Reciprocal Association between the Apical Junctional Complex and AMPK: A Promising Therapeutic Target for Epithelial/Endothelial Barrier Function?


Abstract
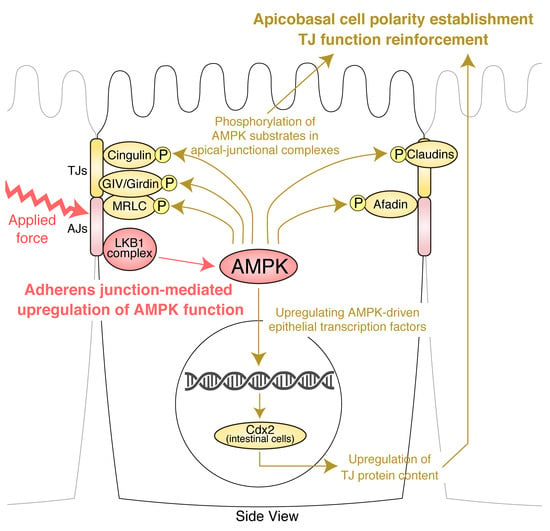
:
1. Introduction
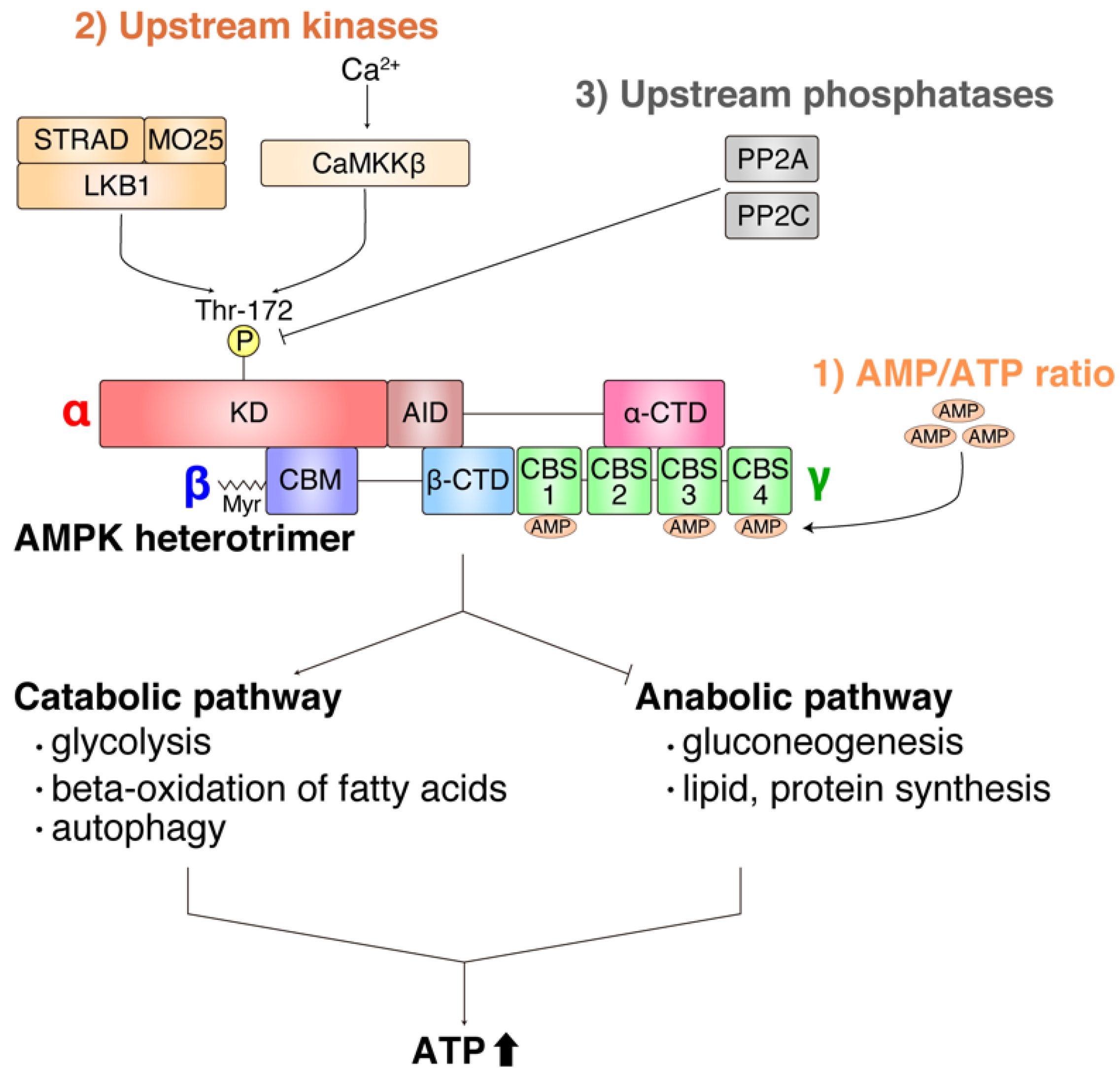
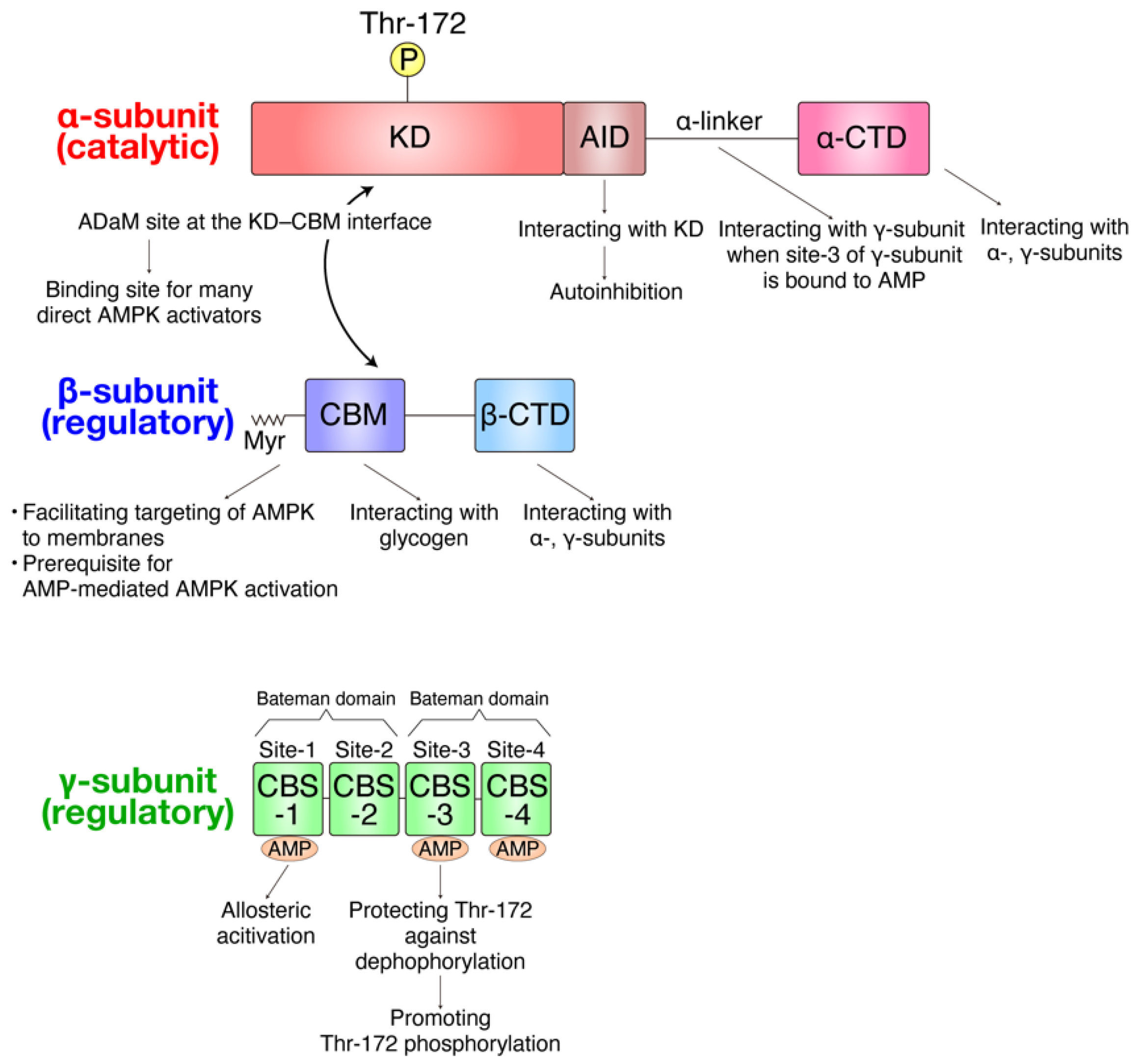
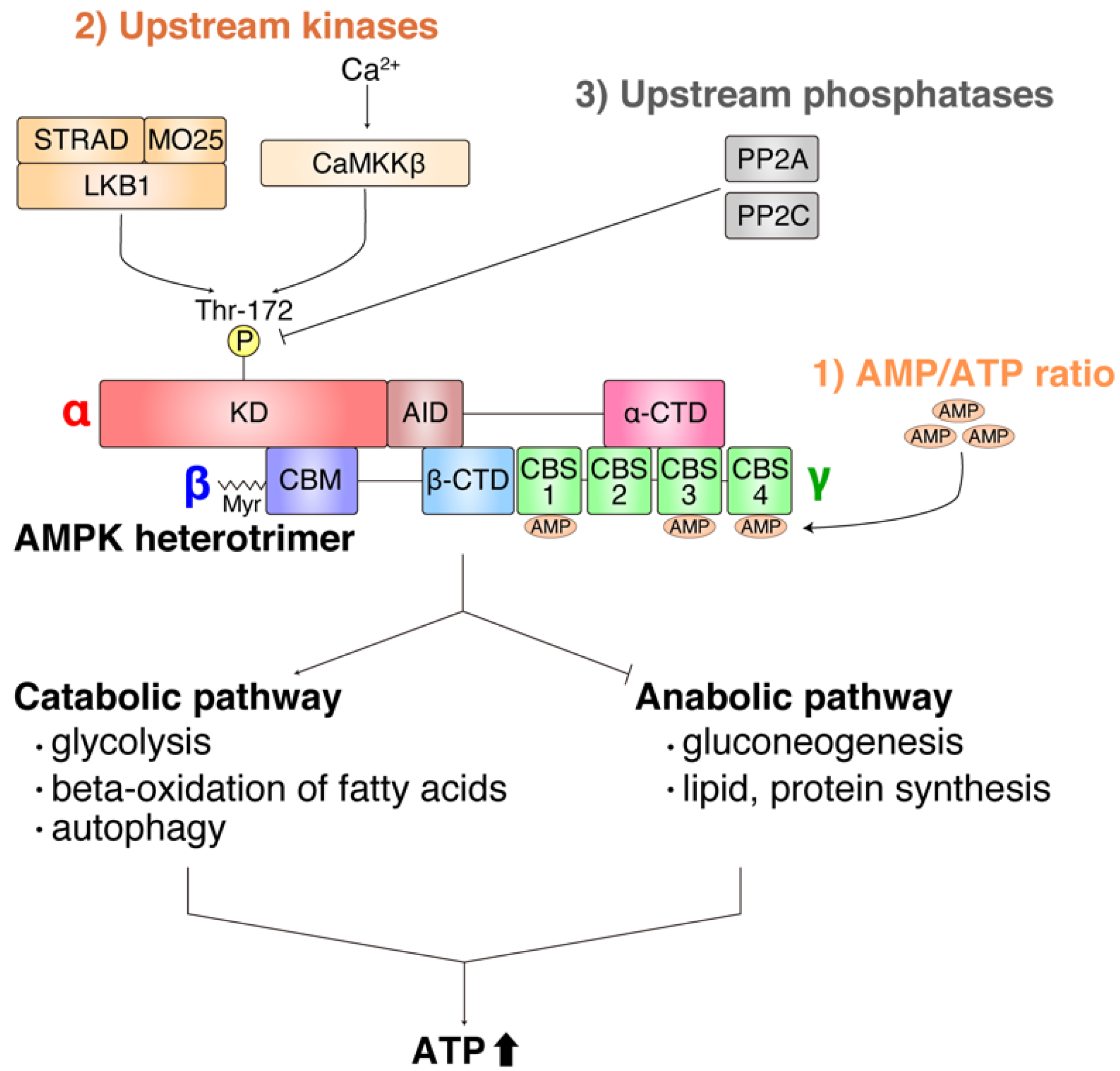
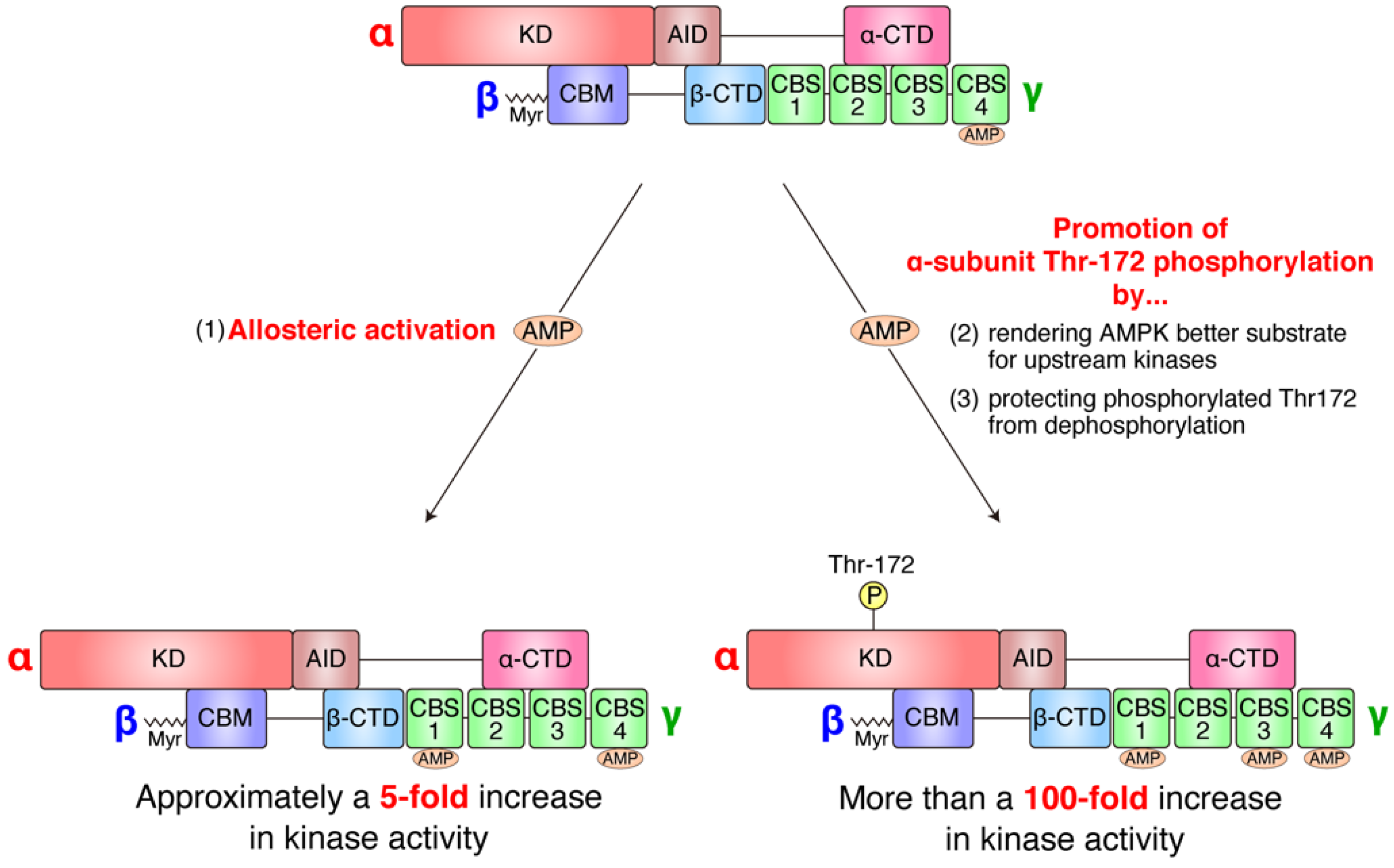
2. Overview of Function and Regulation of AMPK
3. Notable Findings in the Mid-2000s Established the Important Role of the LKB1–AMPK Axis in Apicobasal Epithelial Cell Polarity
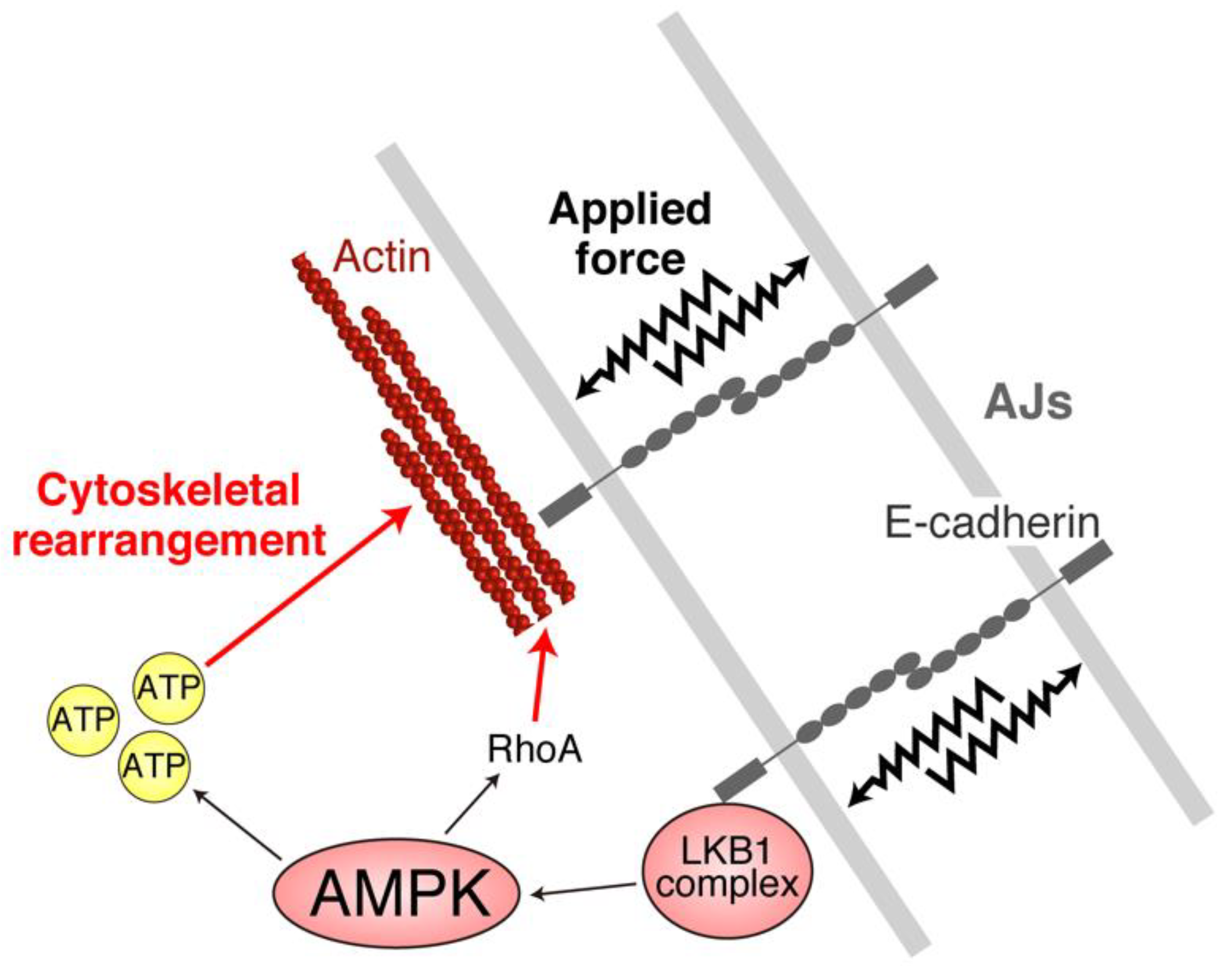
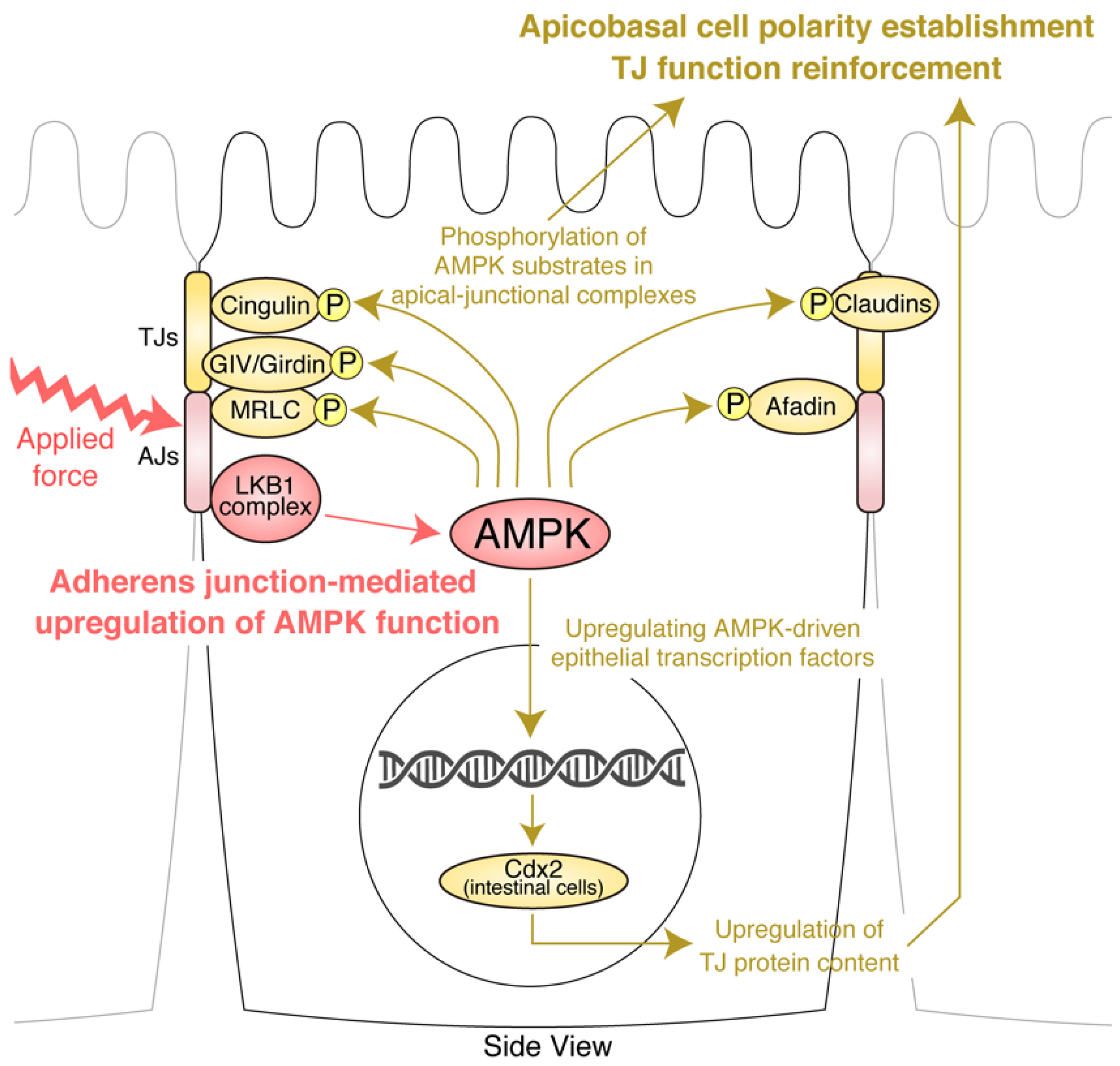
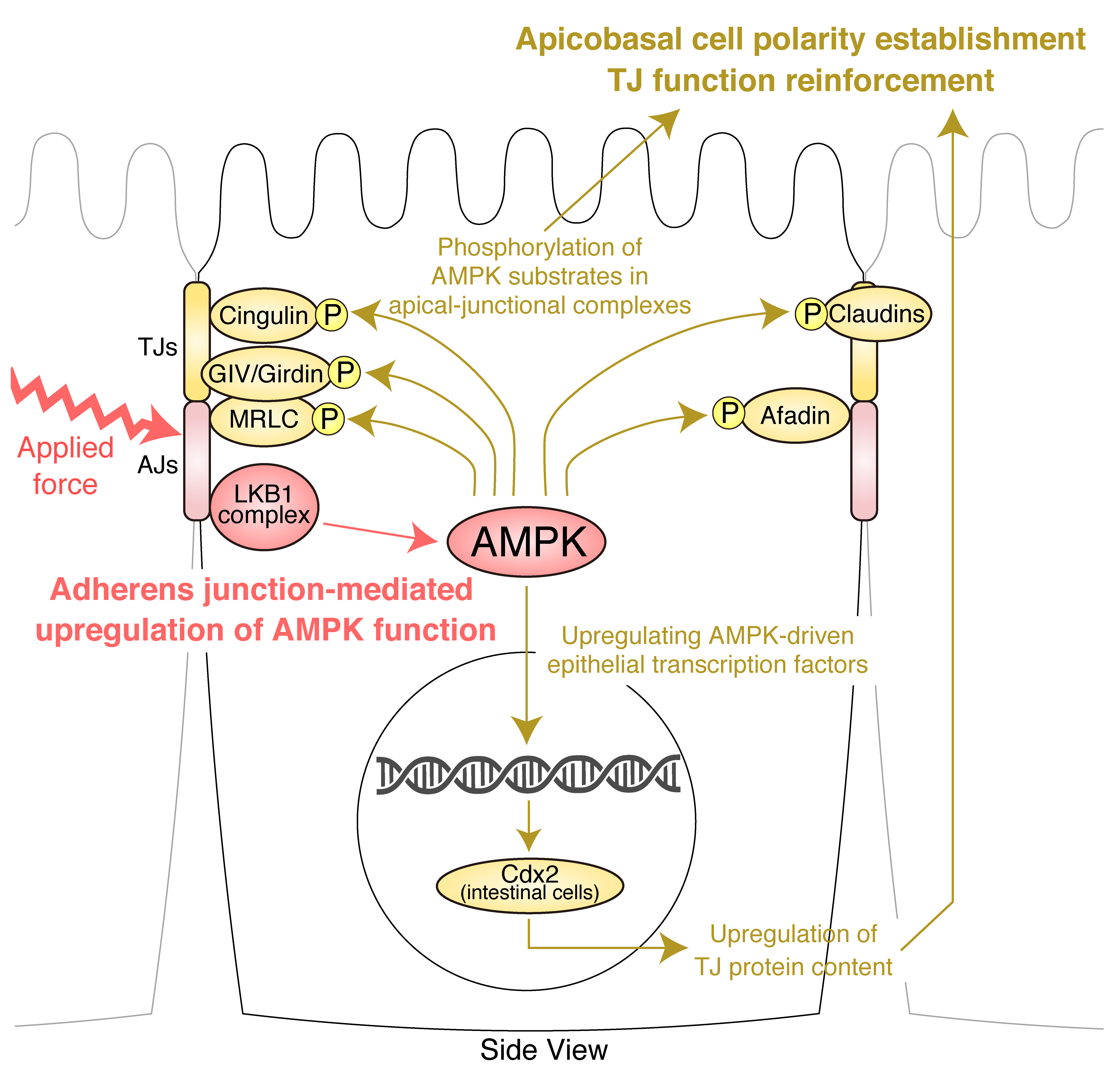
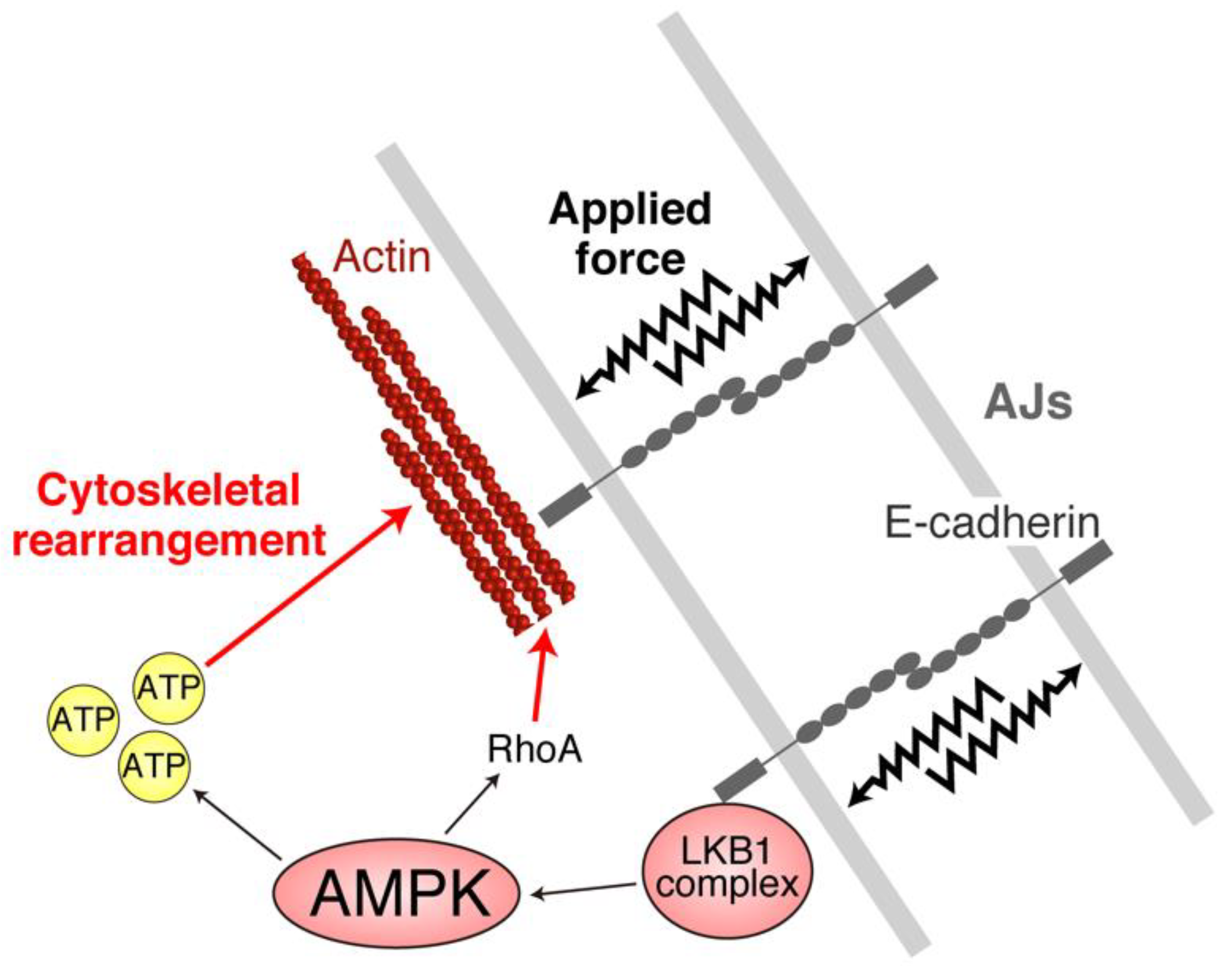
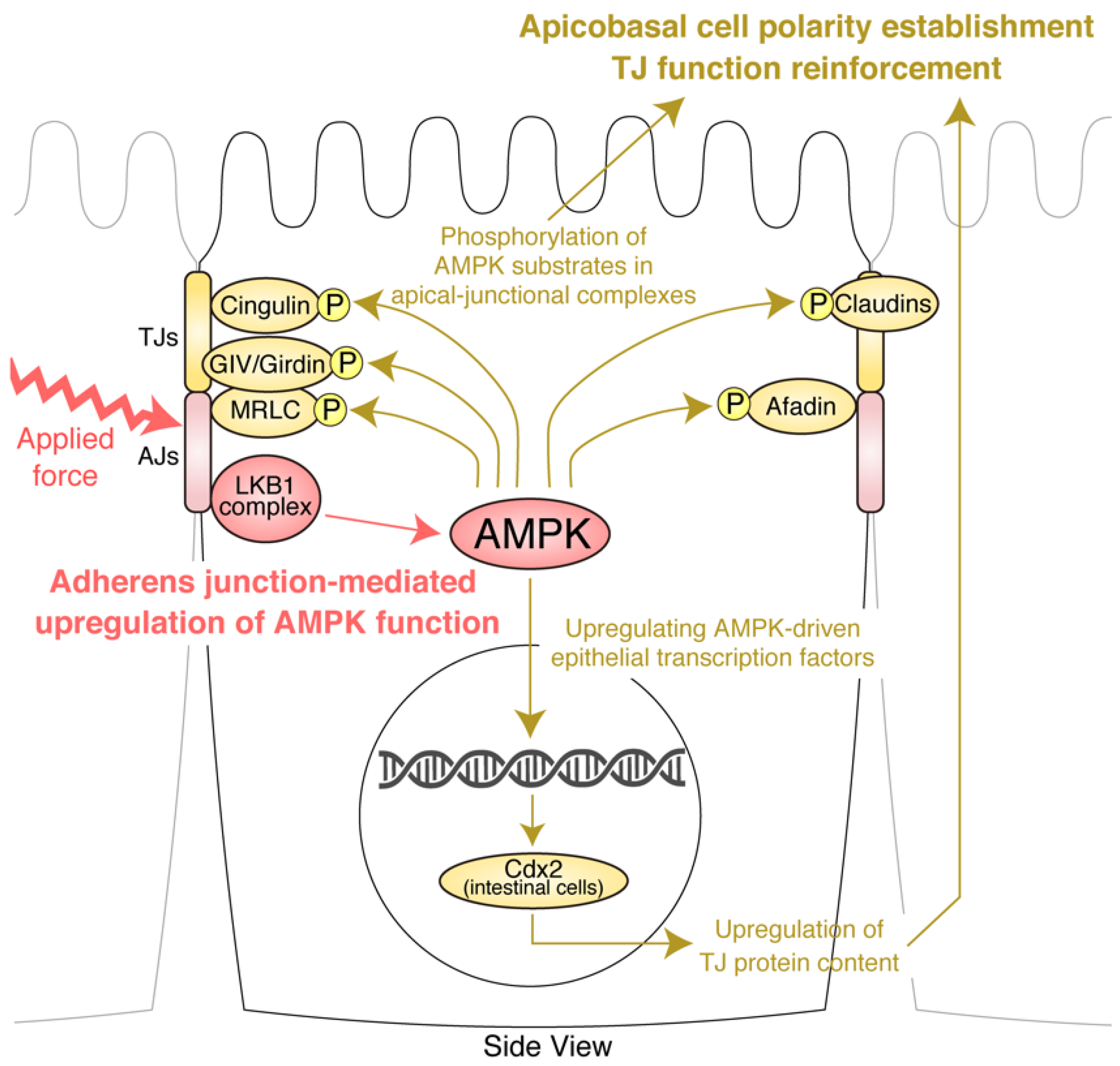
4. AJs and Junctional Tension Promote the LKB1–AMPK Axis to Provide Energy to Resist Applied Force
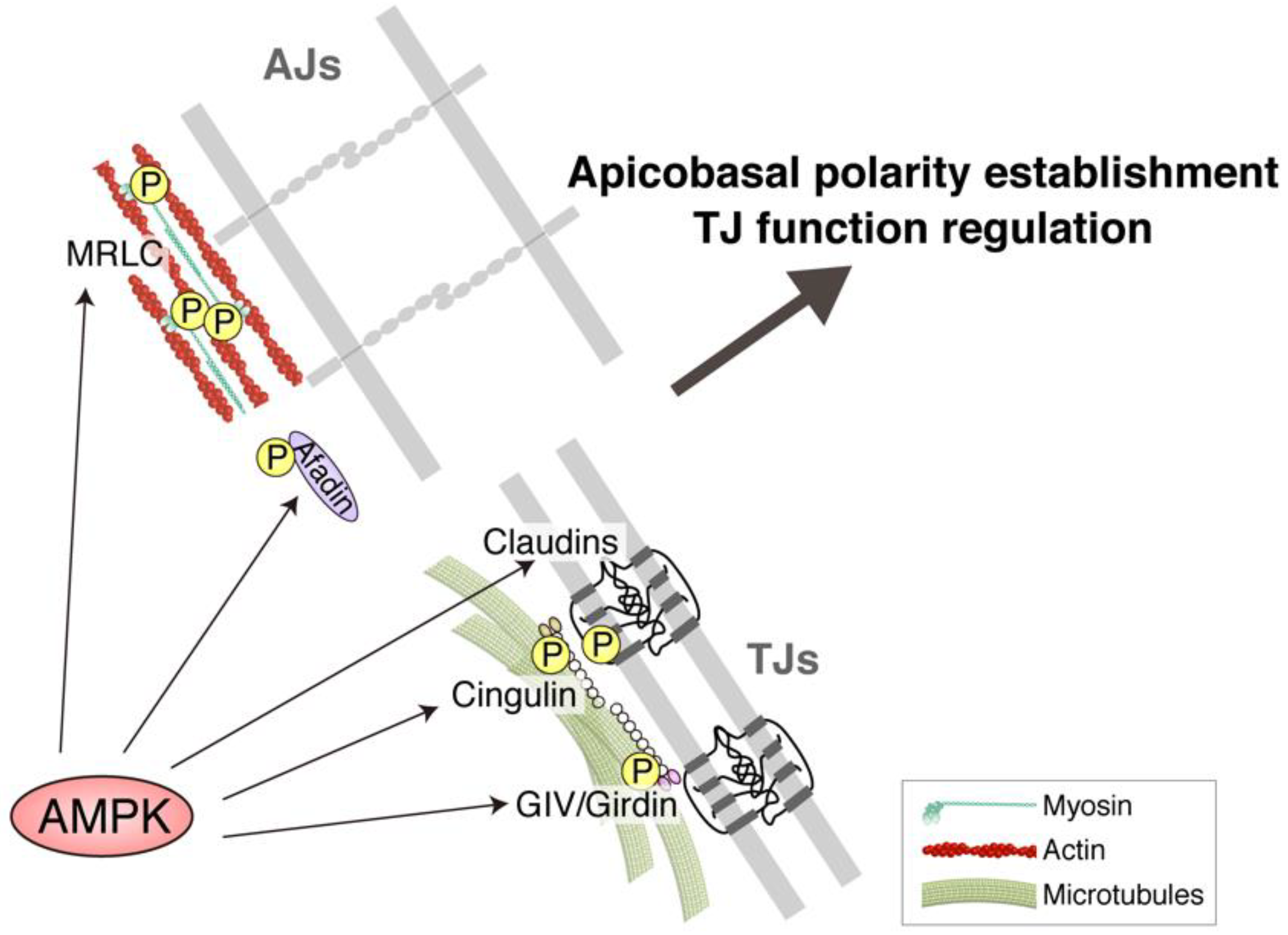
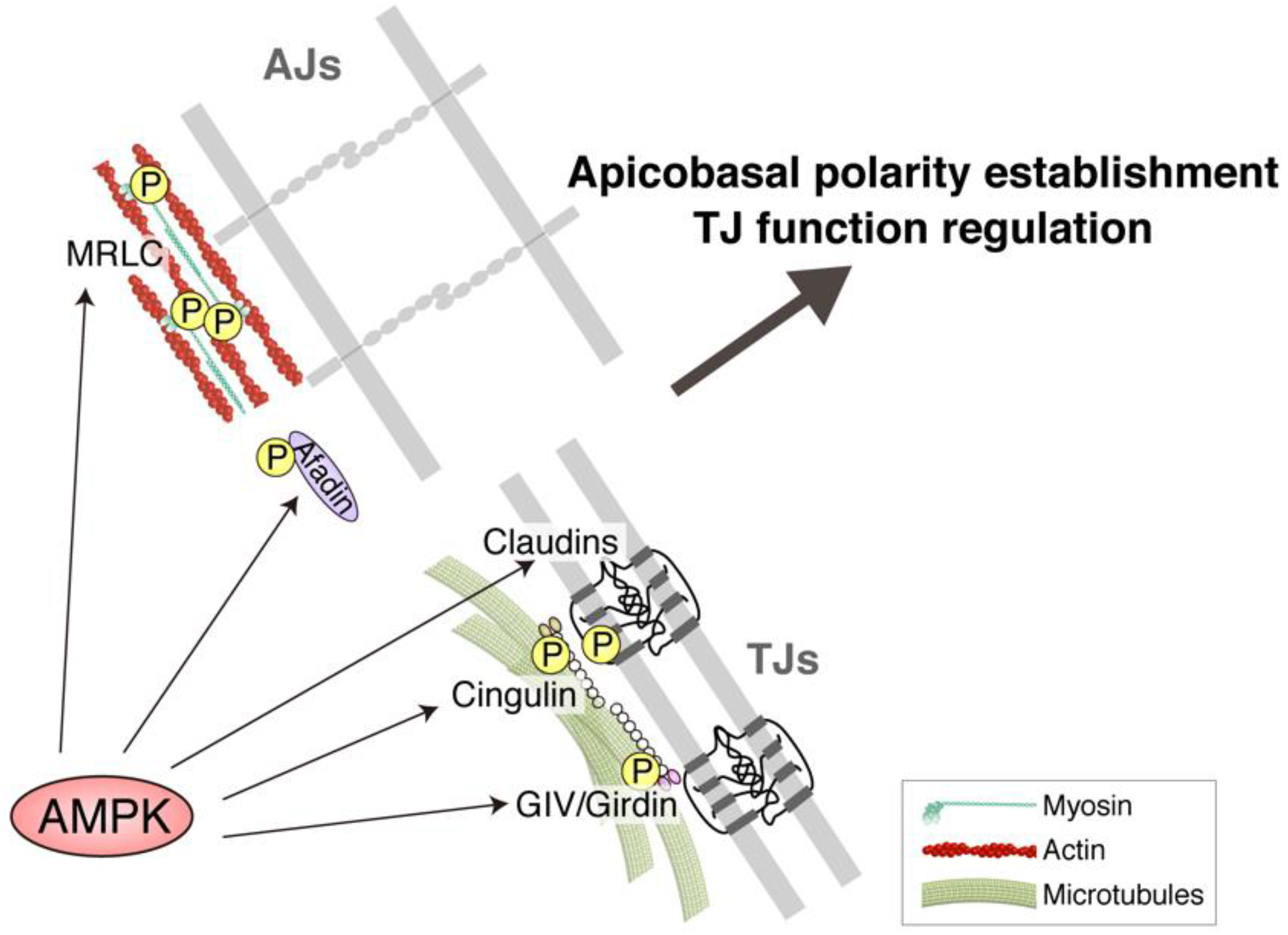
5. TJ Function Is Reinforced by AMPK
6. Molecular Basis of the Effects of AMPK on AJCs: Implication of Novel AMPK Effectors in AJCs and AMPK-Driven Epithelial Transcription Factors
7. Role of AMPK in Endothelial Barrier Function
8. Therapeutic Potential of Targeting AMPK for Manipulation of Epithelial Barrier Function
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TJs | Tight junctions |
| AJs | Adherens junctions |
| AMP | Adenosine monophosphate |
| AMPK | AMP-activated protein kinase |
| HMG-CoA | 3-hydroxy-3-methylglutaryl-coenzyme A |
| acetyl-CoA | Acetyl-coenzyme A |
| ATP | Adenosine triphosphate |
| LKB1 | Liver kinase B1 |
| CaMKKβ | Ca2+/calmodulin-dependent protein kinase kinase β |
| PJS | Peutz–Jeghers syndrome |
| MDCK | Madin–Darby canine kidney |
| FLCN | Folliculin |
| BHD | Birt–Hogg–Dubé |
| AICAR | 5-Aminoimidazole-4-carboxamide ribonucleoside |
| MRLC | Myosin regulatory light chain |
| GIV | G-alpha interacting vesicle associated protein |
| GEF | Guanine-nucleotide-exchange factor |
| CDX2 | Caudal type homeobox 2 |
| PPE | Purple potato extract |
| BBB | Blood–brain barrier |
| VE-cadherin | vascular endothelial-cadherin |
| PMVEC | Pulmonary microvascular endothelial cell |
| ZMP | 5-amino-1-β-d-ribofuranosylimidazole- 4-carboxamide-5′-monophosphate |
| LPS | Lipopolysaccharide |
References
- Tsukita, S.; Furuse, M.; Itoh, M. Multifunctional strands in tight junctions. Nat. Rev. Mol. Cell Biol. 2001, 2, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Tamura, A.; Yamazaki, Y.; Hayashi, D.; Suzuki, K.; Sentani, K.; Yasui, W.; Tsukita, S. Claudin-based paracellular proton barrier in the stomach. Ann. N. Y. Acad. Sci. 2012, 1258, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Tamura, A.; Tsukita, S. Paracellular barrier and channel functions of TJ claudins in organizing biological systems: Advances in the field of barriology revealed in knockout mice. Semin. Cell Dev. Biol. 2014, 36, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Tani, K.; Tamura, A.; Tsukita, S.; Fujiyoshi, Y. Model for the architecture of claudin-based paracellular ion channels through tight junctions. J. Mol. Biol. 2015, 427, 291–297. [Google Scholar] [CrossRef]
- Nakamura, S.; Irie, K.; Tanaka, H.; Nishikawa, K.; Suzuki, H.; Saitoh, Y.; Tamura, A.; Tsukita, S.; Fujiyoshi, Y. Morphologic determinant of tight junctions revealed by claudin-3 structures. Nat. Commun. 2019, 10, 816. [Google Scholar] [CrossRef]
- Tsukita, S.; Tanaka, H.; Tamura, A. The Claudins: From Tight Junctions to Biological Systems. Trends Biochem. Sci. 2019, 44, 141–152. [Google Scholar] [CrossRef]
- Tanaka, H.; Tamura, A.; Suzuki, K.; Tsukita, S. Site-specific distribution of claudin-based paracellular channels with roles in biological fluid flow and metabolism. Ann. N. Y. Acad. Sci. 2017, 1405, 44–52. [Google Scholar] [CrossRef]
- Wada, M.; Tamura, A.; Takahashi, N.; Tsukita, S. Loss of claudins 2 and 15 from mice causes defects in paracellular Na+ flow and nutrient transport in gut and leads to death from malnutrition. Gastroenterology 2013, 144, 369–380. [Google Scholar] [CrossRef]
- Matsumoto, K.; Imasato, M.; Yamazaki, Y.; Tanaka, H.; Watanabe, M.; Eguchi, H.; Nagano, H.; Hikita, H.; Tatsumi, T.; Takehara, T.; et al. Claudin 2 deficiency reduces bile flow and increases susceptibility to cholesterol gallstone disease in mice. Gastroenterology 2014, 147, 1134–1145.e10. [Google Scholar] [CrossRef]
- Tanaka, H.; Takechi, M.; Kiyonari, H.; Shioi, G.; Tamura, A.; Tsukita, S. Intestinal deletion of Claudin-7 enhances paracellular organic solute flux and initiates colonic inflammation in mice. Gut 2015, 64, 1529–1538. [Google Scholar] [CrossRef]
- Tokumasu, R.; Yamaga, K.; Yamazaki, Y.; Murota, H.; Suzuki, K.; Tamura, A.; Bando, K.; Furuta, Y.; Katayama, I.; Tsukita, S. Dose-dependent role of claudin-1 in vivo in orchestrating features of atopic dermatitis. Proc. Natl. Acad. Sci. USA 2016, 113, E4061–E4068. [Google Scholar] [CrossRef] [PubMed]
- Citi, S. Intestinal barriers protect against disease. Science 2018, 359, 1097–1098. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Imasato, M.; Yamazaki, Y.; Matsumoto, K.; Kunimoto, K.; Delpierre, J.; Meyer, K.; Zerial, M.; Kitamura, N.; Watanabe, M.; et al. Claudin-3 regulates bile canalicular paracellular barrier and cholesterol gallstone core formation in mice. J. Hepatol. 2018, 69, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Sentani, K.; Tanaka, H.; Yano, T.; Suzuki, K.; Oshima, M.; Yasui, W.; Tamura, A.; Tsukita, S. Deficiency of Stomach-Type Claudin-18 in Mice Induces Gastric Tumor Formation Independent of H pylori Infection. Cell. Mol. Gastroenterol. Hepatol. 2019, 8, 119–142. [Google Scholar] [CrossRef]
- Nalle, S.C.; Zuo, L.; Ong, M.L.D.M.; Singh, G.; Worthylake, A.M.; Choi, W.; Manresa, M.C.; Southworth, A.P.; Edelblum, K.L.; Baker, G.J.; et al. Graft-versus-host disease propagation depends on increased intestinal epithelial tight junction permeability. J. Clin. Investig. 2019, 129, 902–914. [Google Scholar] [CrossRef]
- Graham, W.V.; He, W.; Marchiando, A.M.; Zha, J.; Singh, G.; Li, H.-S.; Biswas, A.; Ong, M.L.D.M.; Jiang, Z.-H.; Choi, W.; et al. Intracellular MLCK1 diversion reverses barrier loss to restore mucosal homeostasis. Nat. Med. 2019, 25, 690–700. [Google Scholar] [CrossRef]
- Kuo, W.-T.; Shen, L.; Zuo, L.; Shashikanth, N.; Ong, M.L.D.M.; Wu, L.; Zha, J.; Edelblum, K.L.; Wang, Y.; Wang, Y.; et al. Inflammation-induced Occludin Downregulation Limits Epithelial Apoptosis by Suppressing Caspase-3 Expression. Gastroenterology 2019, 157, 1323–1337. [Google Scholar] [CrossRef]
- Hagen, S.J.; Ang, L.-H.; Zheng, Y.; Karahan, S.N.; Wu, J.; Wang, Y.E.; Caron, T.J.; Gad, A.P.; Muthupalani, S.; Fox, J.G. Loss of Tight Junction Protein Claudin 18 Promotes Progressive Neoplasia Development in Mouse Stomach. Gastroenterology 2018, 155, 1852–1867. [Google Scholar] [CrossRef]
- Shah, J.; Rouaud, F.; Guerrera, D.; Vasileva, E.; Popov, L.M.; Kelley, W.L.; Rubinstein, E.; Carette, J.E.; Amieva, M.R.; Citi, S. A Dock-and-Lock Mechanism Clusters ADAM10 at Cell-Cell Junctions to Promote α-Toxin Cytotoxicity. Cell Rep. 2018, 25, 2132–2147.e7. [Google Scholar] [CrossRef]
- King, A.J.; Siegel, M.; He, Y.; Nie, B.; Wang, J.; Koo-McCoy, S.; Minassian, N.A.; Jafri, Q.; Pan, D.; Kohler, J.; et al. Inhibition of sodium/hydrogen exchanger 3 in the gastrointestinal tract by tenapanor reduces paracellular phosphate permeability. Sci. Transl. Med. 2018, 10, eaam6474. [Google Scholar] [CrossRef]
- Clairembault, T.; Leclair-Visonneau, L.; Coron, E.; Bourreille, A.; Le Dily, S.; Vavasseur, F.; Heymann, M.-F.; Neunlist, M.; Derkinderen, P. Structural alterations of the intestinal epithelial barrier in Parkinson’s disease. Acta Neuropathol. Commun. 2015, 3, 12. [Google Scholar] [CrossRef]
- Montagne, A.; Zhao, Z.; Zlokovic, B.V. Alzheimer’s disease: A matter of blood-brain barrier dysfunction? J. Exp. Med. 2017, 214, 3151–3169. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.; Kealy, J.; Humphries, M.M.; Gong, Y.; Hou, J.; Hudson, N.; Cassidy, L.M.; Martiniano, R.; Shashi, V.; Hooper, S.R.; et al. Dose-dependent expression of claudin-5 is a modifying factor in schizophrenia. Mol. Psychiatry 2018, 23, 2156–2166. [Google Scholar] [CrossRef] [PubMed]
- Tsukita, K.; Taguchi, T.; Sakamaki-Tsukita, H.; Tanaka, K.; Suenaga, T. The vagus nerve becomes smaller in patients with Parkinson’s disease: A preliminary cross-sectional study using ultrasonography. Parkinsonism Relat. Disord. 2018, 55, 148–149. [Google Scholar] [CrossRef] [PubMed]
- Tsukita, K.; Sakamaki-Tsukita, H.; Tanaka, K.; Suenaga, T.; Takahashi, R. Value of in vivo α-synuclein deposits in Parkinson’s disease: A systematic review and meta-analysis. Mov. Disord. 2019, 34, 1452–1463. [Google Scholar] [CrossRef] [PubMed]
- Van IJzendoorn, S.C.D.; Derkinderen, P. The Intestinal Barrier in Parkinson’s Disease: Current State of Knowledge. J. Parkinsons Dis. 2019, 9, S323–S329. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, R.; Günzel, D.; Theune, D.; Czichos, C.; Schulzke, J.-D.; Fromm, M. Water channels and barriers formed by claudins. Ann. N. Y. Acad. Sci. 2017, 1397, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Konishi, S.; Yano, T.; Tanaka, H.; Mizuno, T.; Kanoh, H.; Tsukita, K.; Namba, T.; Tamura, A.; Yonemura, S.; Gotoh, S.; et al. Vinculin is critical for the robustness of the epithelial cell sheet paracellular barrier for ions. Life Sci. Alliance 2019, 2, e201900414. [Google Scholar] [CrossRef]
- Yonemura, S.; Wada, Y.; Watanabe, T.; Nagafuchi, A.; Shibata, M. alpha-Catenin as a tension transducer that induces adherens junction development. Nat. Cell Biol. 2010, 12, 533–542. [Google Scholar] [CrossRef]
- Zihni, C.; Mills, C.; Matter, K.; Balda, M.S. Tight junctions: From simple barriers to multifunctional molecular gates. Nat. Rev. Mol. Cell Biol. 2016, 17, 564–580. [Google Scholar] [CrossRef]
- Yano, T.; Kanoh, H.; Tamura, A.; Tsukita, S. Apical cytoskeletons and junctional complexes as a combined system in epithelial cell sheets. Ann. N. Y. Acad. Sci. 2017, 1405, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Citi, S. The mechanobiology of tight junctions. Biophys. Rev. In press. [CrossRef] [PubMed]
- Baas, A.F.; Kuipers, J.; van der Wel, N.N.; Batlle, E.; Koerten, H.K.; Peters, P.J.; Clevers, H.C. Complete polarization of single intestinal epithelial cells upon activation of LKB1 by STRAD. Cell 2004, 116, 457–466. [Google Scholar] [CrossRef] [Green Version]
- Forcet, C.; Etienne-Manneville, S.; Gaude, H.; Fournier, L.; Debilly, S.; Salmi, M.; Baas, A.; Olschwang, S.; Clevers, H.; Billaud, M. Functional analysis of Peutz-Jeghers mutations reveals that the LKB1 C-terminal region exerts a crucial role in regulating both the AMPK pathway and the cell polarity. Hum. Mol. Genet. 2005, 14, 1283–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Li, J.; Young, L.H.; Caplan, M.J. AMP-activated protein kinase regulates the assembly of epithelial tight junctions. Proc. Natl. Acad. Sci. USA 2006, 103, 17272–17277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Koh, H.; Kim, M.; Kim, Y.; Lee, S.Y.; Karess, R.E.; Lee, S.-H.; Shong, M.; Kim, J.-M.; Kim, J.; et al. Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature 2007, 447, 1017–1020. [Google Scholar] [CrossRef]
- Zheng, B.; Cantley, L.C. Regulation of epithelial tight junction assembly and disassembly by AMP-activated protein kinase. Proc. Natl. Acad. Sci. USA 2007, 104, 819–822. [Google Scholar] [CrossRef] [Green Version]
- Sebbagh, M.; Santoni, M.-J.; Hall, B.; Borg, J.-P.; Schwartz, M.A. Regulation of LKB1/STRAD localization and function by E-cadherin. Curr. Biol. 2009, 19, 37–42. [Google Scholar] [CrossRef] [Green Version]
- Bays, J.L.; Campbell, H.K.; Heidema, C.; Sebbagh, M.; DeMali, K.A. Linking E-cadherin mechanotransduction to cell metabolism through force-mediated activation of AMPK. Nat. Cell Biol. 2017, 19, 724–731. [Google Scholar] [CrossRef] [Green Version]
- Rowart, P.; Wu, J.; Caplan, M.J.; Jouret, F. Implications of AMPK in the Formation of Epithelial Tight Junctions. Int. J. Mol. Sci. 2018, 19, 2040. [Google Scholar] [CrossRef] [Green Version]
- Beg, Z.H.; Allmann, D.W.; Gibson, D.M. Modulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity with cAMP and wth protein fractions of rat liver cytosol. Biochem. Biophys. Res. Commun. 1973, 54, 1362–1369. [Google Scholar] [CrossRef]
- Carlson, C.A.; Kim, K.H. Regulation of hepatic acetyl coenzyme A carboxylase by phosphorylation and dephosphorylation. J. Biol. Chem. 1973, 248, 378–380. [Google Scholar] [CrossRef]
- Ferrer, A.; Caelles, C.; Massot, N.; Hegardt, F.G. Activation of rat liver cytosolic 3-hydroxy-3-methylglutaryl coenzyme A reductase kinase by adenosine 5′-monophosphate. Biochem. Biophys. Res. Commun. 1985, 132, 497–504. [Google Scholar] [CrossRef]
- Carling, D.; Zammit, V.A.; Hardie, D.G. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 1987, 223, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Sim, A.T.; Hardie, D.G. The low activity of acetyl-CoA carboxylase in basal and glucagon-stimulated hepatocytes is due to phosphorylation by the AMP-activated protein kinase and not cyclic AMP-dependent protein kinase. FEBS Lett. 1988, 233, 294–298. [Google Scholar] [CrossRef] [Green Version]
- Grahame Hardie, D.; Carling, D.; Sim, A.T.R. The AMP-activated protein kinase: A multisubstrate regulator of lipid metabolism. Trends Biochem. Sci. 1989, 14, 20–23. [Google Scholar] [CrossRef]
- Carling, D. The AMP-activated protein kinase cascade—A unifying system for energy control. Trends Biochem. Sci. 2004, 29, 18–24. [Google Scholar] [CrossRef]
- Xiao, B.; Heath, R.; Saiu, P.; Leiper, F.C.; Leone, P.; Jing, C.; Walker, P.A.; Haire, L.; Eccleston, J.F.; Davis, C.T.; et al. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 2007, 449, 496–500. [Google Scholar] [CrossRef]
- Xiao, B.; Sanders, M.J.; Underwood, E.; Heath, R.; Mayer, F.V.; Carmena, D.; Jing, C.; Walker, P.A.; Eccleston, J.F.; Haire, L.F.; et al. Structure of mammalian AMPK and its regulation by ADP. Nature 2011, 472, 230–233. [Google Scholar] [CrossRef] [Green Version]
- Langendorf, C.G.; Ngoei, K.R.W.; Scott, J.W.; Ling, N.X.Y.; Issa, S.M.A.; Gorman, M.A.; Parker, M.W.; Sakamoto, K.; Oakhill, J.S.; Kemp, B.E. Structural basis of allosteric and synergistic activation of AMPK by furan-2-phosphonic derivative C2 binding. Nat. Commun. 2016, 7, 10912. [Google Scholar] [CrossRef]
- Salt, I.P.; Hardie, D.G. AMP-Activated Protein Kinase: An Ubiquitous Signaling Pathway With Key Roles in the Cardiovascular System. Circ. Res. 2017, 120, 1825–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Jiao, Z.-H.; Zheng, L.-S.; Zhang, Y.-Y.; Xie, S.-T.; Wang, Z.-X.; Wu, J.-W. Structural insight into the autoinhibition mechanism of AMP-activated protein kinase. Nature 2009, 459, 1146–1149. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, L.; Zhou, X.E.; Ke, J.; de Waal, P.W.; Gu, X.; Tan, M.H.E.; Wang, D.; Wu, D.; Xu, H.E.; et al. Structural basis of AMPK regulation by adenine nucleotides and glycogen. Cell Res. 2015, 25, 50–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oakhill, J.S.; Chen, Z.-P.; Scott, J.W.; Steel, R.; Castelli, L.A.; Ling, N.; Macaulay, S.L.; Kemp, B.E. β-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK). Proc. Natl. Acad. Sci. USA 2010, 107, 19237–19241. [Google Scholar] [CrossRef] [Green Version]
- Oligschlaeger, Y.; Miglianico, M.; Chanda, D.; Scholz, R.; Thali, R.F.; Tuerk, R.; Stapleton, D.I.; Gooley, P.R.; Neumann, D. The recruitment of AMP-activated protein kinase to glycogen is regulated by autophosphorylation. J. Biol. Chem. 2015, 290, 11715–11728. [Google Scholar] [CrossRef] [Green Version]
- Xiao, B.; Sanders, M.J.; Carmena, D.; Bright, N.J.; Haire, L.F.; Underwood, E.; Patel, B.R.; Heath, R.B.; Walker, P.A.; Hallen, S.; et al. Structural basis of AMPK regulation by small molecule activators. Nat. Commun. 2013, 4, 3017. [Google Scholar] [CrossRef] [Green Version]
- Carling, D.; Thornton, C.; Woods, A.; Sanders, M.J. AMP-activated protein kinase: New regulation, new roles? Biochem. J. 2012, 445, 11–27. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Boudeau, J.; Reid, J.L.; Mustard, K.J.; Udd, L.; Mäkelä, T.P.; Alessi, D.R.; Hardie, D.G. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2003, 2, 28. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005, 2, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Davies, S.P.; Helps, N.R.; Cohen, P.T.; Hardie, D.G. 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett. 1995, 377, 421–425. [Google Scholar]
- Cheung, P.C.; Salt, I.P.; Davies, S.P.; Hardie, D.G.; Carling, D. Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. Biochem. J. 2000, 346, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-P.; Leiper, F.C.; Woods, A.; Carling, D.; Carlson, M. Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proc. Natl. Acad. Sci. USA 2003, 100, 8839–8843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oakhill, J.S.; Steel, R.; Chen, Z.-P.; Scott, J.W.; Ling, N.; Tam, S.; Kemp, B.E. AMPK is a direct adenylate charge-regulated protein kinase. Science 2011, 332, 1433–1435. [Google Scholar] [CrossRef] [PubMed]
- Sanders, M.J.; Grondin, P.O.; Hegarty, B.D.; Snowden, M.A.; Carling, D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem. J. 2007, 403, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Dite, T.A.; Langendorf, C.G.; Hoque, A.; Galic, S.; Rebello, R.J.; Ovens, A.J.; Lindqvist, L.M.; Ngoei, K.R.W.; Ling, N.X.Y.; Furic, L.; et al. AMP-activated protein kinase selectively inhibited by the type II inhibitor SBI-0206965. J. Biol. Chem. 2018, 293, 8874–8885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wodarz, A. Establishing cell polarity in development. Nat. Cell Biol. 2002, 4, E39–E44. [Google Scholar] [CrossRef] [PubMed]
- Lizcano, J.M.; Göransson, O.; Toth, R.; Deak, M.; Morrice, N.A.; Boudeau, J.; Hawley, S.A.; Udd, L.; Mäkelä, T.P.; Hardie, D.G.; et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004, 23, 833–843. [Google Scholar] [CrossRef] [Green Version]
- Daniel, J.L.; Molish, I.R.; Robkin, L.; Holmsen, H. Nucleotide exchange between cytosolic ATP and F-actin-bound ADP may be a major energy-utilizing process in unstimulated platelets. Eur. J. Biochem. 1986, 156, 677–684. [Google Scholar] [CrossRef]
- Bernstein, B.W.; Bamburg, J.R. Actin-ATP hydrolysis is a major energy drain for neurons. J. Neurosci. 2003, 23, 1–6. [Google Scholar] [CrossRef]
- Borghi, N.; Sorokina, M.; Shcherbakova, O.G.; Weis, W.I.; Pruitt, B.L.; Nelson, W.J.; Dunn, A.R. E-cadherin is under constitutive actomyosin-generated tension that is increased at cell-cell contacts upon externally applied stretch. Proc. Natl. Acad. Sci. USA 2012, 109, 12568–12573. [Google Scholar] [CrossRef] [Green Version]
- Goncharova, E.A.; Goncharov, D.A.; James, M.L.; Atochina-Vasserman, E.N.; Stepanova, V.; Hong, S.-B.; Li, H.; Gonzales, L.; Baba, M.; Linehan, W.M.; et al. Folliculin controls lung alveolar enlargement and epithelial cell survival through E-cadherin, LKB1, and AMPK. Cell Rep. 2014, 7, 412–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, J.M. Twenty-first Bowditch lecture. The epithelial junction: Bridge, gate, and fence. Physiologist 1977, 20, 10–18. [Google Scholar] [PubMed]
- Umeda, K.; Ikenouchi, J.; Katahira-Tayama, S.; Furuse, K.; Sasaki, H.; Nakayama, M.; Matsui, T.; Tsukita, S.; Furuse, M.; Tsukita, S. ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell 2006, 126, 741–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikenouchi, J.; Umeda, K.; Tsukita, S.; Furuse, M.; Tsukita, S. Requirement of ZO-1 for the formation of belt-like adherens junctions during epithelial cell polarization. J. Cell Biol. 2007, 176, 779–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otani, T.; Nguyen, T.P.; Tokuda, S.; Sugihara, K.; Sugawara, T.; Furuse, K.; Miura, T.; Ebnet, K.; Furuse, M. Claudins and JAM-A coordinately regulate tight junction formation and epithelial polarity. J. Cell Biol. 2019, 218, 3372–3396. [Google Scholar] [CrossRef] [Green Version]
- Peng, L.; Li, Z.-R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J. Nutr. 2009, 139, 1619–1625. [Google Scholar] [CrossRef]
- Seo-Mayer, P.W.; Thulin, G.; Zhang, L.; Alves, D.S.; Ardito, T.; Kashgarian, M.; Caplan, M.J. Preactivation of AMPK by metformin may ameliorate the epithelial cell damage caused by renal ischemia. Am. J. Physiol. Renal Physiol. 2011, 301, F1346–F1357. [Google Scholar] [CrossRef] [Green Version]
- Spruss, A.; Kanuri, G.; Stahl, C.; Bischoff, S.C.; Bergheim, I. Metformin protects against the development of fructose-induced steatosis in mice: Role of the intestinal barrier function. Lab. Investig. 2012, 92, 1020–1032. [Google Scholar] [CrossRef] [Green Version]
- Garnett, J.P.; Baker, E.H.; Naik, S.; Lindsay, J.A.; Knight, G.M.; Gill, S.; Tregoning, J.S.; Baines, D.L. Metformin reduces airway glucose permeability and hyperglycaemia-induced Staphylococcus aureus load independently of effects on blood glucose. Thorax 2013, 68, 835–845. [Google Scholar] [CrossRef] [Green Version]
- Yano, T.; Matsui, T.; Tamura, A.; Uji, M.; Tsukita, S. The association of microtubules with tight junctions is promoted by cingulin phosphorylation by AMPK. J. Cell Biol. 2013, 203, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Park, H.-Y.; Kunitake, Y.; Hirasaki, N.; Tanaka, M.; Matsui, T. Theaflavins enhance intestinal barrier of Caco-2 Cell monolayers through the expression of AMP-activated protein kinase-mediated Occludin, Claudin-1, and ZO-1. Biosci. Biotechnol. Biochem. 2015, 79, 130–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Wu, Z.; Ji, Y.; Sun, K.; Dai, Z.; Wu, G. L-Glutamine Enhances Tight Junction Integrity by Activating CaMK Kinase 2-AMP-Activated Protein Kinase Signaling in Intestinal Porcine Epithelial Cells. J. Nutr. 2016, 146, 501–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patkee, W.R.A.; Carr, G.; Baker, E.H.; Baines, D.L.; Garnett, J.P. Metformin prevents the effects of Pseudomonas aeruginosa on airway epithelial tight junctions and restricts hyperglycaemia-induced bacterial growth. J. Cell. Mol. Med. 2016, 20, 758–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Y.; Zhang, H.; Sun, X.; Zhu, M.-J. Metformin Improves Ileal Epithelial Barrier Function in Interleukin-10 Deficient Mice. PLoS ONE 2016, 11, e0168670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Wang, J.; You, Q.; He, S.; Meng, Q.; Gao, J.; Wu, X.; Shen, Y.; Sun, Y.; Wu, X.; et al. Activating AMPK to Restore Tight Junction Assembly in Intestinal Epithelium and to Attenuate Experimental Colitis by Metformin. Front. Pharmacol. 2018, 9, 761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jouret, F.; Rinehart, J.; Sfakianos, J.; Mellman, I.; Lifton, R.P.; Young, L.H.; Caplan, M.J. AMP-activated protein kinase (AMPK) activation and glycogen synthase kinase-3β (GSK-3β) inhibition induce Ca2+-independent deposition of tight junction components at the plasma membrane. J. Biol. Chem. 2011, 286, 16879–16890. [Google Scholar] [CrossRef] [Green Version]
- Yamada, A.; Fujita, N.; Sato, T.; Okamoto, R.; Ooshio, T.; Hirota, T.; Morimoto, K.; Irie, K.; Takai, Y. Requirement of nectin, but not cadherin, for formation of claudin-based tight junctions in annexin II-knockdown MDCK cells. Oncogene 2006, 25, 5085–5102. [Google Scholar] [CrossRef] [Green Version]
- Brakeman, P.R.; Liu, K.D.; Shimizu, K.; Takai, Y.; Mostov, K.E. Nectin proteins are expressed at early stages of nephrogenesis and play a role in renal epithelial cell morphogenesis. Am. J. Physiol. Renal Physiol. 2009, 296, F564–F574. [Google Scholar] [CrossRef]
- Yano, T.; Torisawa, T.; Oiwa, K.; Tsukita, S. AMPK-dependent phosphorylation of cingulin reversibly regulates its binding to actin filaments and microtubules. Sci. Rep. 2018, 8, 15550. [Google Scholar] [CrossRef]
- Citi, S.; Sabanay, H.; Jakes, R.; Geiger, B.; Kendrick-Jones, J. Cingulin, a new peripheral component of tight junctions. Nature 1988, 333, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Mangan, A.J.; Sietsema, D.V.; Li, D.; Moore, J.K.; Citi, S.; Prekeris, R. Cingulin and actin mediate midbody-dependent apical lumen formation during polarization of epithelial cells. Nat. Commun. 2016, 7, 12426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, R.-L.; Mei, M.; Cong, X.; Li, J.; Zhang, Y.; Ding, C.; Wu, L.-L.; Yu, G.-Y. Claudin-4 is required for AMPK-modulated paracellular permeability in submandibular gland cells. J. Mol. Cell Biol. 2014, 6, 486–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiomi, R.; Shigetomi, K.; Inai, T.; Sakai, M.; Ikenouchi, J. CaMKII regulates the strength of the epithelial barrier. Sci. Rep. 2015, 5, 13262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aznar, N.; Patel, A.; Rohena, C.C.; Dunkel, Y.; Joosen, L.P.; Taupin, V.; Kufareva, I.; Farquhar, M.G.; Ghosh, P. AMP-activated protein kinase fortifies epithelial tight junctions during energetic stress via its effector GIV/Girdin. Elife 2016, 5, e20795. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, K.; Kakuwa, T.; Akimoto, K.; Koga, H.; Ohno, S. Regulation of epithelial cell polarity by PAR-3 depends on Girdin transcription and Girdin-Gαi3 signaling. J. Cell Sci. 2015, 128, 2244–2258. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Yang, Q.; Rogers, C.J.; Du, M.; Zhu, M.-J. AMPK improves gut epithelial differentiation and barrier function via regulating Cdx2 expression. Cell Death Differ. 2017, 24, 819–831. [Google Scholar] [CrossRef]
- Sun, X.; Du, M.; Navarre, D.A.; Zhu, M.-J. Purple Potato Extract Promotes Intestinal Epithelial Differentiation and Barrier Function by Activating AMP-Activated Protein Kinase. Mol. Nutr. Food Res. 2018, 62. [Google Scholar] [CrossRef]
- Creighton, J.; Jian, M.; Sayner, S.; Alexeyev, M.; Insel, P.A. Adenosine monophosphate-activated kinase alpha1 promotes endothelial barrier repair. FASEB J. 2011, 25, 3356–3365. [Google Scholar] [CrossRef] [Green Version]
- Castanares-Zapatero, D.; Bouleti, C.; Sommereyns, C.; Gerber, B.; Lecut, C.; Mathivet, T.; Horckmans, M.; Communi, D.; Foretz, M.; Vanoverschelde, J.-L.; et al. Connection between cardiac vascular permeability, myocardial edema, and inflammation during sepsis: Role of the α1AMP-activated protein kinase isoform. Crit. Care Med. 2013, 41, e411–e422. [Google Scholar] [CrossRef]
- Takata, F.; Dohgu, S.; Matsumoto, J.; Machida, T.; Kaneshima, S.; Matsuo, M.; Sakaguchi, S.; Takeshige, Y.; Yamauchi, A.; Kataoka, Y. Metformin induces up-regulation of blood-brain barrier functions by activating AMP-activated protein kinase in rat brain microvascular endothelial cells. Biochem. Biophys. Res. Commun. 2013, 433, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tang, G.; Li, Y.; Wang, Y.; Chen, X.; Gu, X.; Zhang, Z.; Wang, Y.; Yang, G.-Y. Metformin attenuates blood-brain barrier disruption in mice following middle cerebral artery occlusion. J. Neuroinflammation 2014, 11, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komarova, Y.A.; Kruse, K.; Mehta, D.; Malik, A.B. Protein Interactions at Endothelial Junctions and Signaling Mechanisms Regulating Endothelial Permeability. Circ. Res. 2017, 120, 179–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simionescu, M.; Simionescu, N.; Palade, G.E. Segmental differentiations of cell junctions in the vascular endothelium. The microvasculature. J. Cell Biol. 1975, 67, 863–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneeberger, E.E. Structure of intercellular junctions in different segments of the intrapulmonary vasculature. Ann. N. Y. Acad. Sci. 1982, 384, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Navarro, P.; Ruco, L.; Dejana, E. Differential localization of VE- and N-cadherins in human endothelial cells: VE-cadherin competes with N-cadherin for junctional localization. J. Cell Biol. 1998, 140, 1475–1484. [Google Scholar] [CrossRef] [PubMed]
- Gentil-dit-Maurin, A.; Oun, S.; Almagro, S.; Bouillot, S.; Courçon, M.; Linnepe, R.; Vestweber, D.; Huber, P.; Tillet, E. Unraveling the distinct distributions of VE- and N-cadherins in endothelial cells: A key role for p120-catenin. Exp. Cell Res. 2010, 316, 2587–2599. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, J.H.; Yu, Y.S.; Kim, D.H.; Kim, K.-W. Recruitment of pericytes and astrocytes is closely related to the formation of tight junction in developing retinal vessels. J. Neurosci. Res. 2009, 87, 653–659. [Google Scholar] [CrossRef]
- Hellström, M.; Gerhardt, H.; Kalén, M.; Li, X.; Eriksson, U.; Wolburg, H.; Betsholtz, C. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J. Cell Biol. 2001, 153, 543–553. [Google Scholar] [CrossRef] [Green Version]
- Jian, M.-Y.; Liu, Y.; Li, Q.; Wolkowicz, P.; Alexeyev, M.; Zmijewski, J.; Creighton, J. N-cadherin coordinates AMP kinase-mediated lung vascular repair. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L71–L85. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Ross, F.A.; Chevtzoff, C.; Green, K.A.; Evans, A.; Fogarty, S.; Towler, M.C.; Brown, L.J.; Ogunbayo, O.A.; Evans, A.M.; et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010, 11, 554–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fryer, L.G.D.; Parbu-Patel, A.; Carling, D. The Anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J. Biol. Chem. 2002, 277, 25226–25232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, J.; Lee, H.; Kim, S.; Park, J.; Ha, T. The anti-obesity effect of quercetin is mediated by the AMPK and MAPK signaling pathways. Biochem. Biophys. Res. Commun. 2008, 373, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef]
- Shen, Q.W.; Zhu, M.J.; Tong, J.; Ren, J.; Du, M. Ca 2+ /calmodulin-dependent protein kinase kinase is involved in AMP-activated protein kinase activation by α-lipoic acid in C2C12 myotubes. Am. J. Physiol. Cell Physiol. 2007, 293, C1395–C1403. [Google Scholar] [CrossRef]
- Corton, J.M.; Gillespie, J.G.; Hawley, S.A.; Hardie, D.G. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 1995, 229, 558–565. [Google Scholar] [CrossRef]
- Gómez-Galeno, J.E.; Dang, Q.; Nguyen, T.H.; Boyer, S.H.; Grote, M.P.; Sun, Z.; Chen, M.; Craigo, W.A.; van Poelje, P.D.; MacKenna, D.A.; et al. A Potent and Selective AMPK Activator That Inhibits de Novo Lipogenesis. ACS Med. Chem. Lett. 2010, 1, 478–482. [Google Scholar] [CrossRef] [Green Version]
- Cool, B.; Zinker, B.; Chiou, W.; Kifle, L.; Cao, N.; Perham, M.; Dickinson, R.; Adler, A.; Gagne, G.; Iyengar, R.; et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006, 3, 403–416. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, M.F.; Rajamohan, F.; Harris, M.S.; Caspers, N.L.; Magyar, R.; Withka, J.M.; Wang, H.; Borzilleri, K.A.; Sahasrabudhe, P.V.; Hoth, L.R.; et al. Structural Basis for AMPK Activation: Natural and Synthetic Ligands Regulate Kinase Activity from Opposite Poles by Different Molecular Mechanisms. Structure 2014, 22, 1161–1172. [Google Scholar] [CrossRef] [Green Version]
- Zadra, G.; Photopoulos, C.; Tyekucheva, S.; Heidari, P.; Weng, Q.P.; Fedele, G.; Liu, H.; Scaglia, N.; Priolo, C.; Sicinska, E.; et al. A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO Mol. Med. 2014, 6, 519–538. [Google Scholar] [CrossRef] [PubMed]
- Handa, N.; Takagi, T.; Saijo, S.; Kishishita, S.; Takaya, D.; Toyama, M.; Terada, T.; Shirouzu, M.; Suzuki, A.; Lee, S.; et al. Structural basis for compound C inhibition of the human AMP-activated protein kinase α2 subunit kinase domain. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.W.; Galic, S.; Graham, K.L.; Foitzik, R.; Ling, N.X.Y.; Dite, T.A.; Issa, S.M.A.; Langendorf, C.G.; Weng, Q.P.; Thomas, H.E.; et al. Inhibition of AMP-Activated Protein Kinase at the Allosteric Drug-Binding Site Promotes Islet Insulin Release. Chem. Biol. 2015, 22, 705–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Z.; Huang, L.; Yin, P.; Liu, F.; Liu, Y.; Zhang, Z.; Lin, J.; Zou, W.; Li, C. L-Arginine alleviates heat stress-induced intestinal epithelial barrier damage by promoting expression of tight junction proteins via the AMPK pathway. Mol. Biol. Rep. in press. [CrossRef] [PubMed] [Green Version]
- Farbood, Y.; Sarkaki, A.; Khalaj, L.; Khodagholi, F.; Badavi, M.; Ashabi, G. Targeting Adenosine Monophosphate-Activated Protein Kinase by Metformin Adjusts Post-Ischemic Hyperemia and Extracellular Neuronal Discharge in Transient Global Cerebral Ischemia. Microcirculation 2015, 22, 534–541. [Google Scholar] [CrossRef]
- Harmel, E.; Grenier, E.; Bendjoudi Ouadda, A.; El Chebly, M.; Ziv, E.; Beaulieu, J.F.; Sané, A.; Spahis, S.; Laville, M.; Levy, E. AMPK in the small intestine in normal and pathophysiological conditions. Endocrinology 2014, 155, 873–888. [Google Scholar] [CrossRef] [Green Version]
- Wojtaszewski, J.F.P.; Birk, J.B.; Frøsig, C.; Holten, M.; Pilegaard, H.; Dela, F. 5′AMP activated protein kinase expression in human skeletal muscle: Effects of strength training and type 2 diabetes. J. Physiol. 2005, 564, 563–573. [Google Scholar] [CrossRef] [Green Version]
- Birk, J.B.; Wojtaszewski, J.F.P. Predominant alpha2/beta2/gamma3 AMPK activation during exercise in human skeletal muscle. J. Physiol. 2006, 577, 1021–1032. [Google Scholar] [CrossRef]
- Thomson, D.M. The Role of AMPK in the Regulation of Skeletal Muscle Size, Hypertrophy, and Regeneration. Int. J. Mol. Sci. 2018, 19, 3125. [Google Scholar] [CrossRef] [Green Version]
- Tsou, P.; Zheng, B.; Hsu, C.-H.; Sasaki, A.T.; Cantley, L.C. A Fluorescent Reporter of AMPK Activity and Cellular Energy Stress. Cell Metab. 2011, 13, 476–486. [Google Scholar] [CrossRef] [Green Version]
- Konagaya, Y.; Terai, K.; Hirao, Y.; Takakura, K.; Imajo, M.; Kamioka, Y.; Sasaoka, N.; Kakizuka, A.; Sumiyama, K.; Asano, T.; et al. A Highly Sensitive FRET Biosensor for AMPK Exhibits Heterogeneous AMPK Responses among Cells and Organs. Cell Rep. 2017, 21, 2628–2638. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Mechanism | Isoform Selectivity | Reference | |
|---|---|---|---|---|
| Indirect activators | Metformin (biguanides) | Inhibiting the mitochondrial respiratory chain Complex I → Increase in AMP/ATP ratio | No | [111,112] |
| Troglitazone (thiazolidinediones) | Inhibiting the mitochondrial respiratory chain Complex I → Increase in AMP/ATP ratio | No | [112,113] | |
| Resveratrol (polyphenols) | Inhibiting F1F0–ATPase/ATP synthase → Increase in AMP/ATP ratio | No | [112,114] | |
| Quercetin (polyphenols) | Inhibiting F1F0–ATPase/ATP synthase → Increase in AMP/ATP ratio | No | [112,115] | |
| α-Lipoic acid | Increasing calcium level? | No | [116] | |
| Direct activators | 5-Aminoimidazole-4-carboxamide ribonucleoside (AICAR) | Prodrug, converted to 5-amino-1-β-d-ribofuranosylimidazole- 4-carboxamide-5′-monophosphate (ZMP) → ZMP binds to site-3 in the γ-subunit | No | [112,117] |
| Compound-13 (C-13) | Prodrug, converted to Compound-2 (C-2) → C-2 binds to the γ-subunit | α1 > α2 | [118] | |
| A-769662 | Binding to the allosteric drug and metabolite (ADaM) site → Allosteric activation | β1 > β2 | [56,119] | |
| Compound 991 | Binding to the ADaM site → Allosteric activation | β1 > β2 | [56] | |
| Salicylate | Binding to the ADaM site → Allosteric activation | β1 | [120] | |
| MT63-78 | Binding to the ADaM site → Allosteric activation | β1 | [121] | |
| Inhibitors | Compound C (dorsomorphin) | Binding to the α-subunit → Competitive inhibition of ATP | No | [111,122] |
| MT47-100 | Binding to the β2-subunit → Allosteric inhibition | β2 (β1 activation) | [123] | |
| SBI-0206965 | Binding to the α-subunit → Mixed-type inhibition of ATP | No | [65] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsukita, K.; Yano, T.; Tamura, A.; Tsukita, S. Reciprocal Association between the Apical Junctional Complex and AMPK: A Promising Therapeutic Target for Epithelial/Endothelial Barrier Function? Int. J. Mol. Sci. 2019, 20, 6012. https://doi.org/10.3390/ijms20236012
Tsukita K, Yano T, Tamura A, Tsukita S. Reciprocal Association between the Apical Junctional Complex and AMPK: A Promising Therapeutic Target for Epithelial/Endothelial Barrier Function? International Journal of Molecular Sciences. 2019; 20(23):6012. https://doi.org/10.3390/ijms20236012
Chicago/Turabian StyleTsukita, Kazuto, Tomoki Yano, Atsushi Tamura, and Sachiko Tsukita. 2019. "Reciprocal Association between the Apical Junctional Complex and AMPK: A Promising Therapeutic Target for Epithelial/Endothelial Barrier Function?" International Journal of Molecular Sciences 20, no. 23: 6012. https://doi.org/10.3390/ijms20236012
APA StyleTsukita, K., Yano, T., Tamura, A., & Tsukita, S. (2019). Reciprocal Association between the Apical Junctional Complex and AMPK: A Promising Therapeutic Target for Epithelial/Endothelial Barrier Function? International Journal of Molecular Sciences, 20(23), 6012. https://doi.org/10.3390/ijms20236012