The Cutaneous Inflammatory Response to Thermal Burn Injury in a Murine Model

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
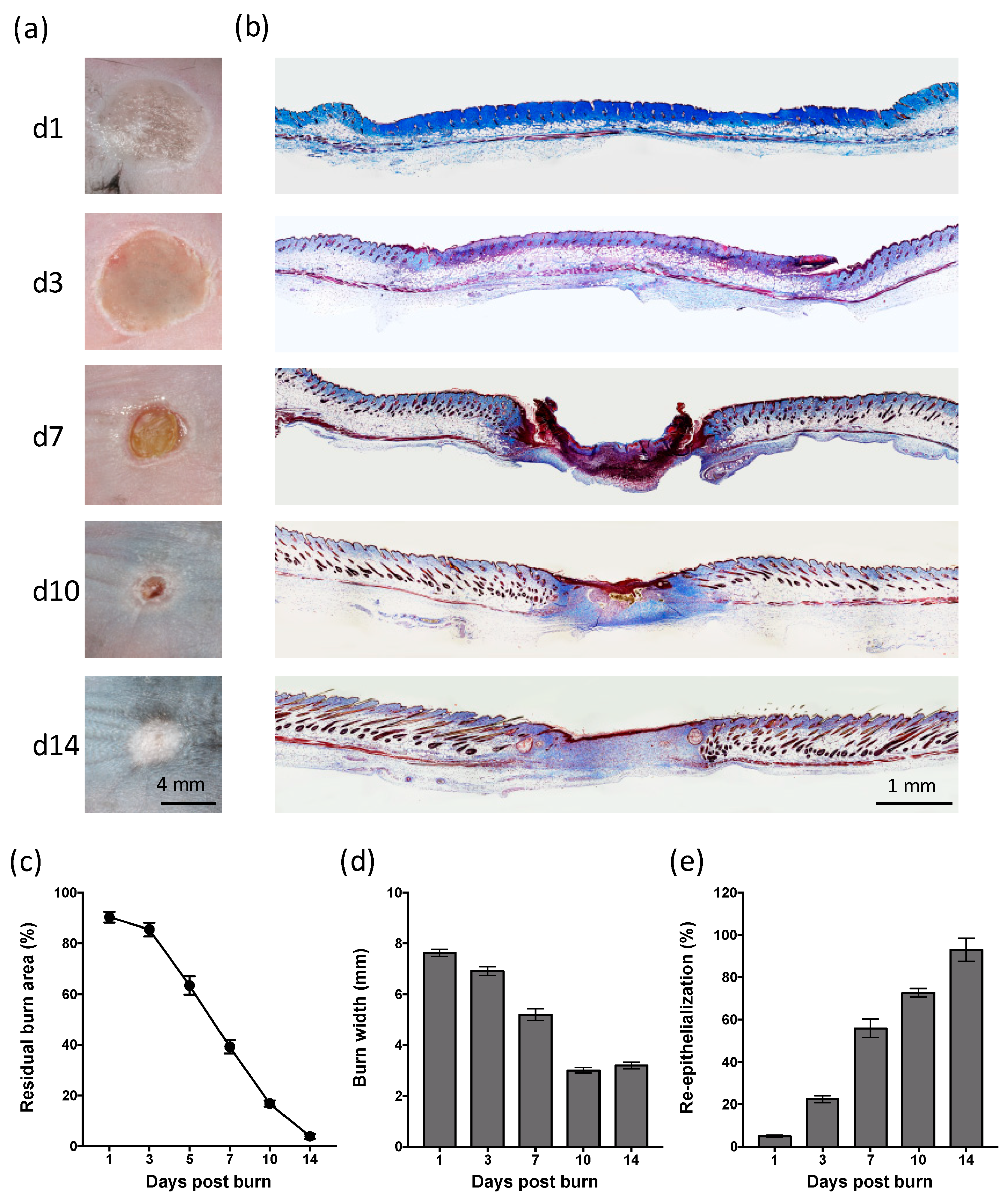
2.1. Partial-Thickness Thermal Burns Heal through Contraction and Re-epithelialization
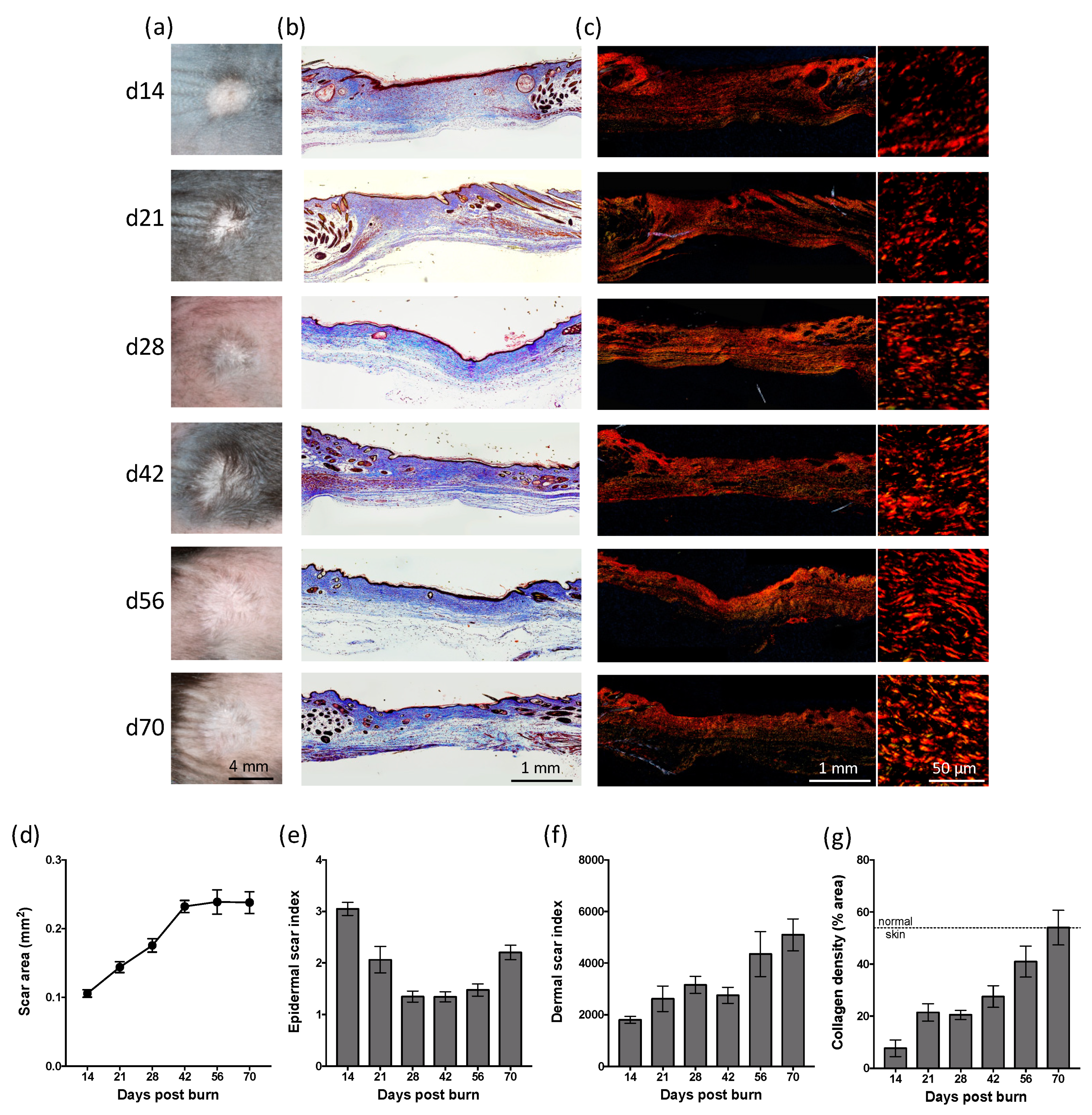
2.2. Partial-Thickness Skin Burns Result in a Persistent Scar
2.3. Partial-Thickness Skin Burns Lead to Changes in the Inflammatory Cell Population during the Healing and Scarring Process
2.4. Partial-Thickness Skin Burns Lead to Changes in Inflammatory Gene Expression that Peak during the Healing Process, with Sustained Effects on Fibrotic Gene Expression
3. Discussion
4. Materials and Methods
4.1. Murine Thermal Burn Model
4.2. Histology
4.3. Immunofluorescent Histochemistry
4.4. Quantitative PCR
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CD | Cluster of differentiation |
| Col | Collagen |
| DC | Dendritic cell |
| IL | Interleukin |
| Gr-1 | granulocyte-differentiation antigen |
| iNOS | inducible nitric oxide synthase |
| LC | Langerhans cell |
| M1 | Inflammatory macrophage |
| M2 | Reparative macrophage |
| MCP | Macrophage chemoattractant protein |
| MHC | Major histocompatibility complex |
| MIP | Macrophage inflammatory protein |
| MSB | Martius, Scarlet and Blue |
| PC | Panniculus carnosus |
| PCR | Polymerase chain reaction |
| SC | Subcutaneous |
| TBP | TATA box binding protein |
| TGF | Transforming growth factor |
| TNF | Tumour necrosis factor |
| TUNEL | Terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labelling |
| VEGF | Vascular endothelial growth factor |
References
- WHO Fact Sheet: Burns. Available online: https://www.who.int/news-room/fact-sheets/detail/burns (accessed on 6 March 2018).
- Wang, Y.; Beekman, J.; Hew, J.; Jackson, S.; Issler-Fisher, A.C.; Parungao, R.; Lajevardi, S.S.; Li, Z.; Maitz, P.K.M. Burn injury: Challenges and advances in burn wound healing, infection, pain and scarring. Adv. Drug Deliv. Rev. 2018, 123, 3–17. [Google Scholar] [CrossRef]
- Monstrey, S.; Hoeksema, H.; Verbelen, J.; Pirayesh, A.; Blondeel, P. Assessment of burn depth and burn wound healing potential. Burns 2008, 34, 761–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.A.; Rawlins, J.; Shenton, A.F.; Sharpe, D.T. The Bradford Burn Study: The epidemiology of burns presenting to an inner city emergency department. Emerg. Med. J. 2007, 24, 564–566. [Google Scholar] [CrossRef] [PubMed]
- Evers, L.H.; Bhavsar, D.; Mailander, P. The biology of burn injury. Exp. Dermatol. 2010, 19, 777–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, L.F.; Chan, R.K. The Burn Wound Microenvironment. Adv. Wound Care 2016, 5, 106–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyler, M.P.; Watts, A.M.; Perry, M.E.; Roberts, A.H.; McGrouther, D.A. Dermal cellular inflammation in burns. an insight into the function of dermal microvascular anatomy. Burns 2001, 27, 433–438. [Google Scholar] [CrossRef]
- Peng, D.; Huang, W.; Ai, S.; Wang, S. Clinical significance of leukocyte infiltrative response in deep wound of patients with major burns. Burns 2006, 32, 946–950. [Google Scholar] [CrossRef] [PubMed]
- Tarran, S.; Langlois, N.E.; Dziewulski, P.; Sztynda, T. Using the inflammatory cell infiltrate to estimate the age of human burn wounds: A review and immunohistochemical study. Med. Sci. Law 2006, 46, 115–126. [Google Scholar] [CrossRef]
- van de Goot, F.; Krijnen, P.A.; Begieneman, M.P.; Ulrich, M.M.; Middelkoop, E.; Niessen, H.W. Acute inflammation is persistent locally in burn wounds: A pivotal role for complement and C-reactive protein. J. Burn Care Res. 2009, 30, 274–280. [Google Scholar] [CrossRef]
- Finnerty, C.C.; Jeschke, M.G.; Herndon, D.N.; Gamelli, R.; Gibran, N.; Klein, M.; Silver, G.; Arnoldo, B.; Remick, D.; Tompkins, R.G. Temporal cytokine profiles in severely burned patients: A comparison of adults and children. Mol. Med. 2008, 14, 553–560. [Google Scholar] [CrossRef]
- Hur, J.; Yang, H.T.; Chun, W.; Kim, J.H.; Shin, S.H.; Kang, H.J.; Kim, H.S. Inflammatory cytokines and their prognostic ability in cases of major burn injury. Ann. Lab. Med. 2015, 35, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, M.G.; Gauglitz, G.G.; Finnerty, C.C.; Kraft, R.; Mlcak, R.P.; Herndon, D.N. Survivors versus nonsurvivors postburn: Differences in inflammatory and hypermetabolic trajectories. Ann. Surg. 2014, 259, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Kim, J.H.; Yim, H.; Kim, D. Changes in the levels of interleukins 6, 8, and 10, tumor necrosis factor alpha, and granulocyte-colony stimulating factor in Korean burn patients: Relation to burn size and postburn time. Ann. Lab. Med. 2012, 32, 339–344. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, L.M.; de Jong, M.A.; Witte, L.; Ulrich, M.M.; Geijtenbeek, T.B. Burn injury suppresses human dermal dendritic cell and Langerhans cell function. Cell Immunol. 2011, 268, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Finnerty, C.C.; Jeschke, M.G.; Branski, L.K.; Barret, J.P.; Dziewulski, P.; Herndon, D.N. Hypertrophic scarring: The greatest unmet challenge after burn injury. Lancet 2016, 388, 1427–1436. [Google Scholar] [CrossRef]
- Kischer, C.W.; Bunce, H., 3rd; Shetlah, M.R. Mast cell analyses in hypertrophic scars, hypertrophic scars treated with pressure and mature scars. J. Investig. Dermatol. 1978, 70, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Niessen, F.B.; Schalkwijk, J.; Vos, H.; Timens, W. Hypertrophic scar formation is associated with an increased number of epidermal Langerhans cells. J. Pathol. 2004, 202, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Abdullahi, A.; Amini-Nik, S.; Jeschke, M.G. Animal models in burn research. Cell. Mol. Life Sci. 2014, 71, 3241–3255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domergue, S.; Jorgensen, C.; Noel, D. Advances in Research in Animal Models of Burn-Related Hypertrophic Scarring. J. Burn Care Res. 2015, 36, e259–e266. [Google Scholar] [CrossRef] [PubMed]
- Calum, H.; Moser, C.; Jensen, P.O.; Christophersen, L.; Maling, D.S.; van Gennip, M.; Bjarnsholt, T.; Hougen, H.P.; Givskov, M.; Jacobsen, G.K.; et al. Thermal injury induces impaired function in polymorphonuclear neutrophil granulocytes and reduced control of burn wound infection. Clin. Exp. Immunol. 2009, 156, 102–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, J.L.; Fourcaudot, A.B.; Sebastian, E.A.; Shankar, R.; Brown, A.W.; Leung, K.P. Standardization of deep partial-thickness scald burns in C57BL/6 mice. Int. J. Burns Trauma 2018, 8, 26–33. [Google Scholar] [PubMed]
- Wong, V.W.; Sorkin, M.; Glotzbach, J.P.; Longaker, M.T.; Gurtner, G.C. Surgical approaches to create murine models of human wound healing. J. Biomed. Biotechnol. 2011, 2011, 969618. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.C.; Rose, L.F.; Christy, R.J.; Leung, K.P.; Chan, R.K. Full-Thickness Thermal Injury Delays Wound Closure in a Murine Model. Adv. Wound Care 2015, 4, 83–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, M.; Yang, Z.; Andl, T.; Cui, C.; Kim, N.; Millar, S.E.; Cotsarelis, G. Wnt-dependent de novo hair follicle regeneration in adult mouse skin after wounding. Nature 2007, 447, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [PubMed]
- Olson, T.S.; Ley, K. Chemokines and chemokine receptors in leukocyte trafficking. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, R7–R28. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kim, H.; Kang, H.W. Comparative evaluations of hypertrophic scar formation in in vivo models. Lasers Surg. Med. 2018, 50, 661–668. [Google Scholar] [CrossRef]
- Cameron, A.M.; Adams, D.H.; Greenwood, J.E.; Anderson, P.J.; Cowin, A.J. A novel murine model of hypertrophic scarring using subcutaneous infusion of bleomycin. Plast. Reconstruct. Surg. 2014, 133, 69–78. [Google Scholar] [CrossRef]
- Aarabi, S.; Bhatt, K.A.; Shi, Y.; Paterno, J.; Chang, E.I.; Loh, S.A.; Holmes, J.W.; Longaker, M.T.; Yee, H.; Gurtner, G.C. Mechanical load initiates hypertrophic scar formation through decreased cellular apoptosis. FASEB J. 2007, 21, 3250–3261. [Google Scholar] [CrossRef]
- Long, K.B.; Artlett, C.M.; Blankenhorn, E.P. Tight skin 2 mice exhibit a novel time line of events leading to increased extracellular matrix deposition and dermal fibrosis. Matrix Biol. 2014, 38, 91–100. [Google Scholar] [CrossRef]
- Zhang, X.; Wei, X.; Liu, L.; Marti, G.P.; Ghanamah, M.S.; Arshad, M.J.; Strom, L.; Spence, R.; Jeng, J.; Milner, S.; et al. Association of increasing burn severity in mice with delayed mobilization of circulating angiogenic cells. Arch. Surg. 2010, 145, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Cai, E.Z.; Ang, C.H.; Raju, A.; Tan, K.B.; Hing, E.C.; Loo, Y.; Wong, Y.C.; Lee, H.; Lim, J.; Moochhala, S.M.; et al. Creation of consistent burn wounds: A rat model. Arch. Plast. Surg. 2014, 41, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Wasiak, J.; Cleland, H.; Campbell, F.; Spinks, A. Dressings for superficial and partial thickness burns. Cochrane Database Syst. Rev. 2013, Cd002106. [Google Scholar] [CrossRef] [PubMed]
- Wise, L.M.; Inder, M.K.; Real, N.C.; Stuart, G.S.; Fleming, S.B.; Mercer, A.A. The vascular endothelial growth factor (VEGF)-E encoded by orf virus regulates keratinocyte proliferation and migration and promotes epidermal regeneration. Cell. Microbiol. 2012, 14, 1376–1390. [Google Scholar] [CrossRef] [PubMed]
- Singer, A.J.; McClain, S.A.; Taira, B.R.; Guerriero, J.L.; Zong, W. Apoptosis and necrosis in the ischemic zone adjacent to third degree burns. Acad. Emerg. Med. 2008, 15, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Yang, C.C.; Chao, S.C.; Wong, T.W. Histopathological differential diagnosis of keloid and hypertrophic scar. Am. J. Dermatopathol. 2004, 26, 379–384. [Google Scholar] [CrossRef]
- Rawlins, J.M.; Lam, W.L.; Karoo, R.O.; Naylor, I.L.; Sharpe, D.T. Quantifying collagen type in mature burn scars: A novel approach using histology and digital image analysis. J. Burn Care Res. 2006, 27, 60–65. [Google Scholar] [CrossRef]
- Verhaegen, P.D.; van Zuijlen, P.P.; Pennings, N.M.; van Marle, J.; Niessen, F.B.; van der Horst, C.M.; Middelkoop, E. Differences in collagen architecture between keloid, hypertrophic scar, normotrophic scar, and normal skin: An objective histopathological analysis. Wound Repair Regen. 2009, 17, 649–656. [Google Scholar] [CrossRef]
- Wise, L.M.; Stuart, G.S.; Real, N.C.; Fleming, S.B.; Mercer, A.A. VEGF Receptor-2 Activation Mediated by VEGF-E Limits Scar Tissue Formation Following Cutaneous Injury. Adv. Wound Care 2018, 7, 283–297. [Google Scholar] [CrossRef]
- Bock, O.; Yu, H.; Zitron, S.; Bayat, A.; Ferguson, M.W.; Mrowietz, U. Studies of transforming growth factors beta 1-3 and their receptors I and II in fibroblast of keloids and hypertrophic scars. Acta Derm Venereol. 2005, 85, 216–220. [Google Scholar] [CrossRef]
- Oliveira, G.V.; Hawkins, H.K.; Chinkes, D.; Burke, A.; Tavares, A.L.; Ramos-e-Silva, M.; Albrecht, T.B.; Kitten, G.T.; Herndon, D.N. Hypertrophic versus non hypertrophic scars compared by immunohistochemistry and laser confocal microscopy: Type I and III collagens. Int. Wound J. 2009, 6, 445–452. [Google Scholar] [CrossRef]
- Wang, R.; Ghahary, A.; Shen, Q.; Scott, P.G.; Roy, K.; Tredget, E.E. Hypertrophic scar tissues and fibroblasts produce more transforming growth factor-beta1 mRNA and protein than normal skin and cells. Wound Repair Regen. 2000, 8, 128–137. [Google Scholar] [CrossRef]
- Daley, J.M.; Brancato, S.K.; Thomay, A.A.; Reichner, J.S.; Albina, J.E. The phenotype of murine wound macrophages. J. Leukoc. Biol. 2010, 87, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Lucas, T.; Waisman, A.; Ranjan, R.; Roes, J.; Krieg, T.; Muller, W.; Roers, A.; Eming, S.A. Differential roles of macrophages in diverse phases of skin repair. J. Immunol. 2010, 184, 3964–3977. [Google Scholar] [CrossRef]
- Zhu, Z.; Ding, J.; Ma, Z.; Iwashina, T.; Tredget, E.E. Systemic depletion of macrophages in the subacute phase of wound healing reduces hypertrophic scar formation. Wound Repair Regen. 2016, 24, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Klinkert, K.; Whelan, D.; Clover, A.J.P.; Leblond, A.L.; Kumar, A.H.S.; Caplice, N.M. Selective M2 Macrophage Depletion Leads to Prolonged Inflammation in Surgical Wounds. Eur. Surg. Res. 2017, 58, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Shook, B.; Xiao, E.; Kumamoto, Y.; Iwasaki, A.; Horsley, V. CD301b+ Macrophages Are Essential for Effective Skin Wound Healing. J. Investig. Dermatol. 2016, 136, 1885–1891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Wang, J.; Li, S.; Yu, Z.; Liu, B.; Song, B.; Su, Y. The clinical dynamic changes of macrophage phenotype and function in different stages of human wound healing and hypertrophic scar formation. Int. Wound J. 2018. [Google Scholar] [CrossRef]
- Kobayashi, M.; Jeschke, M.G.; Shigematsu, K.; Asai, A.; Yoshida, S.; Herndon, D.N.; Suzuki, F. M2b monocytes predominated in peripheral blood of severely burned patients. J. Immunol. 2010, 185, 7174–7179. [Google Scholar] [CrossRef]
- Johnson, K.E.; Wilgus, T.A. Vascular Endothelial Growth Factor and Angiogenesis in the Regulation of Cutaneous Wound Repair. Adv. Wound Care 2014, 3, 647–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, M.W.; O’Kane, S. Scar-free healing: From embryonic mechanisms to adult therapeutic intervention. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2004, 359, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Van De Water, L.; Varney, S.; Tomasek, J.J. Mechanoregulation of the Myofibroblast in Wound Contraction, Scarring, and Fibrosis: Opportunities for New Therapeutic Intervention. Adv. Wound Care 2013, 2, 122–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohannon, J.; Cui, W.; Sherwood, E.; Toliver-Kinsky, T. Dendritic cell modification of neutrophil responses to infection after burn injury. J. Immunol. 2010, 185, 2847–2853. [Google Scholar] [CrossRef]
- Vinish, M.; Cui, W.; Stafford, E.; Bae, L.; Hawkins, H.; Cox, R.; Toliver-Kinsky, T. Dendritic cells modulate burn wound healing by enhancing early proliferation. Wound Repair Regen. 2016, 24, 6–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.; de Almeida, P.E.; Kang, K.H.; Yao, P.; Chan, C.W. Burn injury triggered dysfunction in dendritic cell response to TLR9 activation and resulted in skewed T cell functions. PLoS ONE 2012, 7, e50238. [Google Scholar] [CrossRef] [PubMed]
- Valvis, S.M.; Waithman, J.; Wood, F.M.; Fear, M.W.; Fear, V.S. The Immune Response to Skin Trauma Is Dependent on the Etiology of Injury in a Mouse Model of Burn and Excision. J. Investig. Dermatol. 2015, 135, 2119–2128. [Google Scholar] [CrossRef] [Green Version]
- Wulff, B.C.; Parent, A.E.; Meleski, M.A.; DiPietro, L.A.; Schrementi, M.E.; Wilgus, T.A. Mast cells contribute to scar formation during fetal wound healing. J. Investig. Dermatol. 2012, 132, 458–465. [Google Scholar] [CrossRef]
- Wilgus, T.A.; Wulff, B.C. The Importance of Mast Cells in Dermal Scarring. Adv. Wound Care 2014, 3, 356–365. [Google Scholar] [CrossRef] [Green Version]
- Mak, K.; Manji, A.; Gallant-Behm, C.; Wiebe, C.; Hart, D.A.; Larjava, H.; Hakkinen, L. Scarless healing of oral mucosa is characterized by faster resolution of inflammation and control of myofibroblast action compared to skin wounds in the red Duroc pig model. J. Dermatol. Sci. 2009, 56, 168–180. [Google Scholar] [CrossRef]
- Iba, Y.; Shibata, A.; Kato, M.; Masukawa, T. Possible involvement of mast cells in collagen remodeling in the late phase of cutaneous wound healing in mice. Int. Immunopharmacol. 2004, 4, 1873–1880. [Google Scholar] [CrossRef]
- Harunari, N.; Zhu, K.Q.; Armendariz, R.T.; Deubner, H.; Muangman, P.; Carrougher, G.J.; Isik, F.F.; Gibran, N.S.; Engrav, L.H. Histology of the thick scar on the female, red Duroc pig: Final similarities to human hypertrophic scar. Burns 2006, 32, 669–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, L.M.; Stuart, G.S.; Real, N.C.; Fleming, S.B.; Mercer, A.A. Orf virus IL-10 accelerates wound healing while limiting inflammation and scarring. Wound Repair Regen. 2014, 22, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Turabelidze, A.; Guo, S.; DiPietro, L.A. Importance of housekeeping gene selection for accurate reverse transcription-quantitative polymerase chain reaction in a wound healing model. Wound Repair Regen. 2010, 18, 460–466. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lateef, Z.; Stuart, G.; Jones, N.; Mercer, A.; Fleming, S.; Wise, L. The Cutaneous Inflammatory Response to Thermal Burn Injury in a Murine Model. Int. J. Mol. Sci. 2019, 20, 538. https://doi.org/10.3390/ijms20030538
Lateef Z, Stuart G, Jones N, Mercer A, Fleming S, Wise L. The Cutaneous Inflammatory Response to Thermal Burn Injury in a Murine Model. International Journal of Molecular Sciences. 2019; 20(3):538. https://doi.org/10.3390/ijms20030538
Chicago/Turabian StyleLateef, Zabeen, Gabriella Stuart, Nicola Jones, Andrew Mercer, Stephen Fleming, and Lyn Wise. 2019. "The Cutaneous Inflammatory Response to Thermal Burn Injury in a Murine Model" International Journal of Molecular Sciences 20, no. 3: 538. https://doi.org/10.3390/ijms20030538
APA StyleLateef, Z., Stuart, G., Jones, N., Mercer, A., Fleming, S., & Wise, L. (2019). The Cutaneous Inflammatory Response to Thermal Burn Injury in a Murine Model. International Journal of Molecular Sciences, 20(3), 538. https://doi.org/10.3390/ijms20030538