. [Google Scholar] [CrossRef]
- Lundstrom, K. Special Issue: Gene Therapy with Emphasis on RNA Interference. Viruses 2015, 7, 4482–4487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, L.S.; Searle, P.F.; Onion, D.; Mautner, V. Viral gene therapy strategies: From basic science to clinical application. J. Pathol. 2006, 208, 299–318. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Genes | Enzymes | Substrates | Disease | CRISPR-Cas9 for Disease Models: | CRISPR-Cas9 Gene/Cell Therapy | References | |
|---|---|---|---|---|---|---|---|
| Cell Models | In vivo Models | ||||||
| GBA1 | Glucocerebrosidase | Glucosylceramide | Gaucher disease | - HEK cells - A549 cells | - Zebrafish | X | [53,54] |
| GLA | α-galactosidase A | Globotriaosylceramide | Fabry disease | - HEK cells - hESCs - Podocytes - Human endothelialcells | - Rat - Mouse | - Restoration of GLA enzyme activity in FD patient′s Fibroblasts with mutation knockout | [55,56,57,58,59,60,61] |
| HEXA | α-hexosaminidase s-hexosaminidase | GM2-ganglioside | Tay-Sachs disease | - HEK cells | X | - Gene correction of HEXA 1278+TATC mutation in HEK293T cell using Prime editing | [62] |
| HEXB | α-hexosaminidase β-hexosaminidase | GM2-ganglioside | Sandhoff disease | - iPSCs | X | - No accumulation of GM2 ganglioside in iPSCs generated from fibroblasts of infantile SD patients due to template knockin | [52,63] |
| SMPD | Acid sphingomyelinase | Sphingomyelin | Niemann-pick diseases | - HeLa cells | - Zebrafish | X | [64,65,66] |
| GALC | β-galactocerebrosidase | Galactolipids | Krabbe disease | X | - Zebrafish | - Cell therapy with CRISPR-Cas9 edited hNSCs with correct enzyme activity | [67,68] |
| GLB1 | β-galactosidase | GM1-ganglioside | GM1-gangliosidosis | - iPSCs | - Mouse | X | [69,70] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, R.; Amaral, O. Advances in Sphingolipidoses: CRISPR-Cas9 Editing as an Option for Modelling and Therapy. Int. J. Mol. Sci. 2019, 20, 5897. https://doi.org/10.3390/ijms20235897
Santos R, Amaral O. Advances in Sphingolipidoses: CRISPR-Cas9 Editing as an Option for Modelling and Therapy. International Journal of Molecular Sciences. 2019; 20(23):5897. https://doi.org/10.3390/ijms20235897
Chicago/Turabian StyleSantos, Renato, and Olga Amaral. 2019. "Advances in Sphingolipidoses: CRISPR-Cas9 Editing as an Option for Modelling and Therapy" International Journal of Molecular Sciences 20, no. 23: 5897. https://doi.org/10.3390/ijms20235897