Sphingolipid Metabolism: New Insight into Ceramide-Induced Lipotoxicity in Muscle Cells



Abstract
1. Introduction
2. Type 2 Diabetes Pathophysiology: From Physiological Insulin Signaling to Energy Homeostasis Disruption
3. Lipotoxicity and Muscle Insulin Resistance
3.1. Relationship Between FA and Insulin Resistance: Randle Cycle Hypothesis
3.2. Lack of Direct Link between FA and Insulin Resistance
3.3. Diacylglycerols
3.4. Ceramides
4. Ceramide Metabolism and Muscle Insulin Resistance
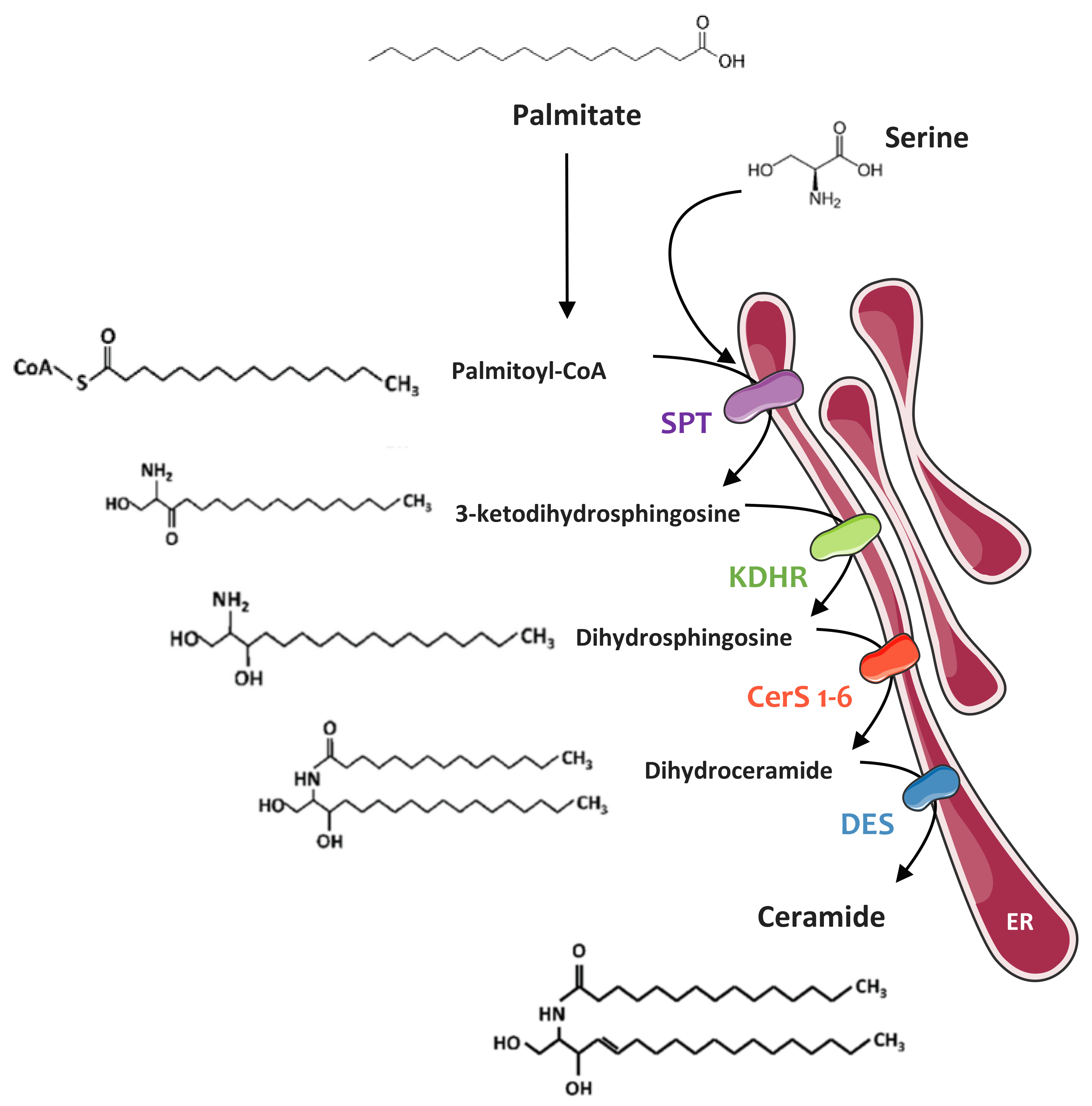
4.1. Sphingolipid Biosynthesis
4.1.1. De Novo Synthesis Pathway
4.1.2. Sphingomyelinase Pathway
4.1.3. Recycling Pathway
4.2. Ceramides and Muscle Insulin Resistance
4.2.1. In Vitro Studies
4.2.2. In Vivo Studies
4.2.3. Human Studies
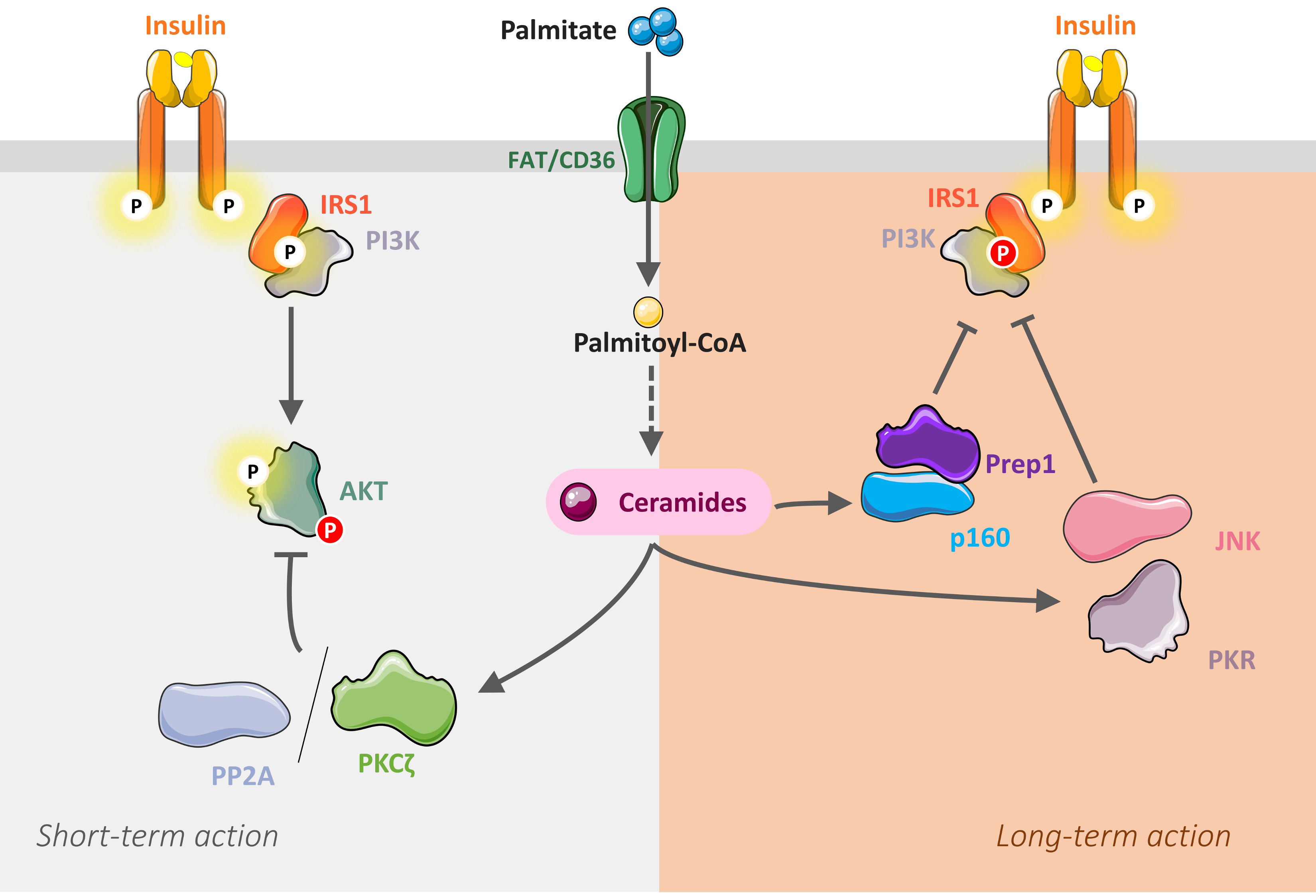
5. Mechanisms of Ceramide-Mediated Muscle Insulin Resistance
5.1. Inhibition of Akt by Ceramides
5.2. Inhibition of IRS1 by Ceramides
5.3. Importance of Ceramide Species in the Onset of Muscle Insulin Resistance
6. Ceramide vs DAG as Modulators of Muscle Insulin Sensitivity
7. Ceramide Lipid Derivatives and Muscle Insulin Resistance
7.1. Ceramide-1-phosphate
7.2. Sphingosine-1-phosphate
7.3. Complex Sphingolipids: Sphingomyelin and Glucosylceramides
7.3.1. Glucosylceramides
7.3.2. Sphingomyelin
8. Circulating Sphingolipids
9. Conclusion
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
Abbreviations
| aSMase | Acid sphingomyelinase |
| C1P | Ceramide-1-phosphate |
| CerS | Ceramide synthase |
| CERT | Ceramide transporter |
| DAG | Diacylglycerols |
| DES | Dihydroceramide Δ4-desaturase |
| FA | Fatty acids |
| FABP | Fatty acid binging protein |
| FAT | Fatty acid translocase |
| FATP | Fatty acid transport protein |
| G6P | Glucose-6-phosphate |
| GlcCer | Glucosylceramides |
| GLUT4 | Glucose transporter 4 |
| GM3 | Monosialodihexosylganglioside |
| GM3S | GM3 synthase |
| GS | Glycogen synthase |
| SK3 | Glycogen synthase kinase 3 |
| IR | Insulin receptor |
| IRS | Insulin receptor substrate |
| JNK | c-Jun NH2-terminal kinase |
| KDHR | 3-Ketodihydrosphingosine reductase |
| nSMase | Neutral sphingomyelinase |
| PC | Phosphatidylcholine |
| PE | Phosphatidylethanolamine |
| PFK | Phospho-fructo-kinase |
| PH | Pleckstrin homology |
| PI3K | Phosphoinositide-3-kinase |
| PKR | Double stranded ARN-activated protein kinase |
| PM | Plasma membrane |
| PP2A | Protein phosphatase 2A |
| ER | Endoplasmic reticulum |
| S1P | Sphingosine-1-phosphate |
| SL | Sphingolipids |
| SM | Sphingomyelin |
| MS | Sphingomyelin synthase |
| SphK | Sphingosine kinase |
| SPT | Serine palmitoyl transferase |
| T1D | Type 1 diabetes |
| T2D | Type 2 diabetes |
| TG | Triglyceride |
| TNFα | Tumor Necrosis Factor α |
References
- Boyle, J.P.; Thompson, T.J.; Gregg, E.W.; Barker, L.E.; Williamson, D.F. Projection of the year 2050 burden of diabetes in the US adult population: Dynamic modeling of incidence, mortality, and prediabetes prevalence. Popul. Health Metr. 2010, 8, 29. [Google Scholar] [CrossRef]
- White, J.R., Jr. Economic considerations in treating patients with type 2 diabetes mellitus. Am. J. Health Syst. Pharm. 2002, 59 (Suppl. S9), S14–S17. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.P.; Mesidor, M.; Winters, K.; Dubbert, P.M.; Wyatt, S.B. Overweight and Obesity: Prevalence, Consequences, and Causes of a Growing Public Health Problem. Curr. Obes. Rep. 2015, 4, 363–370. [Google Scholar] [CrossRef]
- Kwak, S.H.; Park, K.S. Recent progress in genetic and epigenetic research on type 2 diabetes. Exp. Mol. Med. 2016, 48, e220. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Jacot, E.; Jequier, E.; Maeder, E.; Wahren, J.; Felber, J.P. The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes 1981, 30, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Consitt, L.A.; Bell, J.A.; Houmard, J.A. Intramuscular lipid metabolism, insulin action, and obesity. IUBMB. Life 2009, 61, 47–55. [Google Scholar] [CrossRef]
- Katz, L.D.; Glickman, M.G.; Rapoport, S.; Ferrannini, E.; DeFronzo, R.A. Splanchnic and peripheral disposal of oral glucose in man. Diabetes 1983, 32, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.; Price, T.B.; Katz, L.D.; Shulman, R.G.; Shulman, G.I. Direct measurement of change in muscle glycogen concentration after a mixed meal in normal subjects. Am. J. Physiol. 1993, 265, E224–E229. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Ferrannini, E.; Sato, Y.; Felig, P.; Wahren, J. Synergistic interaction between exercise and insulin on peripheral glucose uptake. J. Clin. Investig. 1981, 68, 1468–1474. [Google Scholar] [CrossRef]
- White, M.F. The insulin signalling system and the IRS proteins. Diabetologia 1997, 40 (Suppl. S2), S2–S17. [Google Scholar] [CrossRef] [PubMed]
- White, M.F. The IRS-signalling system: A network of docking proteins that mediate insulin action. Mol. Cell Biochem. 1998, 182, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Lavin, D.P.; White, M.F.; Brazil, D.P. IRS proteins and diabetic complications. Diabetologia 2016, 59, 2280–2291. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef]
- Mardilovich, K.; Pankratz, S.L.; Shaw, L.M. Expression and function of the insulin receptor substrate proteins in cancer. Cell Commun. Signal. 2009, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Fantin, V.R.; Wang, Q.; Lienhard, G.E.; Keller, S.R. Mice lacking insulin receptor substrate 4 exhibit mild defects in growth, reproduction, and glucose homeostasis. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E127–E133. [Google Scholar] [CrossRef] [PubMed]
- Leevers, S.J.; Vanhaesebroeck, B.; Waterfield, M.D. Signalling through phosphoinositide 3-kinases: The lipids take centre stage. Curr. Opin. Cell Biol. 1999, 11, 219–225. [Google Scholar] [CrossRef]
- Lawlor, M.A.; Alessi, D.R. PKB/Akt: A key mediator of cell proliferation, survival and insulin responses? J. Cell Sci. 2001, 114, 2903–2910. [Google Scholar]
- Hajduch, E.; Litherland, G.J.; Hundal, H.S. Protein kinase B (PKB/Akt)—A key regulator of glucose transport? FEBS Lett. 2001, 492, 199–203. [Google Scholar] [CrossRef]
- Huang, X.; Liu, G.; Guo, J.; Su, Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int. J. Biol. Sci. 2018, 14, 1483–1496. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Alessi, D.R. The PI3K-PDK1 connection: More than just a road to PKB. Biochem. J. 2000, 346, 561–576. [Google Scholar]
- Litherland, G.J.; Hajduch, E.; Hundal, H.S. Intracellular signalling mechanisms regulating glucose transport in insulin-sensitive tissues (review). Mol. Membr. Biol. 2001, 18, 195–204. [Google Scholar] [PubMed]
- Coster, A.C.; Govers, R.; James, D.E. Insulin stimulates the entry of GLUT4 into the endosomal recycling pathway by a quantal mechanism. Traffic 2004, 5, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Czech, M.P. The GLUT4 glucose transporter. Cell Metab. 2007, 5, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Thorens, B. GLUT2, glucose sensing and glucose homeostasis. Diabetologia 2015, 58, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Ussher, J.R.; Koves, T.R.; Cadete, V.J.; Zhang, L.; Jaswal, J.S.; Swyrd, S.J.; Lopaschuk, D.G.; Proctor, S.D.; Keung, W.; Muoio, D.M.; et al. Inhibition of de novo ceramide synthesis reverses diet-induced insulin resistance and enhances whole-body oxygen consumption. Diabetes 2010, 59, 2453–2464. [Google Scholar] [CrossRef]
- Bruce, C.R.; Risis, S.; Babb, J.R.; Yang, C.; Kowalski, G.M.; Selathurai, A.; Lee-Young, R.S.; Weir, J.M.; Yoshioka, K.; Takuwa, Y.; et al. Overexpression of sphingosine kinase 1 prevents ceramide accumulation and ameliorates muscle insulin resistance in high-fat diet-fed mice. Diabetes 2012, 61, 3148–3155. [Google Scholar] [CrossRef]
- Hansen, D.; De, S.D.; Calders, P. Impact of Endurance Exercise Training in the Fasted State on Muscle Biochemistry and Metabolism in Healthy Subjects: Can These Effects be of Particular Clinical Benefit to Type 2 Diabetes Mellitus and Insulin-Resistant Patients? Sports Med. 2017, 47, 415–428. [Google Scholar] [CrossRef]
- Yang, J. Enhanced skeletal muscle for effective glucose homeostasis. Prog. Mol. Biol. Transl. Sci. 2014, 121, 133–163. [Google Scholar]
- Belfort, R.; Mandarino, L.; Kashyap, S.; Wirfel, K.; Pratipanawatr, T.; Berria, R.; DeFronzo, R.A.; Cusi, K. Dose-response effect of elevated plasma free fatty acid on insulin signaling. Diabetes 2005, 54, 1640–1648. [Google Scholar] [CrossRef]
- Nicholson, T.; Church, C.; Baker, D.J.; Jones, S.W. The role of adipokines in skeletal muscle inflammation and insulin sensitivity. J. Inflamm. 2018, 15, 9. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, L.K.; Smith, G.C.; Turner, N. Defining lipid mediators of insulin resistance—Controversies and challenges. J. Mol. Endocrinol. 2018, 2018. 62, R65–R82. [Google Scholar] [CrossRef]
- Summers, S.A. Ceramides in insulin resistance and lipotoxicity. Prog. Lipid Res. 2006, 45, 42–72. [Google Scholar] [CrossRef] [PubMed]
- Hage Hassan, R.; Bourron, O.; Hajduch, E. Defect of insulin signal in peripheral tissues: Important role of ceramide. World J. Diabetes 2014, 5, 244–257. [Google Scholar] [CrossRef] [PubMed]
- Goodpaster, B.H.; Theriault, R.; Watkins, S.C.; Kelley, D.E. Intramuscular lipid content is increased in obesity and decreased by weight loss. Metabolism 2000, 49, 467–472. [Google Scholar] [CrossRef]
- Malenfant, P.; Joanisse, D.R.; Theriault, R.; Goodpaster, B.H.; Kelley, D.E.; Simoneau, J.A. Fat content in individual muscle fibers of lean and obese subjects. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 1316–1321. [Google Scholar] [CrossRef] [PubMed]
- Roepstorff, C.; Helge, J.W.; Vistisen, B.; Kiens, B. Studies of plasma membrane fatty acid-binding protein and other lipid-binding proteins in human skeletal muscle. Proc. Nutr. Soc. 2004, 63, 239–244. [Google Scholar] [CrossRef]
- Bonen, A.; Parolin, M.L.; Steinberg, G.R.; Calles-Escandon, J.; Tandon, N.N.; Glatz, J.F.; Luiken, J.J.; Heigenhauser, G.J.; Dyck, D.J. Triacylglycerol accumulation in human obesity and type 2 diabetes is associated with increased rates of skeletal muscle fatty acid transport and increased sarcolemmal FAT/CD36. FASEB J. 2004, 18, 1144–1146. [Google Scholar] [CrossRef]
- Watt, M.J.; Hoy, A.J. Lipid metabolism in skeletal muscle: Generation of adaptive and maladaptive intracellular signals for cellular function. Am. J. Physiol. Endocrinol. Metab 2012, 302, E1315–E1328. [Google Scholar] [CrossRef]
- Martins, A.R.; Nachbar, R.T.; Gorjao, R.; Vinolo, M.A.; Festuccia, W.T.; Lambertucci, R.H.; Cury-Boaventura, M.F.; Silveira, L.R.; Curi, R.; Hirabara, S.M. Mechanisms underlying skeletal muscle insulin resistance induced by fatty acids: Importance of the mitochondrial function. Lipids Health Dis. 2012, 11, 30. [Google Scholar] [CrossRef]
- Rains, J.L.; Jain, S.K. Oxidative stress, insulin signaling, and diabetes. Free Radic. Biol. Med. 2011, 50, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar] [CrossRef] [PubMed]
- Hulver, M.W.; Berggren, J.R.; Cortright, R.N.; Dudek, R.W.; Thompson, R.P.; Pories, W.J.; Macdonald, K.G.; Cline, G.W.; Shulman, G.I.; Dohm, G.L.; et al. Skeletal muscle lipid metabolism with obesity. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E741–E747. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.E.; Parks, E.J. Postprandial metabolism of meal triglyceride in humans. Biochim. Biophys. Acta 2012, 1821, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 1, 785–789. [Google Scholar] [CrossRef]
- Roden, M.; Price, T.B.; Perseghin, G.; Petersen, K.F.; Rothman, D.L.; Cline, G.W.; Shulman, G.I. Mechanism of free fatty acid-induced insulin resistance in humans. J. Clin. Investig. 1996, 97, 2859–2865. [Google Scholar] [CrossRef]
- Anastasiou, C.A.; Kavouras, S.A.; Lentzas, Y.; Gova, A.; Sidossis, L.S.; Melidonis, A. Diabetes mellitus is associated with increased intramyocellular triglyceride, but not diglyceride, content in obese humans. Metabolism 2009, 58, 1636–1642. [Google Scholar] [CrossRef]
- Amati, F.; Dube, J.J.; Carnero, E.A.; Edreira, M.M.; Chomentowski, P.; Coen, P.M.; Switzer, G.E.; Bickel, P.E.; Stefanovic-Racic, M.; Toledo, F.G.; et al. Skeletal-Muscle Triglycerides, Diacylglycerols, and Ceramides in Insulin Resistance: Another Paradox in Endurance-Trained Athletes? Diabetes 2011, 60, 2588–2597. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Roles of Diacylglycerols and Ceramides in Hepatic Insulin Resistance. Trends Pharmacol. Sci. 2017, 38, 649–665. [Google Scholar] [CrossRef]
- Liu, L.; Yu, S.; Khan, R.S.; Ables, G.P.; Bharadwaj, K.G.; Hu, Y.; Huggins, L.A.; Eriksson, J.W.; Buckett, L.K.; Turnbull, A.V.; et al. DGAT1 deficiency decreases PPAR expression and does not lead to lipotoxicity in cardiac and skeletal muscle. J. Lipid Res. 2011, 52, 732–744. [Google Scholar] [CrossRef]
- Chibalin, A.V.; Leng, Y.; Vieira, E.; Krook, A.; Bjornholm, M.; Long, Y.C.; Kotova, O.; Zhong, Z.; Sakane, F.; Steiler, T.; et al. Downregulation of diacylglycerol kinase delta contributes to hyperglycemia-induced insulin resistance. Cell 2008, 132, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Brose, N.; Betz, A.; Wegmeyer, H. Divergent and convergent signaling by the diacylglycerol second messenger pathway in mammals. Curr. Opin. Neurobiol. 2004, 14, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Erion, D.M.; Shulman, G.I. Diacylglycerol-mediated insulin resistance. Nat. Med. 2010, 16, 400–402. [Google Scholar] [CrossRef] [PubMed]
- Turinsky, J.; O’Sullivan, D.M.; Bayly, B.P. 1,2-Diacylglycerol and ceramide levels in insulin-resistant tissues of the rat in vivo. J. Biol. Chem. 1990, 265, 16880–16885. [Google Scholar] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Castro, B.M.; Prieto, M.; Silva, L.C. Ceramide: A simple sphingolipid with unique biophysical properties. Prog. Lipid Res. 2014, 54, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.A.; Halverson-Tamboli, R.A.; Rasenick, M.M. Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosci. 2007, 8, 128–140. [Google Scholar] [CrossRef]
- Mullen, T.D.; Hannun, Y.A.; Obeid, L.M. Ceramide synthases at the centre of sphingolipid metabolism and biology. Biochem. J. 2012, 441, 789–802. [Google Scholar] [CrossRef]
- Colombini, M. Ceramide channels and their role in mitochondria-mediated apoptosis. Biochim. Biophys. Acta 2010, 1797, 1239–1244. [Google Scholar] [CrossRef]
- Spiegel, S.; Merrill, A.H., Jr. Sphingolipid metabolism and cell growth regulation. FASEB J. 1996, 10, 1388–1397. [Google Scholar] [CrossRef]
- Saddoughi, S.A.; Ogretmen, B. Diverse functions of ceramide in cancer cell death and proliferation. Adv. Cancer Res. 2013, 117, 37–58. [Google Scholar] [PubMed]
- Perry, D.K.; Hannun, Y.A. The role of ceramide in cell signaling. Biochim. Biophys. Acta 1998, 1436, 233–243. [Google Scholar] [CrossRef]
- Uchida, Y. Ceramide signaling in mammalian epidermis. Biochim. Biophys. Acta 2014, 1841, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Kollmeyer, J.; Symolon, H.; Momin, A.; Munter, E.; Wang, E.; Kelly, S.; Allegood, J.C.; Liu, Y.; Peng, Q.; et al. Ceramides and other bioactive sphingolipid backbones in health and disease: Lipidomic analysis, metabolism and roles in membrane structure, dynamics, signaling and autophagy. Biochim. Biophys. Acta 2006, 1758, 1864–1884. [Google Scholar] [CrossRef] [PubMed]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar] [PubMed]
- Kitatani, K.; Idkowiak-Baldys, J.; Hannun, Y.A. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell Signal. 2008, 20, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Schmitz-Peiffer, C.; Craig, D.L.; Biden, T.J. Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated PKB pathway in C2C12 skeletal muscle cells pretreated with palmitate. J. Biol. Chem. 1999, 274, 24202–24210. [Google Scholar] [CrossRef]
- Hajduch, E.; Alessi, D.R.; Hemmings, B.A.; Hundal, H.S. Constitutive activation of protein kinase B alpha by membrane targeting promotes glucose and system a amino acid transport, protein synthesis, and inactivation of glycogen synthase kinase 3 in L6 muscle cells. Diabetes 1998, 47, 1006–1013. [Google Scholar] [CrossRef]
- Hajduch, E.; Balendran, A.; Batty, I.H.; Litherland, G.J.; Blair, A.S.; Downes, C.P.; Hundal, H.S. Ceramide impairs the insulin-dependent membrane recruitment of protein kinase B leading to a loss in downstream signalling in L6 skeletal muscle cells. Diabetologia 2001, 44, 173–183. [Google Scholar] [CrossRef]
- Watson, M.L.; Coghlan, M.; Hundal, H.S. Modulating serine palmitoyl transferase (SPT) expression and activity unveils a crucial role in lipid-induced insulin resistance in rat skeletal muscle cells. Biochem. J. 2009, 417, 791–801. [Google Scholar] [CrossRef]
- Cazzolli, R.; Carpenter, L.; Biden, T.J.; Schmitz-Peiffer, C. A role for protein phosphatase 2A-like activity, but not atypical protein kinase Czeta, in the inhibition of protein kinase B/Akt and glycogen synthesis by palmitate. Diabetes 2001, 50, 2210–2218. [Google Scholar] [CrossRef] [PubMed]
- Bourbon, N.A.; Sandirasegarane, L.; Kester, M. Ceramide-induced inhibition of Akt is mediated through protein kinase Czeta: Implications for growth arrest. J. Biol. Chem. 2002, 277, 3286–3292. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.A.; Summers, S.A. Characterizing the effects of saturated fatty acids on insulin signaling and ceramide and diacylglycerol accumulation in 3T3-L1 adipocytes and C2C12 myotubes. Arch. Biochem. Biophys. 2003, 419, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.J.; Hajduch, E.; Kular, G.; Hundal, H.S. Ceramide Disables 3-Phosphoinositide Binding to the Pleckstrin Homology Domain of Protein Kinase B (PKB)/Akt by a PKCzeta-Dependent Mechanism. Mol. Cell Biol. 2003, 23, 7794–7808. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.J.; Turban, S.; Gray, A.; Hajduch, E.; Hundal, H.S. Intracellular ceramide synthesis and protein kinase Czeta activation play an essential role in palmitate-induced insulin resistance in rat L6 skeletal muscle cells. Biochem. J. 2004, 382, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Hajduch, E.; Turban, S.; Le Liepvre, X.; Le Lay, S.; Lipina, C.; Dimopoulos, N.; Dugail, I.; Hundal, H.S. Targeting of PKCzeta and PKB to caveolin-enriched microdomains represents a crucial step underpinning the disruption in PKB-directed signalling by ceramide. Biochem. J. 2008, 410, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Frangioudakis, G.; Diakanastasis, B.; Liao, B.Q.; Saville, J.T.; Hoffman, N.J.; Mitchell, T.W.; Schmitz-Peiffer, C. Ceramide accumulation in L6 skeletal muscle cells due to increased activity of ceramide synthase isoforms has opposing effects on insulin action to those caused by palmitate treatment. Diabetologia 2013, 56, 2697–2701. [Google Scholar] [CrossRef]
- Mahfouz, R.; Khoury, R.; Blachnio-Zabielska, A.; Turban, S.; Loiseau, N.; Lipina, C.; Stretton, C.; Bourron, O.; Ferre, P.; Foufelle, F.; et al. Characterising the Inhibitory Actions of Ceramide upon Insulin Signaling in Different Skeletal Muscle Cell Models: A Mechanistic Insight. PLoS. ONE 2014, 9, e101865. [Google Scholar] [CrossRef]
- Hage Hassan, R.; Pacheco de Sousa, A.C.; Mahfouz, R.; Hainault, I.; Blachnio-Zabielska, A.; Bourron, O.; Koskas, F.; Gorski, J.; Ferre, P.; Foufelle, F.; et al. Sustained Action of Ceramide on the Insulin Signaling Pathway in Muscle Cells: Implication of the double-stranded RNA-activated protein kinase. J. Biol. Chem. 2016, 291, 3019–3029. [Google Scholar] [CrossRef]
- Bandet, C.L.; Mahfouz, R.; Veret, J.; Sotiropoulos, A.; Poirier, M.; Giussani, P.; Campana, M.; Philippe, E.; Blachnio-Zabielska, A.; Ballaire, R.; et al. Ceramide Transporter CERT Is Involved in Muscle Insulin Signaling Defects Under Lipotoxic Conditions. Diabetes 2018, 67, 1258–1271. [Google Scholar] [CrossRef]
- Pillon, N.J.; Frendo-Cumbo, S.; Jacobson, M.R.; Liu, Z.; Milligan, P.L.; Hoang, B.H.; Zierath, J.R.; Bilan, P.J.; Brozinick, J.T.; Klip, A. Sphingolipid changes do not underlie fatty acid-evoked GLUT4 insulin resistance nor inflammation signals in muscle cells. J. Lipid Res. 2018, 59, 1148–1163. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Huang, S.; Wang, X.; Zhang, Q.; Liu, J.; Leng, Y. Downregulation of lipin-1 induces insulin resistance by increasing intracellular ceramide accumulation in C2C12 myotubes. Int. J. Biol. Sci. 2017, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Brozinick, J.T.; Wang, L.P.; Hawkins, E.D.; Sargent, K.M.; Liu, Y.; Narra, K.; Hoehn, K.L.; Knotts, T.A.; Siesky, A.; et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007, 5, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Kowalski, G.M.; Leslie, S.J.; Risis, S.; Yang, C.; Lee-Young, R.S.; Babb, J.R.; Meikle, P.J.; Lancaster, G.I.; Henstridge, D.C.; et al. Distinct patterns of tissue-specific lipid accumulation during the induction of insulin resistance in mice by high-fat feeding. Diabetologia 2013, 56, 1638–1648. [Google Scholar] [CrossRef] [PubMed]
- Blachnio-Zabielska, A.U.; Chacinska, M.; Vendelbo, M.H.; Zabielski, P. The Crucial Role of C18-Cer in Fat-Induced Skeletal Muscle Insulin Resistance. Cell Physiol. Biochem. 2016, 40, 1207–1220. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Lim, X.Y.; Toop, H.D.; Osborne, B.; Brandon, A.E.; Taylor, E.N.; Fiveash, C.E.; Govindaraju, H.; Teo, J.D.; McEwen, H.P.; et al. A selective inhibitor of ceramide synthase 1 reveals a novel role in fat metabolism. Nat. Commun. 2018, 9, 3165. [Google Scholar] [CrossRef] [PubMed]
- Turpin-Nolan, S.M.; Hammerschmidt, P.; Chen, W.; Jais, A.; Timper, K.; Awazawa, M.; Brodesser, S.; Bruning, J.C. CerS1-Derived C18:0 Ceramide in Skeletal Muscle Promotes Obesity-Induced Insulin Resistance. Cell Rep. 2019, 26, 1–10. [Google Scholar] [CrossRef]
- Adams, J.M.; Pratipanawatr, T.; Berria, R.; Wang, E.; DeFronzo, R.A.; Sullards, M.C.; Mandarino, L.J. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes 2004, 53, 25–31. [Google Scholar] [CrossRef]
- Straczkowski, M.; Kowalska, I.; Baranowski, M.; Nikolajuk, A.; Otziomek, E.; Zabielski, P.; Adamska, A.; Blachnio, A.; Gorski, J.; Gorska, M. Increased skeletal muscle ceramide level in men at risk of developing type 2 diabetes. Diabetologia 2007, 50, 2366–2373. [Google Scholar] [CrossRef]
- Coen, P.M.; Dube, J.J.; Amati, F.; Stefanovic-Racic, M.; Ferrell, R.E.; Toledo, F.G.; Goodpaster, B.H. Insulin resistance is associated with higher intramyocellular triglycerides in type I but not type II myocytes concomitant with higher ceramide content. Diabetes 2010, 59, 80–88. [Google Scholar] [CrossRef]
- Coen, P.M.; Hames, K.C.; Leachman, E.M.; Delany, J.P.; Ritov, V.B.; Menshikova, E.V.; Dube, J.J.; Stefanovic-Racic, M.; Toledo, F.G.; Goodpaster, B.H. Reduced skeletal muscle oxidative capacity and elevated ceramide but not diacylglycerol content in severe obesity. Obesity 2013, 21, 2362–2371. [Google Scholar] [CrossRef] [PubMed]
- Bergman, B.C.; Brozinick, J.T.; Strauss, A.; Bacon, S.; Kerege, A.; Bui, H.H.; Sanders, P.; Siddall, P.; Wei, T.; Thomas, M.K.; et al. Muscle sphingolipids during rest and exercise: A C18:0 signature for insulin resistance in humans. Diabetologia 2016, 59, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Broskey, N.T.; Obanda, D.N.; Burton, J.H.; Cefalu, W.T.; Ravussin, E. Skeletal muscle ceramides and daily fat oxidation in obesity and diabetes. Metabolism 2018, 82, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Perreault, L.; Newsom, S.A.; Strauss, A.; Kerege, A.; Kahn, D.E.; Harrison, K.A.; Snell-Bergeon, J.K.; Nemkov, T.; D’Alessandro, A.; Jackman, M.R.; et al. Intracellular localization of diacylglycerols and sphingolipids influences insulin sensitivity and mitochondrial function in human skeletal muscle. JCI. Insight. 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, R.N.; Yu, C.; Hoofnagle, A.; Hari, N.; Jensen, P.N.; Fretts, A.M.; Umans, J.G.; Howard, B.V.; Sitlani, C.M.; Siscovick, D.S.; et al. Circulating Sphingolipids, Insulin, HOMA-IR, and HOMA-B: The Strong Heart Family Study. Diabetes 2018, 67, 1663–1672. [Google Scholar] [CrossRef]
- Stratford, S.; Hoehn, K.L.; Liu, F.; Summers, S.A. Regulation of insulin action by ceramide: Dual mechanisms linking ceramide accumulation to the inhibition of Akt/protein kinase B. J. Biol. Chem. 2004, 279, 36608–36615. [Google Scholar] [CrossRef] [PubMed]
- Turban, S.; Hajduch, E. Protein kinase C isoforms: Mediators of reactive lipid metabolites in the development of insulin resistance. FEBS Lett. 2011, 585, 269–274. [Google Scholar] [CrossRef]
- Doornbos, R.P.; Theelen, M.; van der Hoeven, P.C.; van Blitterswijk, W.J.; Verkleij, A.J.; en Henegouwen, P.M.V.B. Protein kinase Czeta is a negative regulator of protein kinase B activity. J. Biol. Chem. 1999, 274, 8589–8596. [Google Scholar] [CrossRef]
- Konishi, H.; Kuroda, S.; Kikkawa, U. The pleckstrin homology domain of RAC protein kinase associates with the regulatory domain of protein kinase C zeta. Biochem. Biophys. Res. Commun. 1994, 205, 1770–1775. [Google Scholar] [CrossRef]
- Mao, M.; Fang, X.; Lu, Y.; Lapushin, R.; Bast, R.C., Jr.; Mills, G.B. Inhibition of growth-factor-induced phosphorylation and activation of protein kinase B/Akt by atypical protein kinase C in breast cancer cells. Biochem. J. 2000, 352 Pt 2, 475–482. [Google Scholar] [CrossRef]
- Razani, B.; Woodman, S.E.; Lisanti, M.P. Caveolae: From cell biology to animal physiology. Pharmacol. Rev. 2002, 54, 431–467. [Google Scholar] [CrossRef] [PubMed]
- Le Lay, S.; Hajduch, E.; Lindsay, M.R.; Le Liepvre, X.; Thiele, C.; Ferre, P.; Parton, R.G.; Kurzchalia, T.; Simons, K.; Dugail, I. Cholesterol-induced caveolin targeting to lipid droplets in adipocytes: A role for caveolar endocytosis. Traffic 2006, 7, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Oka, N.; Yamamoto, M.; Schwencke, C.; Kawabe, J.; Ebina, T.; Ohno, S.; Couet, J.; Lisanti, M.P.; Ishikawa, Y. Caveolin interaction with protein kinase C. Isoenzyme-dependent regulation of kinase activity by the caveolin scaffolding domain peptide. J. Biol. Chem. 1997, 272, 33416–33421. [Google Scholar] [CrossRef] [PubMed]
- Blouin, C.M.; Prado, C.; Takane, K.K.; Lasnier, F.; Garcia-Ocana, A.; Ferre, P.; Dugail, I.; Hajduch, E. Plasma membrane subdomain compartmentalization contributes to distinct mechanisms of ceramide action on insulin signaling. Diabetes 2010, 59, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Silveira, L.R.; Fiamoncini, J.; Hirabara, S.M.; Procopio, J.; Cambiaghi, T.D.; Pinheiro, C.H.; Lopes, L.R.; Curi, R. Updating the effects of fatty acids on skeletal muscle. J. Cell Physiol. 2008, 217, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Shulman, G.I. Etiology of insulin resistance. Am. J. Med. 2006, 119, S10–S16. [Google Scholar] [CrossRef] [PubMed]
- Tanti, J.F.; Gual, P.; Gremeaux, T.; Gonzalez, T.; Barres, R.; Le Marchand-Brustel, Y. Alteration in insulin action: Role of IRS-1 serine phosphorylation in the retroregulation of insulin signalling. Ann. Endocrinol. 2004, 65, 43–48. [Google Scholar] [CrossRef]
- Carvalho, B.M.; Oliveira, A.G.; Ueno, M.; Araujo, T.G.; Guadagnini, D.; Carvalho-Filho, M.A.; Geloneze, B.; Lima, M.M.; Pareja, J.C.; Carvalheira, J.B.; et al. Modulation of Double-Stranded RNA-Activated Protein Kinase in Insulin Sensitive Tissues of Obese Humans. Obesity 2013, 21, 2452–2457. [Google Scholar] [CrossRef]
- Carvalho-Filho, M.A.; Carvalho, B.M.; Oliveira, A.G.; Guadagnini, D.; Ueno, M.; Dias, M.M.; Tsukumo, D.M.; Hirabara, S.M.; Reis, L.F.; Curi, R.; et al. Double-Stranded RNA-Activated Protein Kinase Is a Key Modulator of Insulin Sensitivity in Physiological Conditions and in Obesity in Mice. Endocrinology 2012, 153, 5261–5274. [Google Scholar] [CrossRef]
- Nakamura, T.; Arduini, A.; Baccaro, B.; Furuhashi, M.; Hotamisligil, G.S. Small molecule inhibitors of PKR improve glucose homeostasis in obese, diabetic mice. Diabetes 2013, 63, 526–534. [Google Scholar] [CrossRef]
- Cimmino, I.; Lorenzo, V.; Fiory, F.; Doti, N.; Ricci, S.; Cabaro, S.; Liotti, A.; Vitagliano, L.; Longo, M.; Miele, C.; et al. A peptide antagonist of Prep1-p160 interaction improves ceramide-induced insulin resistance in skeletal muscle cells. Oncotarget 2017, 8, 71845–71858. [Google Scholar] [CrossRef] [PubMed]
- Oriente, F.; Fernandez Diaz, L.C.; Miele, C.; Iovino, S.; Mori, S.; Diaz, V.M.; Troncone, G.; Cassese, A.; Formisano, P.; Blasi, F.; et al. Prep1 deficiency induces protection from diabetes and increased insulin sensitivity through a p160-mediated mechanism. Mol. Cell Biol. 2008, 28, 5634–5645. [Google Scholar] [CrossRef] [PubMed]
- Oriente, F.; Cabaro, S.; Liotti, A.; Longo, M.; Parrillo, L.; Pagano, T.B.; Raciti, G.A.; Penkov, D.; Paciello, O.; Miele, C.; et al. PREP1 deficiency downregulates hepatic lipogenesis and attenuates steatohepatitis in mice. Diabetologia 2013, 56, 2713–2722. [Google Scholar] [CrossRef] [PubMed]
- Turpin, S.M.; Nicholls, H.T.; Willmes, D.M.; Mourier, A.; Brodesser, S.; Wunderlich, C.M.; Mauer, J.; Xu, E.; Hammerschmidt, P.; Bronneke, H.S.; et al. Obesity-Induced CerS6-Dependent C16:0 Ceramide Production Promotes Weight Gain and Glucose Intolerance. Cell Metab. 2014, 20, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Raichur, S.; Wang, S.T.; Chan, P.W.; Li, Y.; Ching, J.; Chaurasia, B.; Dogra, S.; Ohman, M.K.; Takeda, K.; Sugii, S.; et al. CerS2 Haploinsufficiency Inhibits beta-oxidation and Confers Susceptibility to Diet-Induced Steatohepatitis and Insulin Resistance. Cell Metab. 2014, 20, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Hla, T.; Kolesnick, R. C16:0-ceramide signals insulin resistance. Cell Metab. 2014, 20, 703–705. [Google Scholar] [CrossRef] [PubMed]
- Teruel, T.; Hernandez, R.; Lorenzo, M. Ceramide mediates insulin resistance by tumor necrosis factor-alpha in brown adipocytes by maintaining Akt in an inactive dephosphorylated state. Diabetes 2001, 50, 2563–2571. [Google Scholar] [CrossRef]
- Selathurai, A.; Kowalski, G.M.; Burch, M.L.; Sepulveda, P.; Risis, S.; Lee-Young, R.S.; Lamon, S.; Meikle, P.J.; Genders, A.J.; McGee, S.L.; et al. The CDP-Ethanolamine Pathway Regulates Skeletal Muscle Diacylglycerol Content and Mitochondrial Biogenesis without Altering Insulin Sensitivity. Cell Metab. 2015, 21, 718–730. [Google Scholar] [CrossRef]
- Timmers, S.; de Vogel-van den Bosch, J.; Hesselink, M.K.; van Beurden, D.; Schaart, G.; Ferraz, M.J.; Losen, M.; Martinez-Martinez, P.; de Baets, M.H.; Aerts, J.M.; et al. Paradoxical increase in TAG and DAG content parallel the insulin sensitizing effect of unilateral DGAT1 overexpression in rat skeletal muscle. PLoS ONE 2011, 6, e14503. [Google Scholar] [CrossRef]
- Timmers, S.; Nabben, M.; Bosma, M.; van Bree, B.; Lenaers, E.; van Beurden, D.; Schaart, G.; Westerterp-Plantenga, M.S.; Langhans, W.; Hesselink, M.K.; et al. Augmenting muscle diacylglycerol and triacylglycerol content by blocking fatty acid oxidation does not impede insulin sensitivity. Proc. Natl. Acad. Sci. USA 2012, 109, 11711–11716. [Google Scholar] [CrossRef]
- Perreault, L.; Bergman, B.C.; Hunerdosse, D.M.; Eckel, R.H. Altered intramuscular lipid metabolism relates to diminished insulin action in men, but not women, in progression to diabetes. Obesity 2010, 18, 2093–2100. [Google Scholar] [CrossRef] [PubMed]
- Jocken, J.W.; Moro, C.; Goossens, G.H.; Hansen, D.; Mairal, A.; Hesselink, M.K.; Langin, D.; van Loon, L.J.; Blaak, E.E. Skeletal muscle lipase content and activity in obesity and type 2 diabetes. J. Clin. Endocrinol. Metab. 2010, 95, 5449–5453. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Chen, Y.; Cline, G.W.; Zhang, D.; Zong, H.; Wang, Y.; Bergeron, R.; Kim, J.K.; Cushman, S.W.; Cooney, G.J.; et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem. 2002, 277, 50230–50236. [Google Scholar] [CrossRef] [PubMed]
- Itani, S.I.; Ruderman, N.B.; Schmieder, F.; Boden, G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes 2002, 51, 2005–2011. [Google Scholar] [CrossRef] [PubMed]
- Vistisen, B.; Hellgren, L.I.; Vadset, T.; Scheede-Bergdahl, C.; Helge, J.W.; Dela, F.; Stallknecht, B. Effect of gender on lipid-induced insulin resistance in obese subjects. Eur. J. Endocrinol. 2008, 158, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Yuzefovych, L.; Wilson, G.; Rachek, L. Different effects of oleate vs. palmitate on mitochondrial function, apoptosis, and insulin signaling in L6 skeletal muscle cells: Role of oxidative stress. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E1096–E1105. [Google Scholar] [CrossRef] [PubMed]
- Mitsutake, S.; Date, T.; Yokota, H.; Sugiura, M.; Kohama, T.; Igarashi, Y. Ceramide kinase deficiency improves diet-induced obesity and insulin resistance. FEBS Lett. 2012, 586, 1300–1305. [Google Scholar] [CrossRef] [PubMed]
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012, 22, 50–60. [Google Scholar] [CrossRef]
- Fayyaz, S.; Japtok, L.; Kleuser, B. Divergent role of sphingosine 1-phosphate on insulin resistance. Cell Physiol. Biochem. 2014, 34, 134–147. [Google Scholar] [CrossRef]
- Hanada, K. Intracellular trafficking of ceramide by ceramide transfer protein. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 426–437. [Google Scholar] [CrossRef]
- Giussani, P.; Brioschi, L.; Bassi, R.; Riboni, L.; Viani, P. Phosphatidylinositol 3-kinase/AKT pathway regulates the endoplasmic reticulum to golgi traffic of ceramide in glioma cells: A link between lipid signaling pathways involved in the control of cell survival. J. Biol. Chem. 2009, 284, 5088–5096. [Google Scholar] [CrossRef] [PubMed]
- Alpy, F.; Tomasetto, C. Give lipids a START: The StAR-related lipid transfer (START) domain in mammals. J. Cell Sci. 2005, 118, 2791–2801. [Google Scholar] [CrossRef]
- Langeveld, M.; Aerts, J.M. Glycosphingolipids and insulin resistance. Prog. Lipid Res. 2009, 48, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Hashiramoto, A.; Haluzik, M.; Mizukami, H.; Beck, S.; Norton, A.; Kono, M.; Tsuji, S.; Daniotti, J.L.; Werth, N.; et al. Enhanced insulin sensitivity in mice lacking ganglioside GM3. Proc. Natl. Acad. Sci. USA 2003, 100, 3445–3449. [Google Scholar] [CrossRef]
- Chavez, J.A.; Siddique, M.M.; Wang, S.T.; Ching, J.; Shayman, J.A.; Summers, S.A. Ceramides and glucosylceramides are independent antagonists of insulin signaling. J. Biol. Chem. 2014, 289, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Przybylska, M.; Wu, I.H.; Zhang, J.; Siegel, C.; Komarnitsky, S.; Yew, N.S.; Cheng, S.H. Inhibiting glycosphingolipid synthesis improves glycemic control and insulin sensitivity in animal models of type 2 diabetes. Diabetes 2007, 56, 1210–1218. [Google Scholar] [CrossRef] [PubMed]
- Sekimoto, J.; Kabayama, K.; Gohara, K.; Inokuchi, J. Dissociation of the insulin receptor from caveolae during TNFalpha-induced insulin resistance and its recovery by D-PDMP. FEBS Lett. 2012, 586, 191–195. [Google Scholar] [CrossRef]
- Kabayama, K.; Sato, T.; Saito, K.; Loberto, N.; Prinetti, A.; Sonnino, S.; Kinjo, M.; Igarashi, Y.; Inokuchi, J. Dissociation of the insulin receptor and caveolin-1 complex by ganglioside GM3 in the state of insulin resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 13678–13683. [Google Scholar] [CrossRef]
- Floegel, A.; Stefan, N.; Yu, Z.; Muhlenbruch, K.; Drogan, D.; Joost, H.G.; Fritsche, A.; Haring, H.U.; de Angelis, M.H.; Peters, A.; et al. Identification of serum metabolites associated with risk of type 2 diabetes using a targeted metabolomic approach. Diabetes 2013, 62, 639–648. [Google Scholar] [CrossRef]
- Park, M.; Kaddai, V.; Ching, J.; Fridianto, K.T.; Sieli, R.J.; Sugii, S.; Summers, S.A. A Role for Ceramides, but NOT Sphingomyelins, as antagonists of insulin signaling and mitochondrial metabolism in C2C12 myotubes. J. Biol. Chem. 2016, 291, 23978–23988. [Google Scholar] [CrossRef]
- Haus, J.M.; Kashyap, S.R.; Kasumov, T.; Zhang, R.; Kelly, K.R.; DeFronzo, R.A.; Kirwan, J.P. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes 2009, 58, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Watt, M.J.; Barnett, A.C.; Bruce, C.R.; Schenk, S.; Horowitz, J.F.; Hoy, A.J. Regulation of plasma ceramide levels with fatty acid oversupply: Evidence that the liver detects and secretes de novo synthesised ceramide. Diabetologia 2012, 55, 2741–2746. [Google Scholar] [CrossRef] [PubMed]
- Boon, J.; Hoy, A.J.; Stark, R.; Brown, R.D.; Meex, R.C.; Henstridge, D.C.; Schenk, S.; Meikle, P.J.; Horowitz, J.F.; Kingwell, B.A.; et al. Ceramides contained in LDL are elevated in type 2 diabetes and promote inflammation and skeletal muscle insulin resistance. Diabetes 2013, 62, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Kakazu, E.; Mauer, A.S.; Yin, M.; Malhi, H. Hepatocytes release ceramide-enriched pro-inflammatory extracellular vesicles in an IRE1alpha-dependent manner. J. Lipid Res. 2016, 57, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.Y.; Holland, W.L.; Kusminski, C.M.; Sun, K.; Sharma, A.X.; Pearson, M.J.; Sifuentes, A.J.; McDonald, J.G.; Gordillo, R.; Scherer, P.E. Targeted Induction of Ceramide Degradation Leads to Improved Systemic Metabolism and Reduced Hepatic Steatosis. Cell Metab. 2015, 22, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Wigger, L.; Cruciani-Guglielmacci, C.; Nicolas, A.; Denom, J.; Fernandez, N.; Fumeron, F.; Marques-Vidal, P.; Ktorza, A.; Kramer, W.; Schulte, A.; et al. Plasma Dihydroceramides Are Diabetes Susceptibility Biomarker Candidates in Mice and Humans. Cell Rep. 2017, 18, 2269–2279. [Google Scholar] [CrossRef]
- Szpigel, A.; Hainault, I.; Carlier, A.; Venteclef, N.; Batto, A.F.; Hajduch, E.; Bernard, C.; Ktorza, A.; Gautier, J.F.; Ferre, P.; et al. Lipid environment induces ER stress, TXNIP expression and inflammation in immune cells of individuals with type 2 diabetes. Diabetologia 2018, 61, 399–412. [Google Scholar] [CrossRef]
- Summers, S.A. Could Ceramides Become the New Cholesterol? Cell Metab. 2018, 27, 276–280. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| First Author (Year) | Ref. | Model (Vitro) | Intervention | Ceramide Content | Change in Insulin Resistance |
|---|---|---|---|---|---|
| Schmitz-Peiffer (1999) | [67] | C2C12 myotubes | Pal / C2-cer | ↗ [total Cer] | ↗ |
| Hajduch (2001) | [69] | L6 myotubes | C2-cer | ND | ↗ |
| Cazzolli (2001) | [71] | C2C12 myotubes | Pal | ND | ↗ |
| Pal + PP2A inhibition | ↘ | ||||
| Bourbon (2002) | [72] | Smooth muscles cells (a7r5) | C2-cer / C6-cer | ND | ↗ |
| C2-cer / C6-cer + PKCζ inhibition | ND | ↘ | |||
| Chavez (2003) | [73] | C2C12 myotubes | Pal | ↗ [long chain Cer, total Cer] | ↗ |
| Powell (2003) | [74] | L6 myotubes | C2-cer | ND | ↗ |
| C2-cer + PKCζ inhibition | ND | ↘ | |||
| Powell (2004) | [75] | L6 myotubes | Pal / C2-cer | ↗ [total Cer] | ↗ |
| Pal / C2-cer + SPT inhibition | ↘ [total Cer] | ↘ | |||
| Pal/C2-cer + PP2A/PKCζ inhibition | ND | ↘ | |||
| Hajduch (2008) | [76] | L6 myotubes | C2-cer | ND | ↗ |
| C2-cer + PKCζ inhibition | ND | ↘ | |||
| Watson (2009) | [70] | L6 myotubes | Pal | ↗ [total Cer] | ↗ |
| Pal + SPT inhibition | ↘ [total Cer] | ↘ | |||
| Frangioudakis (2013) | [77] | L6 myotubes | Pal + CerS (1,2,4,5,6) overexpression | ↗ [some species] depending on CerS overexpressed | ↘ |
| Pal + CerS(1,2,4,5,6) knockdown | ↘ [some species] depending on CerS Knockdowned | No effect on insulin signaling | |||
| Mahfouz (2014) | [78] | C2C12 myotubes / L6 myotubes | Pal / C2-cer | ND | ↗ |
| Pal / C2-cer + PP2A/PKCζ inhibition | ND | ↘ | |||
| Human myotubes | Pal | ↗ [C16:0, C18:0, C20:0] | ↗ | ||
| Pal + SPT inhibition | ↘ [C16:0, C18:0, C20:0] | ↘ | |||
| Hage Hassan (2016) | [79] | C2C12 myotubes | Pal / C2-cer | ↗ [total Cer] when Pal / ND when C2-cer | ↗ |
| Pal / C2-cer + SPT inhibition | ↘ [total Cer] when Pal / ND when C2-cer | ↘ | |||
| Human myotubes | Pal | ND | ↗ | ||
| Pal + PKR inhibition | ND | ↘ | |||
| Bandet (2018) | [80] | C2C12 | Pal | ↗ [total Cer] | ↗ |
| Pal + SPT inhibition | ↘ [total Cer] | ↘ | |||
| Pillon (2018) | [81] | L6 myotubes | Pal | ↗ [total Cer] | ↗ |
| Pal + SPT / CerS inhibition | ↘ [total Cer] | No effect on insulin signaling | |||
| Huang (2016) | [82] | C2C12 myotubes | Lipin-1 inhibition | ↗ [C16:0, C22:0, C24:0] | ↗ |
| First author (year) | Ref. | Model (rodents) | Intervention | Muscle ceramide content | Change in insulin resistance |
| Turinsky (1990) | [54] | Zucker rats | / | ↗ [total Cer] | ↗ |
| Holland (2007) | [83] | Mice | DES1 haploinsufficiency | ↘ [total Cer] | ↘ |
| Rats | Dexamethasone + SPT inhibition | ND | ↘ | ||
| Lipids infusion + SPT inhibition | ↘ [total Cer] | ↘ | |||
| Zucker rats | SPT inhibition | ↘ [total Cer] | ↘ | ||
| Ussher (2010) | [26] | Mice | HFD | ↗ [total Cer] | ↗ |
| HFD + SPT inhibition | ↘ [total Cer] | ↘ | |||
| Turner (2013) | [84] | Mice | HFD | 3weeks: ↗ [C18:0]; 16weeks: ↗ [C16:0, C18:0] | ↗ |
| Blachnio-Zabielska (2016) | [85] | Rats | HFD | ↗ [C14:0, C18:0, C18:1, C24:1, C24:0, total Cer] | ↗ |
| HFD + SPT inhibition | ↘ [C16:0, C18:0, C18:1, C20:0, total Cer] | ↘ | |||
| Hage Hassan (2016) | [79] | Mice | HFD | ND | ↗ |
| HFD + SPT inhibition | ND | ↘ | |||
| Turner (2018) | [86] | Mice | HFD + CerS1 inhibition | ↘ [C18:0, C18:1]; ↗ [C22:0, C24:0, C24:1, total Cer] | ↗ |
| Bandet (2018) | [80] | Mice | HFD | ↗ [total Cer] | ↗ |
| HFD + CERT overexpression | ↘ [C16:0, C22:0, C24:0, C24:1] | ↘ | |||
| Turpin-Nolan (2019) | [87] | Mice | HFD | ↗ [C14:0, C18:0]; ↘ [C26:0] | ↗ |
| HFD + CerS1 KO | ↗ [C16:0, C22:0, C22:1, C24:0, C24:1] ↘ [C18:0] | ↘ | |||
| HFD + CerS1 KO muscle specific | ↗ [C22:1, C24:0, C24:1]; ↘ [C18:0, C18:1, C22:0] | ↘ | |||
| First author (year) | Ref. | Model (human) | Intervention | Muscle ceramide content | Change in insulin resistance |
| Adams (2004) | [88] | Lean (n = 10) and obese (n = 10) | / | ↗ [C16:0, C18:0, C20:0, C22:0, C24:0, C24:1, total Cer] compared to lean | ↗ (total and in muscle) in obese compared to lean |
| Straczkowski (2007) | [89] | Lean (n = 12), NGT (n = 12) or IGT (n = 9) obese, healthy offspring of T2D people (n = 12) | / | ↗ [total Cer] in offspring and IGT obese compared to lean; ↗ [total Cer] in ITG obese compared to others | ND |
| Coen (2010) | [90] | Women obese insulin resistant (n = 12) or insulin sensitive (n = 10) | / | ↗ [C14:0, C16:0, C18:0, total Cer, saturated Cer, unsaturated Cer] | ↗ in insulin resistant obese compared to insulin sensitive obese |
| Amati (2011) | [48] | Lean (n = 7), athletes (n = 14), IGT obese (n = 21) | / | ↗ [C18:1, C24:0, C24:1, total]; ↘ [C14:0] | ↗ in obese compared to others; ↘ in athletes compared to others |
| Coen (2013) | [91] | Women lean (n = 8) or obese (2 groups: 30<BMI<34,9 (n = 7) and BMI > 35 (n = 15)) | / | ↗ [C14:0, C20:1, C22:1, C24:0, C24:1] in the two groups of obese | ↗ in obese (30 < BMI < 34.9) compared to lean; ↗ in obese (BMI > 35) to others |
| Bergman (2016) | [92] | Obeses (n = 14) / T2D (n = 15) / athletes (n = 15) | / | ↗ [C18:0] in T2D vs obese and athletes; ↗ [C24:0] in athletes vs obese and T2D | ↘ in muscle of athletes compared to others; |
| Broskey (2018) | [93] | Obese without T2D (n = 62) and obese with T2D (n = 44) | / | ↗ [C18:1, C20:0, C22:0, C24:0, C24:1 total Cer] | ↗ in obese with T2D compared to obese without T2D |
| Perreault (2018) | [94] | Lean (n = 15) / athletes (n = 16) / obese without T2D (n = 15) / obese with T2D (n = 12) | / | ↗ [Cer total] in total muscle of T2D compared to others; ↗ [C16:0, C18:0, Cer total] in sarcolemma of T2D compared to others; ↗ [C18:0, Cer total] in nucleus of T2D compared to others | ↗ in T2D compared to others; ↗ in obese compared to lean and athletes |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bandet, C.L.; Tan-Chen, S.; Bourron, O.; Le Stunff, H.; Hajduch, E. Sphingolipid Metabolism: New Insight into Ceramide-Induced Lipotoxicity in Muscle Cells. Int. J. Mol. Sci. 2019, 20, 479. https://doi.org/10.3390/ijms20030479
Bandet CL, Tan-Chen S, Bourron O, Le Stunff H, Hajduch E. Sphingolipid Metabolism: New Insight into Ceramide-Induced Lipotoxicity in Muscle Cells. International Journal of Molecular Sciences. 2019; 20(3):479. https://doi.org/10.3390/ijms20030479
Chicago/Turabian StyleBandet, Cécile L., Sophie Tan-Chen, Olivier Bourron, Hervé Le Stunff, and Eric Hajduch. 2019. "Sphingolipid Metabolism: New Insight into Ceramide-Induced Lipotoxicity in Muscle Cells" International Journal of Molecular Sciences 20, no. 3: 479. https://doi.org/10.3390/ijms20030479
APA StyleBandet, C. L., Tan-Chen, S., Bourron, O., Le Stunff, H., & Hajduch, E. (2019). Sphingolipid Metabolism: New Insight into Ceramide-Induced Lipotoxicity in Muscle Cells. International Journal of Molecular Sciences, 20(3), 479. https://doi.org/10.3390/ijms20030479