TGFB1-Mediated Gliosis in Multiple Sclerosis Spinal Cords Is Favored by the Regionalized Expression of HOXA5 and the Age-Dependent Decline in Androgen Receptor Ligands


Abstract
1. Introduction
2. Results
2.1. The Human Spinal Cord Genomic Signature Retrieved from the ARCHS4 Database Is Specifically Enriched in Homeobox Genes
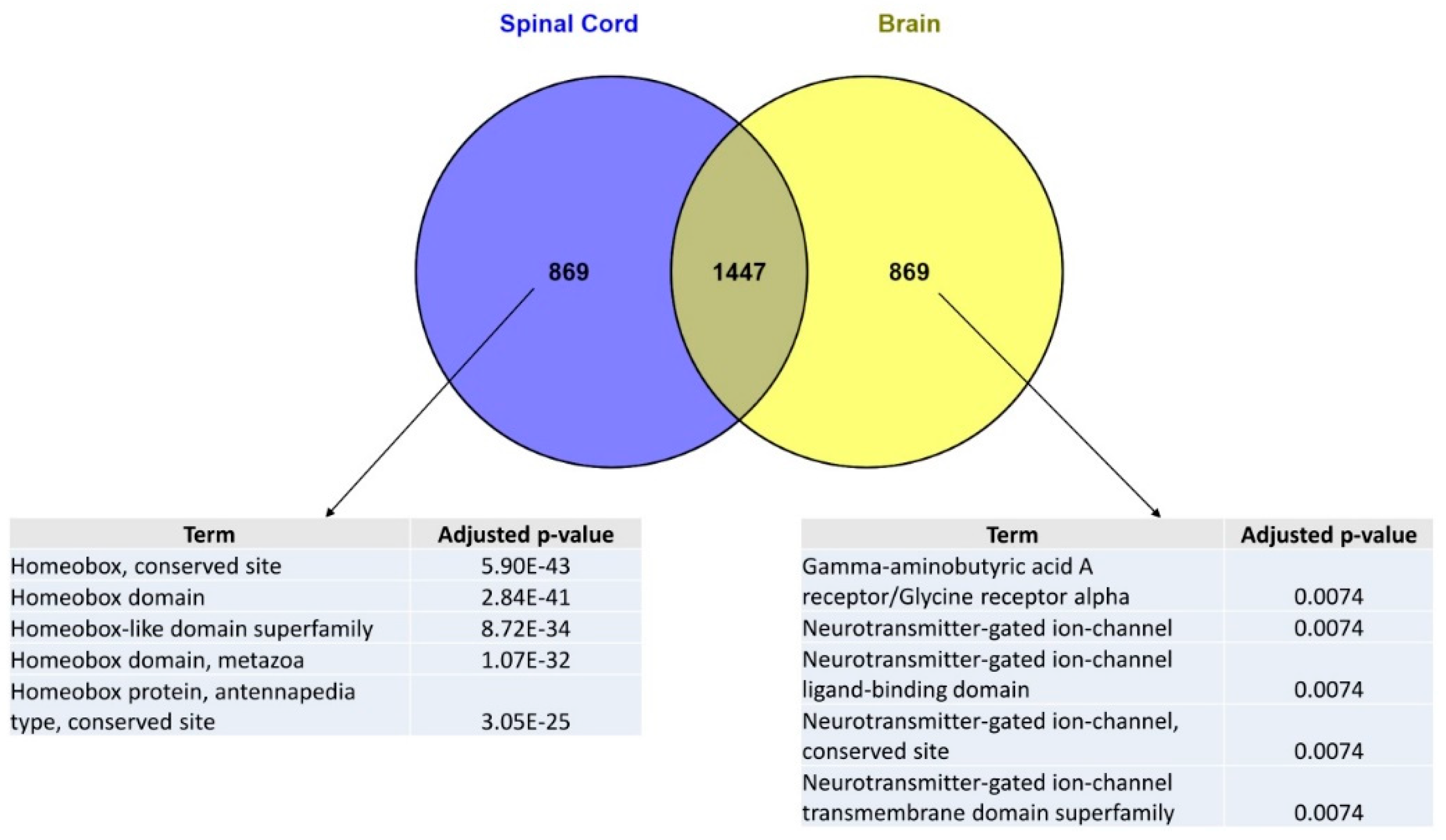
2.2. In the Normal Adult Human Central Nervous System, a Set of Homeobox Genes Are Uniquely Overexpressed in the Spinal Cord as Compared to Other CNS Regions
2.3. The Spinal Cord-Overexpressed HOXA5 Homeobox Protein Interacts with the Gliosis-Associated Transcription Factors SOX2 (SRY-box 2) and SMAD1.
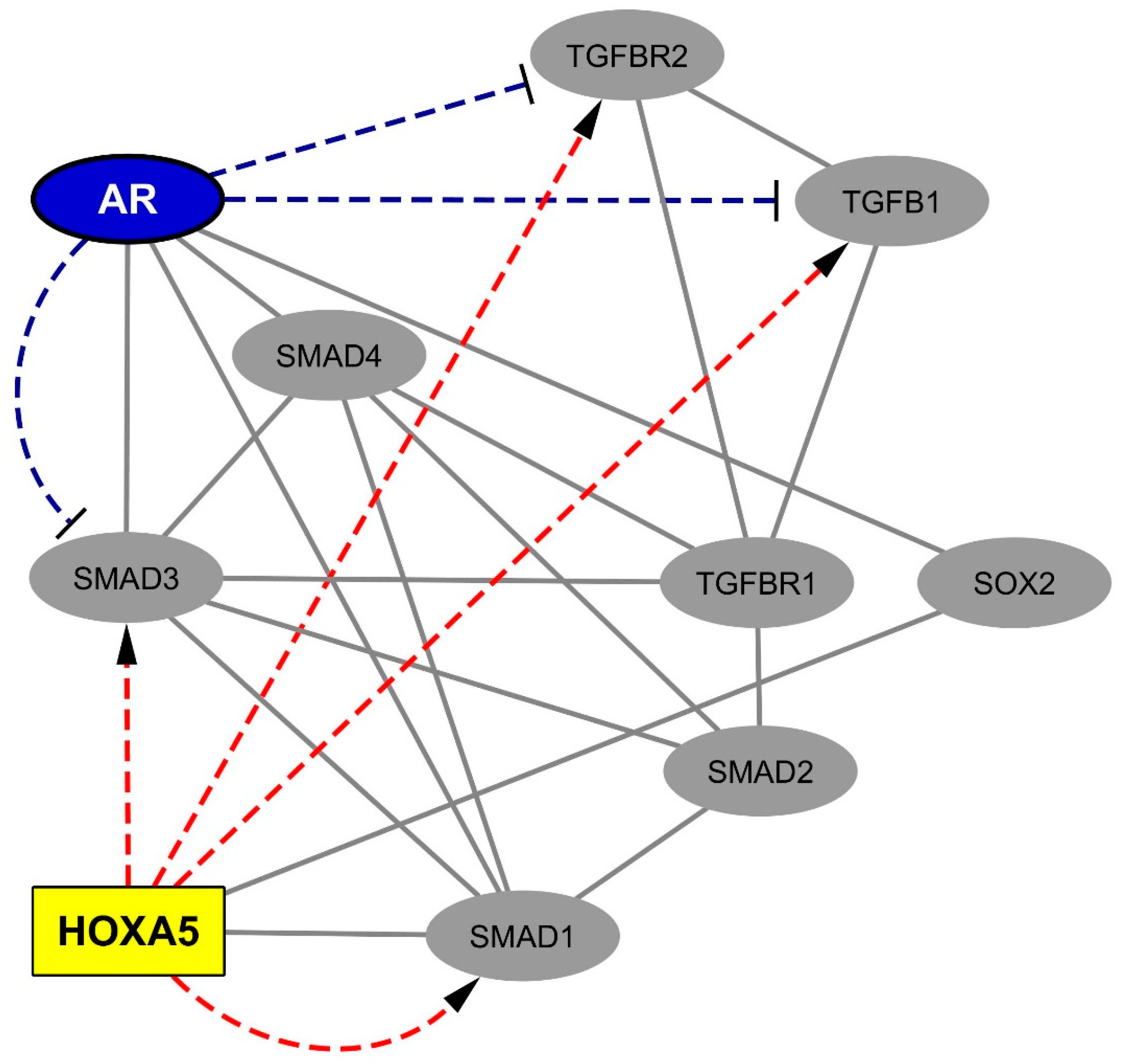
2.4. Data Mining Analysis Indicates that the Progliotic Pathway Associated with SC-Overexpressed HOX Proteins Is Negatively Regulated by the Androgen Receptor
3. Discussion
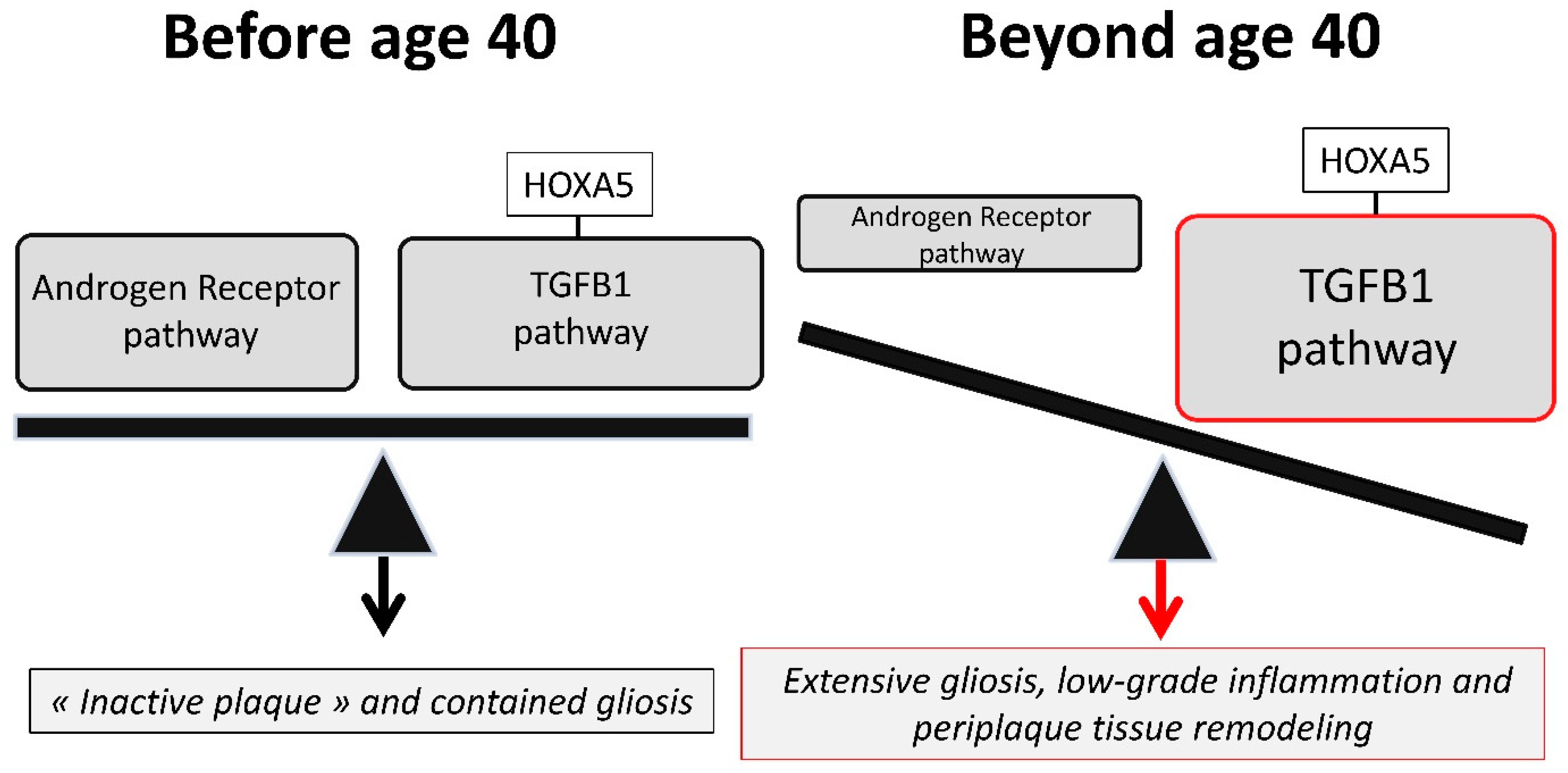
3.1. The Spinal Cord-Overexpressed HOXA5 Gene May Amplify the TGFB1 Progliotic Pathway
3.2. Androgens, via Transcriptional Silencing and SMAD-Interfering Mechanisms, May Antagonize the Progliotic Effects of TGFB1
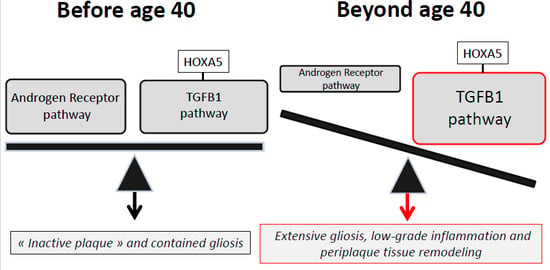
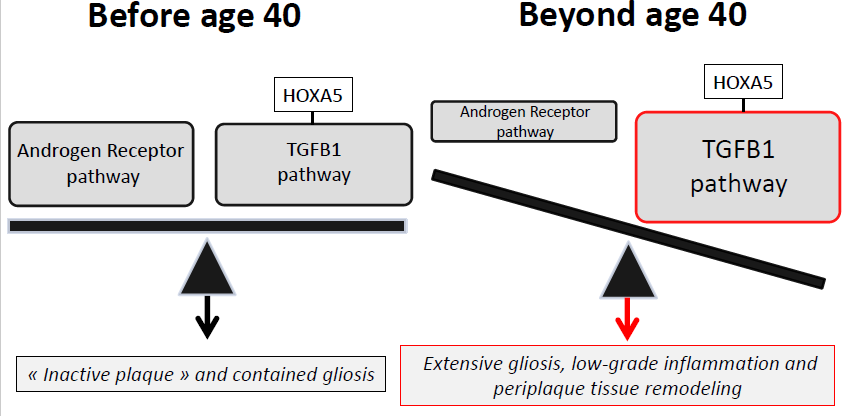
3.3. An Imbalance between the Androgen Receptor Pathway and the HOXA5-Promoted TGFB1 Pathway May Explain the Impact of Age and Gender on MS Spinal Cord Gliosis
3.4. Therapeutic Implications
4. Materials and Method
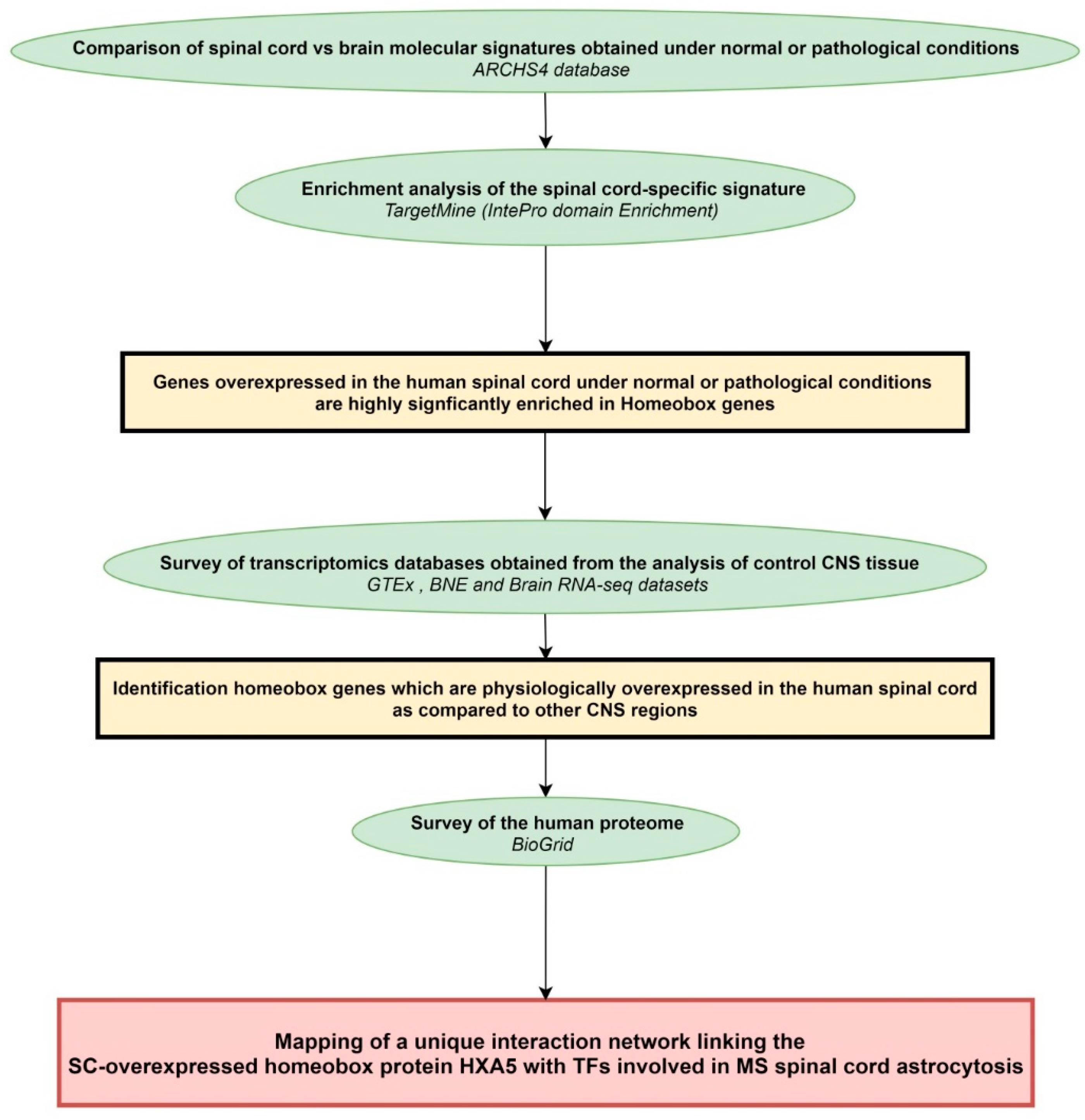
4.1. Workflow of the Study
4.2. Mining of Transcriptomics Databases Obtained from the Analysis of Human CNS Tissue Samples
4.3. Mining of the Proteomics Database BioGrid
4.4. Enrichment Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kearney, H.; Miller, D.H.; Ciccarelli, O. Spinal cord MRI in multiple sclerosis—diagnostic, prognostic and clinical value. Nat. Rev. Neurol. 2015, 11, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Cawley, N.; Tur, C.; Prados, F.; Plantone, D.; Kearney, H.; Abdel-Aziz, K.; Ourselin, S.; Wheeler-Kingshott, C.A.G.; Miller, D.H.; Thompson, A.J.; et al. Spinal cord atrophy as a primary outcome measure in phase II trials of progressive multiple sclerosis. Mult. Scler. J. 2018, 24, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Valsasina, P.; Aboulwafa, M.; Preziosa, P.; Messina, R.; Falini, A.; Comi, G.; Filippi, M.; Rocca, M.A. Cervical Cord T1-weighted Hypointense Lesions at MR Imaging in Multiple Sclerosis: Relationship to Cord Atrophy and Disability. Radiology 2018, 288, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Confavreux, C.; Vukusic, S. Age at disability milestones in multiple sclerosis. Brain 2006, 129, 595–605. [Google Scholar] [CrossRef]
- Confavreux, C.; Vukusic, S. Natural history of multiple sclerosis: A unifying concept. Brain 2006, 129, 606–616. [Google Scholar] [CrossRef]
- Lassmann, H. Pathogenic Mechanisms Associated with Different Clinical Courses of Multiple Sclerosis. Front. Immunol. 2019, 9, 3116. [Google Scholar] [CrossRef]
- Kutzelnigg, A.; Lucchinetti, C.F.; Stadelmann, C.; Brück, W.; Rauschka, H.; Bergmann, M.; Schmidbauer, M.; Parisi, J.E.; Lassmann, H. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 2005, 128, 2705–2712. [Google Scholar] [CrossRef]
- Lieury, A.; Chanal, M.; Androdias, G.; Reynolds, R.; Cavagna, S.; Giraudon, P.; Confavreux, C.; Nataf, S. Tissue remodeling in periplaque regions of multiple sclerosis spinal cord lesions. Glia 2014, 62, 1645–1658. [Google Scholar] [CrossRef]
- Nataf, S.; Barritault, M.; Pays, L. A Unique TGFB1-Driven Genomic Program Links Astrocytosis, Low-Grade Inflammation and Partial Demyelination in Spinal Cord Periplaques from Progressive Multiple Sclerosis Patients. Int. J. Mol. Sci. 2017, 18, 2097. [Google Scholar] [CrossRef]
- Frischer, J.M.; Weigand, S.D.; Guo, Y.; Kale, N.; Parisi, J.E.; Pirko, I.; Mandrekar, J.; Bramow, S.; Metz, I.; Brück, W.; et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann. Neurol. 2015, 78, 710–721. [Google Scholar] [CrossRef]
- Bramow, S.; Frischer, J.M.; Lassmann, H.; Koch-Henriksen, N.; Lucchinetti, C.F.; Sørensen, P.S.; Laursen, H. Demyelination versus remyelination in progressive multiple sclerosis. Brain 2010, 133, 2983–2998. [Google Scholar] [CrossRef]
- Taylor, R.A.; Chang, C.-F.; Goods, B.A.; Hammond, M.D.; Grory, B.M.; Ai, Y.; Steinschneider, A.F.; Renfroe, S.C.; Askenase, M.H.; McCullough, L.D.; et al. TGF-β1 modulates microglial phenotype and promotes recovery after intracerebral hemorrhage. J. Clin. Investig. 2016, 127, 280–292. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, H.-Q.; Huang, Y.; Qiu, Y.-H.; Peng, Y.-P. Transforming growth factor-β1 acts via TβR-I on microglia to protect against MPP+-induced dopaminergic neuronal loss. Brain. Behav. Immun. 2016, 51, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.M.; Hale, J.H. Macrophage/Microglia regulation of astrocytic tenascin: Synergistic action of transforming growth factor-beta and basic fibroblast growth factor. J. Neurosci. 1997, 17, 9624–9633. [Google Scholar] [CrossRef] [PubMed]
- Baghdassarian, D.; Toru-Delbauffe, D.; Gavaret, J.M.; Pierre, M. Effects of transforming growth factor-beta 1 on the extracellular matrix and cytoskeleton of cultured astrocytes. Glia 1993, 7, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.M.; Strunz, C. Growth factor and cytokine regulation of chondroitin sulfate proteoglycans by astrocytes. Glia 2005, 52, 209–218. [Google Scholar] [CrossRef]
- Baror, R.; Neumann, B.; Segel, M.; Chalut, K.J.; Fancy, S.P.J.; Schafer, D.P.; Franklin, R.J.M. Transforming growth factor-beta renders ageing microglia inhibitory to oligodendrocyte generation by CNS progenitors. Glia 2019, 67, 1374–1384. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Lachmann, A.; Torre, D.; Keenan, A.B.; Jagodnik, K.M.; Lee, H.J.; Wang, L.; Silverstein, M.C.; Ma’ayan, A. Massive mining of publicly available RNA-seq data from human and mouse. Nat. Commun. 2018, 9, 1366. [Google Scholar] [CrossRef]
- Chen, Y.-A.; Tripathi, L.P.; Fujiwara, T.; Kameyama, T.; Itoh, M.N.; Mizuguchi, K. The TargetMine Data Warehouse: Enhancement and Updates. Front. Genet. 2019, 10, 934. [Google Scholar] [CrossRef]
- Mitchell, A.; Chang, H.-Y.; Daugherty, L.; Fraser, M.; Hunter, S.; Lopez, R.; McAnulla, C.; McMenamin, C.; Nuka, G.; Pesseat, S.; et al. The InterPro protein families database: The classification resource after 15 years. Nucleic Acids Res. 2015, 43, D213–D221. [Google Scholar] [CrossRef] [PubMed]
- The GTEx Consortium. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [PubMed]
- eGTEx Project. Enhancing GTEx by bridging the gaps between genotype, gene expression, and disease. Nat. Genet. 2017, 49, 1664–1670. [Google Scholar] [CrossRef] [PubMed]
- The GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.B.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef]
- Searle, B.C.; Gittelman, R.M.; Manor, O.; Akey, J.M. Detecting Sources of Transcriptional Heterogeneity in Large-Scale RNA-Seq Data Sets. Genetics 2016, 204. [Google Scholar] [CrossRef][Green Version]
- Durrenberger, P.F.; Fernando, F.S.; Kashefi, S.N.; Bonnert, T.P.; Seilhean, D.; Nait-Oumesmar, B.; Schmitt, A.; Gebicke-Haerter, P.J.; Falkai, P.; Grünblatt, E.; et al. Common mechanisms in neurodegeneration and neuroinflammation: A BrainNet Europe gene expression microarray study. J. Neural Transm. 2015, 122, 1055–1068. [Google Scholar] [CrossRef]
- Witschi, R.; Johansson, T.; Morscher, G.; Scheurer, L.; Deschamps, J.; Zeilhofer, H.U. Hoxb8-Cre mice: A tool for brain-sparing conditional gene deletion. Genesis 2010, 48, 596–602. [Google Scholar] [CrossRef]
- Sugiyama, K.; Tanaka, K. Spinal cord-specific deletion of the glutamate transporter GLT1 causes motor neuron death in mice. Biochem. Biophys. Res. Commun. 2018, 497, 689–693. [Google Scholar] [CrossRef]
- Schmidt, E.F.; Kus, L.; Gong, S.; Heintz, N. BAC transgenic mice and the GENSAT database of engineered mouse strains. Cold Spring Harb. Protoc. 2013, 2013, 73692. [Google Scholar] [CrossRef]
- Gerfen, C.R.; Paletzki, R.; Heintz, N. GENSAT BAC cre-recombinase driver lines to study the functional organization of cerebral cortical and basal ganglia circuits. Neuron 2013, 80, 1368–1383. [Google Scholar] [CrossRef] [PubMed]
- Molino, Y.; Jabès, F.; Bonnet, A.; Gaudin, N.; Bernard, A.; Benech, P.; Khrestchatisky, M. Gene expression comparison reveals distinct basal expression of HOX members and differential TNF-induced response between brain- and spinal cord-derived microvascular endothelial cells. J. Neuroinflamm. 2016, 13, 290. [Google Scholar] [CrossRef] [PubMed]
- Rhee, D.Y.; Cho, D.-Y.; Zhai, B.; Slattery, M.; Ma, L.; Mintseris, J.; Wong, C.Y.; White, K.P.; Celniker, S.E.; Przytycka, T.M.; et al. Transcription factor networks in Drosophila melanogaster. Cell Rep. 2014, 8, 2031–2043. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, J.A.; Wright, K.J.; Tjian, R. Shifting players and paradigms in cell-specific transcription. Mol. Cell 2009, 36, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Grove, C.A.; Walhout, A.J.M. Transcription factor functionality and transcription regulatory networks. Mol. Biosyst. 2008, 4, 309–314. [Google Scholar] [CrossRef]
- Sonawane, A.R.; Platig, J.; Fagny, M.; Chen, C.-Y.; Paulson, J.N.; Lopes-Ramos, C.M.; DeMeo, D.L.; Quackenbush, J.; Glass, K.; Kuijjer, M.L. Understanding Tissue-Specific Gene Regulation. Cell Rep. 2017, 21, 1077–1088. [Google Scholar] [CrossRef]
- Oughtred, R.; Stark, C.; Breitkreutz, B.-J.; Rust, J.; Boucher, L.; Chang, C.; Kolas, N.; O’Donnell, L.; Leung, G.; McAdam, R.; et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 2019, 47, D529–D541. [Google Scholar] [CrossRef]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef]
- Shijo, T.; Warita, H.; Suzuki, N.; Ikeda, K.; Mitsuzawa, S.; Akiyama, T.; Ono, H.; Nishiyama, A.; Izumi, R.; Kitajima, Y.; et al. Antagonizing bone morphogenetic protein 4 attenuates disease progression in a rat model of amyotrophic lateral sclerosis. Exp. Neurol. 2018, 307, 164–179. [Google Scholar] [CrossRef]
- Fuller, M.L.; DeChant, A.K.; Rothstein, B.; Caprariello, A.; Wang, R.; Hall, A.K.; Miller, R.H. Bone morphogenetic proteins promote gliosis in demyelinating spinal cord lesions. Ann. Neurol. 2007, 62, 288–300. [Google Scholar] [CrossRef]
- Chen, C.; Zhong, X.; Smith, D.K.; Tai, W.; Yang, J.; Zou, Y.; Wang, L.-L.; Sun, J.; Qin, S.; Zhang, C.-L. Astrocyte-Specific Deletion of Sox2 Promotes Functional Recovery After Traumatic Brain Injury. Cereb. Cortex 2019, 29, 54–69. [Google Scholar] [CrossRef] [PubMed]
- Luo, K. Signaling Cross Talk between TGF-β/Smad and Other Signaling Pathways. Cold Spring Harb. Perspect. Biol. 2017, 9, a022137. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, K.; Miyazono, K. Regulation of TGF-β Family Signaling by Inhibitory Smads. Cold Spring Harb. Perspect. Biol. 2017, 9, a022095. [Google Scholar] [CrossRef] [PubMed]
- Nurgazieva, D.; Mickley, A.; Moganti, K.; Ming, W.; Ovsyi, I.; Popova, A.; Sachindra; Awad, K.; Wang, N.; Bieback, K.; et al. TGF-β1, but Not Bone Morphogenetic Proteins, Activates Smad1/5 Pathway in Primary Human Macrophages and Induces Expression of Proatherogenic Genes. J. Immunol. 2015, 194, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Vizán, P.; Das, D.; Chakravarty, P.; Vogt, J.; Rogers, K.W.; Müller, P.; Hinck, A.P.; Sapkota, G.P.; Hill, C.S. TGF-β uses a novel mode of receptor activation to phosphorylate SMAD1/5 and induce epithelial-to-mesenchymal transition. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Dummula, K.; Vinukonda, G.; Chu, P.; Xing, Y.; Hu, F.; Mailk, S.; Csiszar, A.; Chua, C.; Mouton, P.; Kayton, R.J.; et al. Bone Morphogenetic Protein Inhibition Promotes Neurological Recovery after Intraventricular Hemorrhage. J. Neurosci. 2011, 31, 12068–12082. [Google Scholar] [CrossRef]
- Luo, J.; Lin, A.H.; Masliah, E.; Wyss-Coray, T. Bioluminescence imaging of Smad signaling in living mice shows correlation with excitotoxic neurodegeneration. Proc. Natl. Acad. Sci. USA 2006, 103, 18326–18331. [Google Scholar] [CrossRef]
- Arevalo, M.A.; Santos-Galindo, M.; Acaz-Fonseca, E.; Azcoitia, I.; Garcia-Segura, L.M. Gonadal hormones and the control of reactive gliosis. Horm. Behav. 2013, 63, 216–221. [Google Scholar] [CrossRef]
- Barreto, G.; Veiga, S.; Azcoitia, I.; Garcia-Segura, L.M.; Garcia-Ovejero, D. Testosterone decreases reactive astroglia and reactive microglia after brain injury in male rats: Role of its metabolites, oestradiol and dihydrotestosterone. Eur. J. Neurosci. 2007, 25, 3039–3046. [Google Scholar] [CrossRef]
- Garcia-Estrada, J.; Del Rio, J.A.; Luquin, S.; Soriano, E.; Garcia-Segura, L.M. Gonadal hormones down-regulate reactive gliosis and astrocyte proliferation after a penetrating brain injury. Brain Res. 1993, 628, 271–278. [Google Scholar] [CrossRef]
- Giatti, S.; Rigolio, R.; Romano, S.; Mitro, N.; Viviani, B.; Cavaletti, G.; Caruso, D.; Garcia-Segura, L.M.; Melcangi, R.C. Dihydrotestosterone as a Protective Agent in Chronic Experimental Autoimmune Encephalomyelitis. Neuroendocrinology 2015, 101, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Cornelius, S.C.; Pultz, N.J.; Jorgensen, J.S.; Bonham, M.J.; Kim, S.-J.; Danielpour, D. The Androgen Receptor Represses Transforming Growth Factor-β Signaling through Interaction with Smad3. J. Biol. Chem. 2002, 277, 1240–1248. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Gao, S.; Wang, Z. Transcriptional regulation of the TGF-beta1 promoter by androgen receptor. Biochem. J. 2008, 416, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Gao, S.; Chu, J.; Zhou, L.; Wang, Z. Negative androgen-response elements mediate androgen-dependent transcriptional inhibition of TGF-β1 and CDK2 promoters in the prostate gland. J. Androl. 2012, 33, 27–36. [Google Scholar] [CrossRef]
- Huntley, R.P.; Binns, D.; Dimmer, E.; Barrell, D.; O’Donovan, C.; Apweiler, R. QuickGO: A user tutorial for the web-based Gene Ontology browser. Database 2009, 2009, bap010. [Google Scholar] [CrossRef]
- Binns, D.; Dimmer, E.; Huntley, R.; Barrell, D.; O’Donovan, C.; Apweiler, R. QuickGO: A web-based tool for Gene Ontology searching. Bioinformatics 2009, 25, 3045–3046. [Google Scholar] [CrossRef]
- The UniProt Consortium UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [CrossRef]
- Mathelier, A.; Zhao, X.; Zhang, A.W.; Parcy, F.; Worsley-Hunt, R.; Arenillas, D.J.; Buchman, S.; Chen, C.; Chou, A.; Ienasescu, H.; et al. JASPAR 2014: An extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 2014, 42, D142–D147. [Google Scholar] [CrossRef]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, 2016. [Google Scholar] [CrossRef]
- Cao, W.; Huang, H.; Xia, T.; Liu, C.; Muhammad, S.; Sun, C. Homeobox a5 Promotes White Adipose Tissue Browning Through Inhibition of the Tenascin C/Toll-Like Receptor 4/Nuclear Factor Kappa B Inflammatory Signaling in Mice. Front. Immunol. 2018, 9, 647. [Google Scholar] [CrossRef]
- Cieply, B.; Farris, J.; Denvir, J.; Ford, H.L.; Frisch, S.M. Epithelial-mesenchymal transition and tumor suppression are controlled by a reciprocal feedback loop between ZEB1 and Grainyhead-like-2. Cancer Res. 2013, 73, 6299–6309. [Google Scholar] [CrossRef] [PubMed]
- Itoh, N.; Itoh, Y.; Tassoni, A.; Ren, E.; Kaito, M.; Ohno, A.; Ao, Y.; Farkhondeh, V.; Johnsonbaugh, H.; Burda, J.; et al. Cell-specific and region-specific transcriptomics in the multiple sclerosis model: Focus on astrocytes. Proc. Natl. Acad. Sci. USA 2018, 115, E302–E309. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.E.; Liddelow, S.A.; Chakraborty, C.; Münch, A.E.; Heiman, M.; Barres, B.A. Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. USA 2018, 115, E1896–E1905. [Google Scholar] [CrossRef] [PubMed]
- Emsley, J.G.; Macklis, J.D. Astroglial heterogeneity closely reflects the neuronal-defined anatomy of the adult murine CNS. Neuron Glia Biol. 2006, 2, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Buosi, A.S.; Matias, I.; Araujo, A.P.B.; Batista, C.; Gomes, F.C.A. Heterogeneity in Synaptogenic Profile of Astrocytes from Different Brain Regions. Mol. Neurobiol. 2018, 55, 751–762. [Google Scholar] [CrossRef]
- Schitine, C.; Nogaroli, L.; Costa, M.R.; Hedin-Pereira, C. Astrocyte heterogeneity in the brain: From development to disease. Front. Cell. Neurosci. 2015, 9, 76. [Google Scholar] [CrossRef]
- Hussain, R.; Ghoumari, A.M.; Bielecki, B.; Steibel, J.; Boehm, N.; Liere, P.; Macklin, W.B.; Kumar, N.; Habert, R.; Mhaouty-Kodja, S.; et al. The neural androgen receptor: A therapeutic target for myelin repair in chronic demyelination. Brain 2013, 136, 132–146. [Google Scholar] [CrossRef]
- Bielecki, B.; Mattern, C.; Ghoumari, A.M.; Javaid, S.; Smietanka, K.; Abi Ghanem, C.; Mhaouty-Kodja, S.; Ghandour, M.S.; Baulieu, E.-E.; Franklin, R.J.M.; et al. Unexpected central role of the androgen receptor in the spontaneous regeneration of myelin. Proc. Natl. Acad. Sci. USA 2016, 113, 14829–14834. [Google Scholar] [CrossRef]
- Gubbels Bupp, M.R.; Jorgensen, T.N. Androgen-Induced Immunosuppression. Front. Immunol. 2018, 9, 794. [Google Scholar] [CrossRef]
- Giefing-Kröll, C.; Berger, P.; Lepperdinger, G.; Grubeck-Loebenstein, B. How sex and age affect immune responses, susceptibility to infections, and response to vaccination. Aging Cell 2015, 14, 309–321. [Google Scholar] [CrossRef]
- Atallah, A.; Mhaouty-Kodja, S.; Grange-Messent, V. Chronic depletion of gonadal testosterone leads to blood–brain barrier dysfunction and inflammation in male mice. J. Cereb. Blood Flow Metab. 2017, 37, 3161–3175. [Google Scholar] [CrossRef] [PubMed]
- Laffont, S.; Blanquart, E.; Savignac, M.; Cénac, C.; Laverny, G.; Metzger, D.; Girard, J.-P.; Belz, G.T.; Pelletier, L.; Seillet, C.; et al. Androgen signaling negatively controls group 2 innate lymphoid cells. J. Exp. Med. 2017, 214, 1581–1592. [Google Scholar] [CrossRef] [PubMed]
- Chitnis, T. The role of testosterone in MS risk and course. Mult. Scler. J. 2018, 24, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Bove, R.; Musallam, A.; Healy, B.; Raghavan, K.; Glanz, B.; Bakshi, R.; Weiner, H.; De Jager, P.; Miller, K.; Chitnis, T. Low testosterone is associated with disability in men with multiple sclerosis. Mult. Scler. J. 2014, 20, 1584–1592. [Google Scholar] [CrossRef] [PubMed]
- Baulieu, E.-E.; Thomas, G.; Legrain, S.; Lahlou, N.; Roger, M.; Debuire, B.; Faucounau, V.; Girard, L.; Hervy, M.-P.; Latour, F.; et al. Dehydroepiandrosterone (DHEA), DHEA sulfate, and aging: Contribution of the DHEAge Study to a sociobiomedical issue. Proc. Natl. Acad. Sci. USA 2000, 97, 4279–4284. [Google Scholar] [CrossRef]
- Parker, L.; Gral, T.; Perrigo, V.; Skowsky, R. Decreased adrenal androgen sensitivity to ACTH during aging. Metabolism 1981, 30, 601–604. [Google Scholar] [CrossRef]
- Samaras, N.; Samaras, D.; Frangos, E.; Forster, A.; Philippe, J. A review of age-related dehydroepiandrosterone decline and its association with well-known geriatric syndromes: Is treatment beneficial? Rejuvenation Res. 2013, 16, 285–294. [Google Scholar] [CrossRef]
- Diotel, N.; Charlier, T.D.; Lefebvre d’Hellencourt, C.; Couret, D.; Trudeau, V.L.; Nicolau, J.C.; Meilhac, O.; Kah, O.; Pellegrini, E. Steroid Transport, Local Synthesis, and Signaling within the Brain: Roles in Neurogenesis, Neuroprotection, and Sexual Behaviors. Front. Neurosci. 2018, 12, 84. [Google Scholar] [CrossRef]
- Rollnik, J.D.; Sindern, E.; Schweppe, C.; Malin, J.P. Biologically active TGF-β1 is increased in cerebrospinal fluid while it is reduced in serum in multiple sclerosis patients. Acta Neurol. Scand. 2009, 96, 101–105. [Google Scholar] [CrossRef]
- Itman, C.; Wong, C.; Hunyadi, B.; Ernst, M.; Jans, D.A.; Loveland, K.L. Smad3 dosage determines androgen responsiveness and sets the pace of postnatal testis development. Endocrinology 2011, 152, 2076–2089. [Google Scholar] [CrossRef]
- Kang, H.-Y.; Huang, K.-E.; Chang, S.Y.; Ma, W.-L.; Lin, W.-J.; Chang, C. Differential Modulation of Androgen Receptor-mediated Transactivation by Smad3 and Tumor Suppressor Smad4. J. Biol. Chem. 2002, 277, 43749–43756. [Google Scholar] [CrossRef] [PubMed]
- Hayes, S.A.; Zarnegar, M.; Sharma, M.; Yang, F.; Peehl, D.M.; ten Dijke, P.; Sun, Z. SMAD3 represses androgen receptor-mediated transcription. Cancer Res. 2001, 61, 2112–2118. [Google Scholar] [PubMed]
- Song, K.; Wang, H.; Krebs, T.L.; Wang, B.; Kelley, T.J.; Danielpour, D. DHT selectively reverses Smad3-mediated/TGF-beta-induced responses through transcriptional down-regulation of Smad3 in prostate epithelial cells. Mol. Endocrinol. 2010, 24, 2019–2029. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Yu, P.; Chen, H.; Geller, H.M. Targeted inhibition of KCa3.1 attenuates TGF-β-induced reactive astrogliosis through the Smad2/3 signaling pathway. J. Neurochem. 2014, 130, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Tatomir, A.; Tegla, C.A.; Martin, A.; Boodhoo, D.; Nguyen, V.; Sugarman, A.J.; Mekala, A.; Anselmo, F.; Talpos-Caia, A.; Cudrici, C.; et al. RGC-32 regulates reactive astrocytosis and extracellular matrix deposition in experimental autoimmune encephalomyelitis. Immunol. Res. 2018, 66, 445–461. [Google Scholar] [CrossRef] [PubMed]
- Coers, S.; Tanzer, L.; Jones, K.J. Testosterone treatment attenuates the effects of facial nerve transection on glial fibrillary acidic protein (GFAP) levels in the hamster facial motor nucleus. Metab. Brain Dis. 2002, 17, 55–63. [Google Scholar] [CrossRef]
- Lanz, T.V.; Ding, Z.; Ho, P.P.; Luo, J.; Agrawal, A.N.; Srinagesh, H.; Axtell, R.; Zhang, H.; Platten, M.; Wyss-Coray, T.; et al. Angiotensin II sustains brain inflammation in mice via TGF-β. J. Clin. Investig. 2010, 120, 2782–2794. [Google Scholar] [CrossRef]
- Luo, J.; Ho, P.P.; Buckwalter, M.S.; Hsu, T.; Lee, L.Y.; Zhang, H.; Kim, D.-K.; Kim, S.-J.; Gambhir, S.S.; Steinman, L.; et al. Glia-dependent TGF-β signaling, acting independently of the TH17 pathway, is critical for initiation of murine autoimmune encephalomyelitis. J. Clin. Investig. 2007, 117, 3306–3315. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Borrow, P.; Brooker, M.J.; Mucke, L. Astroglial overproduction of TGF-β1 enhances inflammatory central nervous system disease in transgenic mice. J. Neuroimmunol. 1997, 77, 45–50. [Google Scholar] [CrossRef]
- Ziehn, M.O.; Avedisian, A.A.; Dervin, S.M.; Umeda, E.A.; O’Dell, T.J.; Voskuhl, R.R. Therapeutic testosterone administration preserves excitatory synaptic transmission in the hippocampus during autoimmune demyelinating disease. J. Neurosci. 2012, 32, 12312–12324. [Google Scholar] [CrossRef]
- Palaszynski, K.M.; Loo, K.K.; Ashouri, J.F.; Liu, H.; Voskuhl, R.R. Androgens are protective in experimental autoimmune encephalomyelitis: Implications for multiple sclerosis. J. Neuroimmunol. 2004, 146, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Dalal, M.; Kim, S.; Voskuhl, R.R. Testosterone therapy ameliorates experimental autoimmune encephalomyelitis and induces a T helper 2 bias in the autoantigen-specific T lymphocyte response. J. Immunol. 1997, 159, 3–6. [Google Scholar] [PubMed]
- Du, C.; Khalil, M.W.; Sriram, S. Administration of dehydroepiandrosterone suppresses experimental allergic encephalomyelitis in SJL/J mice. J. Immunol. 2001, 167, 7094–7101. [Google Scholar] [CrossRef] [PubMed]
- Offner, H.; Zamora, A.; Drought, H.; Matejuk, A.; Auci, D.L.; Morgan, E.E.; Vandenbark, A.A.; Reading, C.L. A synthetic androstene derivative and a natural androstene metabolite inhibit relapsing–remitting EAE. J. Neuroimmunol. 2002, 130, 128–139. [Google Scholar] [CrossRef]
- Lafyatis, R. Transforming growth factor β--at the centre of systemic sclerosis. Nat. Rev. Rheumatol. 2014, 10, 706–719. [Google Scholar] [CrossRef] [PubMed]
- Rice, L.M.; Padilla, C.M.; McLaughlin, S.R.; Mathes, A.; Ziemek, J.; Goummih, S.; Nakerakanti, S.; York, M.; Farina, G.; Whitfield, M.L.; et al. Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients. J. Clin. Investig. 2015, 125, 2795–2807. [Google Scholar] [CrossRef]
- Taroni, J.N.; Martyanov, V.; Mahoney, J.M.; Whitfield, M.L. A Functional Genomic Meta-Analysis of Clinical Trials in Systemic Sclerosis: Toward Precision Medicine and Combination Therapy. J. Investig. Dermatol. 2017, 137, 1033–1041. [Google Scholar] [CrossRef]
- Den Hollander, M.W.; Bensch, F.; Glaudemans, A.W.J.M.; Oude Munnink, T.H.; Enting, R.H.; den Dunnen, W.F.A.; Heesters, M.A.A.M.; Kruyt, F.A.E.; Lub-de Hooge, M.N.; Cees de Groot, J.; et al. TGF-β Antibody Uptake in Recurrent High-Grade Glioma Imaged with 89Zr-Fresolimumab PET. J. Nucl. Med. 2015, 56, 1310–1314. [Google Scholar] [CrossRef]
- De Gramont, A.; Faivre, S.; Raymond, E. Novel TGF-β inhibitors ready for prime time in onco-immunology. Oncoimmunology 2017, 6, e1257453. [Google Scholar] [CrossRef]
- Peters, H.; Border, W.A.; Noble, N.A. Angiotensin II blockade and low-protein diet produce additive therapeutic effects in experimental glomerulonephritis. Kidney Int. 2000, 57, 1493–1501. [Google Scholar] [CrossRef]
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013, 4, 2516. [Google Scholar] [CrossRef] [PubMed]
- Shin, G.T.; Kim, S.J.; Ma, K.A.; Kim, H.S.; Kim, D. ACE inhibitors attenuate expression of renal transforming growth factor-beta1 in humans. Am. J. Kidney Dis. 2000, 36, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Song, J.H.; Lee, S.W.; Suh, J.H.; Kim, E.S.; Hong, S.B.; Kim, K.A.; Kim, M.-J. The effects of dual blockade of the renin- angiotensin system on urinary protein and transforming growth factor-b excretion in 2 groups of patients with IgA and diabetic nephropathy. Clin. Nephrol. 2003, 60, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Pokharel, S.; van Geel, P.P.; Sharma, U.C.; Cleutjens, J.P.M.; Bohnemeier, H.; Tian, X.-L.; Schunkert, H.; Crijns, H.J.G.M.; Paul, M.; Pinto, Y.M. Increased Myocardial Collagen Content in Transgenic Rats Overexpressing Cardiac Angiotensin-Converting Enzyme Is Related to Enhanced Breakdown of N -Acetyl-Ser-Asp-Lys-Pro and Increased Phosphorylation of Smad2/3. Circulation 2004, 110, 3129–3135. [Google Scholar] [CrossRef]
- Bar-Klein, G.; Cacheaux, L.P.; Kamintsky, L.; Prager, O.; Weissberg, I.; Schoknecht, K.; Cheng, P.; Kim, S.Y.; Wood, L.; Heinemann, U.; et al. Losartan prevents acquired epilepsy via TGF-β signaling suppression. Ann. Neurol. 2014, 75, 864–875. [Google Scholar] [CrossRef]
- Papadopoulos, P.; Tong, X.-K.; Imboden, H.; Hamel, E. Losartan improves cerebrovascular function in a mouse model of Alzheimer’s disease with combined overproduction of amyloid-β and transforming growth factor-β1. J. Cereb. Blood Flow Metab. 2017, 37, 1959–1970. [Google Scholar] [CrossRef]
- Kurth, F.; Luders, E.; Sicotte, N.L.; Gaser, C.; Giesser, B.S.; Swerdloff, R.S.; Montag, M.J.; Voskuhl, R.R.; Mackenzie-Graham, A. Neuroprotective effects of testosterone treatment in men with multiple sclerosis. NeuroImage Clin. 2014, 4, 454–460. [Google Scholar] [CrossRef]
- Gold, S.M.; Chalifoux, S.; Giesser, B.S.; Voskuhl, R.R. Immune modulation and increased neurotrophic factor production in multiple sclerosis patients treated with testosterone. J. Neuroinflamm. 2008, 5, 32. [Google Scholar] [CrossRef]
- Sicotte, N.L.; Giesser, B.S.; Tandon, V.; Klutch, R.; Steiner, B.; Drain, A.E.; Shattuck, D.W.; Hull, L.; Wang, H.-J.; Elashoff, R.M.; et al. Testosterone treatment in multiple sclerosis: A pilot study. Arch. Neurol. 2007, 64, 683–688. [Google Scholar] [CrossRef]
- Kritz-Silverstein, D.; von Mühlen, D.; Laughlin, G.A.; Bettencourt, R. Effects of dehydroepiandrosterone supplementation on cognitive function and quality of life: The DHEA and Well-Ness (DAWN) Trial. J. Am. Geriatr. Soc. 2008, 56, 1292–1298. [Google Scholar] [CrossRef]
- Corona, G.; Rastrelli, G.; Giagulli, V.A.; Sila, A.; Sforza, A.; Forti, G.; Mannucci, E.; Maggi, M. Dehydroepiandrosterone supplementation in elderly men: A meta-analysis study of placebo-controlled trials. J. Clin. Endocrinol. Metab. 2013, 98, 3615–3626. [Google Scholar] [CrossRef] [PubMed]
- Scheffers, C.S.; Armstrong, S.; Cantineau, A.E.; Farquhar, C.; Jordan, V. Dehydroepiandrosterone for women in the peri- or postmenopausal phase. Cochrane Database Syst. Rev. 2015, 1, CD011066. [Google Scholar] [CrossRef] [PubMed]
- Matejuk, A.; Hopke, C.; Vandenbark, A.A.; Hurn, P.D.; Offner, H. Middle-age male mice have increased severity of experimental autoimmune encephalomyelitis and are unresponsive to testosterone therapy. J. Immunol. 2005, 174, 2387–2395. [Google Scholar] [CrossRef] [PubMed]
- Nataf, S. Cord-Age-Gender Connections Shape the Pathophysiology of Multiple Sclerosis Progressive Forms. Int. J. Mol. Sci. 2019, 20, 5103. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spinal Cord (n =159) | Cerebellum (n = 241) | Hippocampus (n = 197) | Brain Cortex (n = 255) | |
|---|---|---|---|---|
| HOXA2 | 8.14 | 0 | 0 | 0 |
| HOXA4 | 29.14 | 0.03 | 0 | 0.01 |
| HOXA5 | 23.01 | 0.08 | 0.01 | 0.02 |
| HOXB2 | 20.31 | 0.54 | 0.19 | 0.10 |
| HOXB3 | 6.16 | 0.17 | 0.05 | 0.04 |
| HOXB5 | 8.52 | 0 | 0 | 0 |
| HOXB6 | 5.37 | 0 | 0 | 0 |
| HOXB7 | 15.82 | 0 | 0.05 | 0.02 |
| HOXB8 | 17.91 | 0 | 0 | 0 |
| HOXD8 | 5.41 | 0 | 0 | 0.01 |
| Gene Symbol | SC/BC Ratio | Adjusted p-Value |
|---|---|---|
| HOXA2 | 15.49 | 4.57 × 10−7 |
| HOXA4 | 58.62 | 2.62 × 10−6 |
| HOXA5 | 234.90 | 9.03 × 10−6 |
| HOXB2 | 41.46 | 1.14 × 10−7 |
| HOXB3 | 2.58 | 0.01 |
| HOXB5 | 52.41 | 0.002 |
| HOXB6 | 1.74 | 0.06 |
| HOXB7 | 102.29 | 2.47 × 10−6 |
| HOXB8 | 407.52 | 0.009 |
| HOXD8 | 13.42 | 0.001 |
| SC-Overexpressed HOX Genes | Gene Symbols (Bold) and Names of TFs Interacting with SC-Overexpressed HOX Proteins |
|---|---|
| HOXA5 | DDIT3 (DNA damage inducible transcript 3) |
| HOXA5 | FOXA2 (forkhead box A2) |
| HOXA5 | FOXO1 (forkhead box O1) |
| HOXA5 | SMAD1 (SMAD family member 1) |
| HOXA5 | SOX2 (SRY-box 2) |
| HOXA5 | TWIST1 (twist family bHLH transcription factor 21) |
| HOXA5 | ZNF707 (zinc finger protein 707) |
| HOXB5 | HOXC4 (homeobox C4) |
| HOXB5 | HRAS (HRas proto-oncogene, GTPase) |
| HOXB5 | NKX2-1 (NK2 homeobox 1) |
| HOXB6 | ALX4 (ALX Homeobox 4) |
| HOXB6 | TCF21 (transcription Factor 21) |
| HOXB7, HOXB8 | PBX1 (PBX homeobox 1) |
| HOXB8 | PBX3 (PBX homeobox 3) |
| Queried GO Term | Relevant Overlapping GO Term | Rank of Similarity |
|---|---|---|
| Positive regulation of SMAD protein signal transduction (GO:0060391) | Negative regulation of androgen receptor signaling pathway (GO:0060766) | 23rd from 743 overlapping GO terms |
| Negative regulation of SMAD protein signal transduction (GO:0060392) | No overlap with androgen receptor-related GO-term | Not applicable |
| Negative regulation of androgen receptor signaling pathway (GO:0060766) | Positive regulation of SMAD protein signal transduction (GO:0060391) | 4th from 562 overlapping GO terms |
| Positive regulation of androgen receptor activity (GO:2000825) | No overlap with SMAD-related GO-term | Not applicable |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nataf, S.; Guillen, M.; Pays, L. TGFB1-Mediated Gliosis in Multiple Sclerosis Spinal Cords Is Favored by the Regionalized Expression of HOXA5 and the Age-Dependent Decline in Androgen Receptor Ligands. Int. J. Mol. Sci. 2019, 20, 5934. https://doi.org/10.3390/ijms20235934
Nataf S, Guillen M, Pays L. TGFB1-Mediated Gliosis in Multiple Sclerosis Spinal Cords Is Favored by the Regionalized Expression of HOXA5 and the Age-Dependent Decline in Androgen Receptor Ligands. International Journal of Molecular Sciences. 2019; 20(23):5934. https://doi.org/10.3390/ijms20235934
Chicago/Turabian StyleNataf, Serge, Marine Guillen, and Laurent Pays. 2019. "TGFB1-Mediated Gliosis in Multiple Sclerosis Spinal Cords Is Favored by the Regionalized Expression of HOXA5 and the Age-Dependent Decline in Androgen Receptor Ligands" International Journal of Molecular Sciences 20, no. 23: 5934. https://doi.org/10.3390/ijms20235934
APA StyleNataf, S., Guillen, M., & Pays, L. (2019). TGFB1-Mediated Gliosis in Multiple Sclerosis Spinal Cords Is Favored by the Regionalized Expression of HOXA5 and the Age-Dependent Decline in Androgen Receptor Ligands. International Journal of Molecular Sciences, 20(23), 5934. https://doi.org/10.3390/ijms20235934