The Potential of Combining Tubulin-Targeting Anticancer Therapeutics and Immune Therapy

Abstract
:1. A Brief Overview of Microtubules in the Context of Mammalian Cell Cycle
2. The Immune System as the Body’s Security Guard
3. Tubulin Inhibitors Effectively Target Cycling Cancer Cells
4. Regulation of T Cells
5. Responses of T Cells to Microtubule Inhibitors
6. Regulation of Monocytes
7. Responses of Monocytes and Macrophages to Microtubule Inhibitors
8. Regulation of NK Cells
9. Responses of NK Cells to Microtubule Inhibitors
10. Regulation of Dendritic Cells
11. Responses of Dendritic Cells to Microtubule Inhibitors
12. The Art of Combining Tubulin Inhibitors and Immunotherapy
13. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Barnum, K.J.; O’Connell, M.J. Cell cycle regulation by checkpoints. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2014; pp. 29–40. [Google Scholar]
- O’Connor, C. Cell division: Stages of mitosis. Nat. Educ. 2008, 1, 188. [Google Scholar]
- Barisic, M.; Maiato, H. Cracking the (tubulin) code of mitosis. Oncotarget 2015, 6, 19356–19357. [Google Scholar] [CrossRef] [PubMed]
- Lodish, H.; Berk, A.; Lawrence Zipursky, S.; Matsudaira, P.; Baltimore, D.; Darnell, J. Microtubule structures. In Molecular Cell Biology, 4th ed.; W. H. Freeman: New York, NY, USA, 2000; pp. 190–193. [Google Scholar]
- Parker, A.L.; Kavallaris, M.; McCarroll, J.A. Microtubules and their role in cellular stress in cancer. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Tang, Y.; Chen, W.-W.; Wang, Y.-L.; Yang, L.; Li, X.; Song, G.-L.; Kuang, J. Tubb3 regulation by the erk and akt signaling pathways: A mechanism involved in the effect of arginine adp-ribosyltransferase 1 (art1) on apoptosis of colon carcinoma ct26 cells. Tumour Biol. 2016, 37, 2353–2363. [Google Scholar] [CrossRef] [PubMed]
- Janssen, L.M.E.; Ramsay, E.E.; Logsdon, C.D.; Overwijk, W.W. The immune system in cancer metastasis: Friend or foe? J. Immunother. Cancer 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berraondo, P.; Minute, L.; Ajona, D.; Corrales, L.; Melero, I.; Pio, R. Innate immune mediators in cancer: Between defense and resistance. Immunol. Rev. 2016, 274, 290–306. [Google Scholar] [CrossRef] [PubMed]
- Gradishar, W.J. Taxanes for the treatment of metastatic breast cancer. Breast Cancer 2012, 6, 159. [Google Scholar] [CrossRef] [PubMed]
- Anampa, J.; Makower, D.; Sparano, J.A. Progress in adjuvant chemotherapy for breast cancer: An overview. BMC Med. 2015, 13, 195. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.M.; Bouvet, M. Nanoparticle Albumin-Bound-Paclitaxel: A Limited Improvement under the Current Therapeutic Paradigm of Pancreatic Cancer; Taylor & Francis: Abingdon, UK, 2015. [Google Scholar]
- Ringel, I.; Horwitz, S.B. Studies with rp 56976 (taxotere): A semisynthetic analogue of taxol. J. Natl. Cancer Inst. 1991, 83, 288–291. [Google Scholar] [CrossRef] [PubMed]
- Riou, J.-F.; Naudin, A.; Lavelle, F.J.B. Effects of taxotere on murine and human tumor cell lines. Biochem. Biophys. Res. Commun. 1992, 187, 164–170. [Google Scholar] [CrossRef]
- Bissery, M.-C.; Nohynek, G.; Sanderink, G.-J.; Lavelie, F. Docetaxel (taxotere®) a review of preclinical and clinical experience. Part i. Anticancer Drugs 1995, 6, 339–355. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Khuri, F.R. Mode of action of docetaxel – a basis for combination with novel anticancer agents. Cancer Treat. Rev. 2003, 29, 407–415. [Google Scholar] [CrossRef]
- Moudi, M.; Go, R.; Yien, C.Y.S.; Nazre, M. Vinca alkaloids. Int. J. Prev. Med. 2013, 4, 1231. [Google Scholar] [PubMed]
- Rowinsky, E. The vinca alkaloids. In Holland-Frei Cancer Medicine, 6th ed.; BC Decker: Hamilton, ON, Canada, 2003. [Google Scholar]
- Leung, Y.Y.; Hui, L.L.Y.; Kraus, V.B. Colchicine—Update on mechanisms of action and therapeutic uses. Semin. Arthritis Rheum. 2015, 45, 341. [Google Scholar] [CrossRef] [PubMed]
- Weaver, B.A. How taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 2014, 25, 2677. [Google Scholar] [CrossRef]
- Pienta, K.J. Preclinical mechanisms of action of docetaxel and docetaxel combinations in prostate cancer. In Seminars in Oncology; Elsevier: New York, NY, USA, 2001. [Google Scholar]
- Engels, F.K.; Sparreboom, A.; Mathot, R.A.A.; Verweij, J. Potential for improvement of docetaxel-based chemotherapy: a pharmacological review. Br. J. Cancer 2005, 93, 173. [Google Scholar] [CrossRef]
- Lin, Z.-Y.; Wu, C.C.; Chuang, Y.H.; Chuang, W.L. Anti-cancer mechanisms of clinically acceptable colchicine concentrations on hepatocellular carcinoma. Life Sci. 2013, 93, 323–328. [Google Scholar] [CrossRef]
- Fakih, M.; Replogle, T.; Lehr, J.E.; Pienta, K.J.; Yagoda, A. Inhibition of prostate cancer growth by estramustine and colchicine. Prostate 1995, 26, 310–315. [Google Scholar] [CrossRef]
- Kuo, M.-C.; Chang, S.-J.; Hsieh, M.-C.J.M. Colchicine significantly reduces incident cancer in gout male patients: A 12-year cohort study. Medicine 2015, 94. [Google Scholar] [CrossRef]
- Gigant, B.; Wang, C.; Ravelli, R.B.G.; Roussi, F.; Steinmetz, M.O.; Curmi, P.A.; Sobel, A.; Knossow, M. Structural basis for the regulation of tubulin by vinblastine. Nature 2005, 435, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J.; Morgan, D.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell; Garland Science: New York, NY, USA; Abingdon, UK, 2014. [Google Scholar]
- Pennock, N.D.; White, J.T.; Cross, E.W.; Cheney, E.E.; Tamburini, B.A.; Kedl, R.M. T cell responses: Naive to memory and everything in between. Adv. Physiol. Educ. 2013, 37, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Kondĕlková, K.; Vokurková, D.; Krejsek, J.; Borská, L.; Fiala, Z.; Ctirad, A. Regulatory t cells (treg) and their roles in immune system with respect to immunopathological disorders. Acta Med. 2010, 53, 73–77. [Google Scholar] [CrossRef]
- Turk, M.J.; Guevara-Patiño, J.A.; Rizzuto, G.A.; Engelhorn, M.E.; Sakaguchi, S.; Houghton, A.N. Concomitant tumor immunity to a poorly immunogenic melanoma is prevented by regulatory t cells. J. Exp. Med. 2004, 200, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Ilfeld, D.; Feierman, E.; Kuperman, O.; Kivity, S.; Topilsky, M.; Netzer, L.; Pecht, M.; Trainin, N. Effect of colchicine on t cell subsets of healthy volunteers. Immunology 1984, 53, 595–598. [Google Scholar]
- Vicari, A.P.; Luu, R.; Zhang, N.; Patel, S.; Makinen, S.R.; Hanson, D.C.; Weeratna, R.D.; Krieg, A.M. Paclitaxel reduces regulatory t cell numbers and inhibitory function and enhances the anti-tumor effects of the tlr9 agonist pf-3512676 in the mouse. Cancer Immunol. Immunother. 2008, 58, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Mullins, D.W.; Koci, M.D.; Burger, C.J.; Elgert, K.D. Interleukin-12 overcomes paclitaxel-mediated suppression of t-cell proliferation. Immunopharmacol. Immunotoxicol. 1998, 20, 473–492. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Y.; Duan, X.-F.; Wang, L.-P.; Xu, Y.-J.; Huang, L.; Zhang, T.-F.; Liu, J.-Y.; Li, F.; Zhang, Z.; Yue, D.-L.; et al. Selective depletion of regulatory t cell subsets by docetaxel treatment in patients with nonsmall cell lung cancer. J. Immunol. Res. 2014, 2014, 286170. [Google Scholar] [CrossRef]
- North, R.J.; Awwad, M. Elimination of cycling cd4+ suppressor t cells with an anti-mitotic drug releases non-cycling cd8+ t cells to cause regression of an advanced lymphoma. Immunology 1990, 71, 90–95. [Google Scholar]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of monocytes, macrophages, and dendritic cells. Science 2010, 327, 656–661. [Google Scholar] [CrossRef]
- Nichols, B.A.; Bainton, D.F.; Farquhar, M.G. Differentiation of monocytes. Origin, nature, and fate of their azurophil granules. J. Cell Biol. 1971, 50, 498–515. [Google Scholar] [CrossRef] [PubMed]
- Ziegler-Heitbrock, L.; Ancuta, P.; Crowe, S.; Dalod, M.; Grau, V.; Hart, D.N.; Leenen, P.J.M.; Liu, Y.-J.; MacPherson, G.; Randolph, G.J.; et al. Nomenclature of monocytes and dendritic cells in blood. Blood 2010, 116, e74–e80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghattas, A.; Griffiths, H.R.; Devitt, A.; Lip, G.Y.H.; Shantsila, E. Monocytes in coronary artery disease and atherosclerosis: Where are we now? J. Am. Coll. Cardiol. 2013, 62, 1541–1551. [Google Scholar] [CrossRef] [PubMed]
- Mills, C. M1 and M2 macrophages: Oracles of health and disease. Crit. Rev. Immunol. 2012, 32, 463–488. [Google Scholar] [CrossRef] [PubMed]
- Richards, D.M.; Hettinger, J.; Feuerer, M. Monocytes and macrophages in cancer: Development and functions. Cancer Microenviron. 2013, 6, 179. [Google Scholar] [CrossRef] [PubMed]
- Manié, S.; Schmid-Alliana, A.; Kubar, J.; Ferrua, B.; Rossi, B. Disruption of microtubule network in human monocytes induces expression of interleukin-1 but not that of interleukin-6 nor tumor necrosis factor-alpha. Involvement of protein kinase a stimulation. J. Biol. Chem. 1993, 268, 13675–13681. [Google Scholar]
- Schwarz, N.; Toledo-Flores, D.; Fernando, S.; Di Bartolo, B.; Nicholls S, J.; Psaltis P, J. Pro-inflammatory effects of colchicine on macrophages stimulated with atherogenic stimuli in vitro. Heart Lung Circ. 2016, 25, S89. [Google Scholar] [CrossRef]
- Millrud, C.R.; Mehmeti, M.; Leandersson, K. Docetaxel promotes the generation of anti-tumorigenic human macrophages. Exp. Cell Res. 2018, 362, 525–531. [Google Scholar] [CrossRef]
- Zingoni, A.; Fionda, C.; Borrelli, C.; Cippitelli, M.; Santoni, A.; Soriani, A. Natural killer cell response to chemotherapy-stressed cancer cells: Role in tumor immunosurveillance. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Markasz, L.; Stuber, G.; Vanherberghen, B.; Flaberg, E.; Olah, E.; Carbone, E.; Eksborg, S.; Klein, E.; Skribek, H.; Szekely, L. Effect of frequently used chemotherapeutic drugs on the cytotoxic activity of human natural killer cells. Mol. Cancer Ther. 2007, 6, 644–654. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, S.E.; Keating, N.; Belz, G.T. Natural killer cells and anti-tumor immunity. Mol. Immunol. 2017. [Google Scholar] [CrossRef]
- Larsen, S.K.; Gao, Y.; Basse, P.H. Nk cells in the tumor microenvironment. Crit. Rev. Oncog. 2014, 19, 91. [Google Scholar] [CrossRef] [PubMed]
- Glasner, A.; Levi, A.; Enk, J.; Isaacson, B.; Viukov, S.; Orlanski, S.; Scope, A.; Neuman, T.; Enk, C.D.; Hanna, J.H.; et al. Nkp46 receptor-mediated interferon-γ production by natural killer cells increases fibronectin 1 to alter tumor architecture and control metastasis. Immunity 2018, 48, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Kubo, M.; Morisaki, T.; Matsumoto, K.; Tasaki, A.; Yamanaka, N.; Nakashima, H.; Kuroki, H.; Nakamura, K.; Nakamura, M.; Katano, M. Paclitaxel probably enhances cytotoxicity of natural killer cells against breast carcinoma cells by increasing perforin production. Cancer Immunol. Immunother. 2005, 54, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Di Modica, M.; Sfondrini, L.; Regondi, V.; Varchetta, S.; Oliviero, B.; Mariani, G.; Bianchi, G.V.; Generali, D.; Balsari, A.; Triulzi, T.; et al. Taxanes enhance trastuzumab-mediated adcc on tumor cells through nkg2d-mediated nk cell recognition. Oncotarget 2016, 7, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Chuang, L.T.; Lotzová, E.; Heath, J.; Cook, K.R.; Munkarah, A.; Morris, M.; Wharton, J.T. Alteration of lymphocyte microtubule assembly, cytotoxicity, and activation by the anticancer drug taxol. Cancer Res. 1994, 54, 1286–1291. [Google Scholar] [PubMed]
- Orange, J.S.; Eliza Harris, K.; Andzelm, M.M.; Valter, M.M.; Geha, R.S.; Strominger, J.L. The mature activating natural killer cell immunologic synapse is formed in distinct stages. Proc. Natl. Acad. Sci. USA 2003, 100, 14151. [Google Scholar] [CrossRef] [PubMed]
- Katz, P.; Zaytoun, A.; Lee, J.J.T.J.O.I. Mechanisms of human cell-mediated cytotoxicity. III. Dependence of natural killing on microtubule and microfilament integrity. J. Immunol. 1982, 129, 2816–2825. [Google Scholar] [PubMed]
- Barber, D.F.; Long, E.O. Coexpression of cd58 or cd48 with intercellular adhesion molecule 1 on target cells enhances adhesion of resting nk cells. J. Immunol. 2003, 170, 294–299. [Google Scholar] [CrossRef]
- Davis, D.M.; Chiu, I.; Fassett, M.; Cohen, G.B.; Mandelboim, O.; Strominger, J.L. The human natural killer cell immune synapse. Proc. Natl. Acad. Sci. USA 1999, 96, 15062. [Google Scholar] [CrossRef]
- Nolte-’t Hoen, E.N.M.; Almeida, C.R.; Cohen, N.R.; Nedvetzki, S.; Yarwood, H.; Davis, D.M. Increased surveillance of cells in mitosis by human nk cells suggests a novel strategy for limiting tumor growth and viral replication. Blood 2007, 109, 670–673. [Google Scholar] [CrossRef] [PubMed]
- Carpén, O.; Virtanen, I.; Saksela, E. The cytotoxic activity of human natural killer cells requires an intact secretory apparatus. Cell. Immunol. 1981, 58, 97–106. [Google Scholar] [CrossRef]
- Kang, H.J.; Kim, K.H.; Rhew, H.Y.; Park, K.-Y. Effects of mitomycin-c, cisplatin, and vinblastine on natural killer cell activity in mice. J. Korean Assoc. Cancer Prev. 2000, 5, 144–153. [Google Scholar]
- Howard, C.J.; Charleston, B.; Stephens, S.A.; Sopp, P.; Hope, J.C. The role of dendritic cells in shaping the immune response. Anim. Health Res. Rev. 2004, 5, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Collin, M.; McGovern, N.; Haniffa, M. Human dendritic cell subsets. Immunology 2013, 140, 22. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. In Interaction of Immune and Cancer Cells; Springer-Verlag Wien: Vienna, Austria, 2013; pp. 75–89. [Google Scholar]
- Donnadieu, E. Defects in t Cell Trafficking and Resistance to Cancer Immunotherapy; Springer: Basel, Switzerland, 2016. [Google Scholar]
- Aspord, C.; Pedroza-Gonzalez, A.; Gallegos, M.; Tindle, S.; Burton, E.C.; Su, D.; Marches, F.; Banchereau, J.; Palucka, A.K. Breast cancer instructs dendritic cells to prime interleukin 13-secreting cd4+ t cells that facilitate tumor development. J. Exp. Med. 2007, 204, 1037–1047. [Google Scholar] [CrossRef]
- Ferrari, S.; Rovati, B.; Porta, C.; Alessandrino, P.E.; Bertolini, A.; Collovà, E.; Riccardi, A.; Danova, M. Lack of dendritic cell mobilization into the peripheral blood of cancer patients following standard- or high-dose chemotherapy plus granulocyte-colony stimulating factor. Cancer Immunol. Immunother. 2003, 52, 359–366. [Google Scholar] [PubMed]
- Tsavaris, N.; Kosmas, C.; Vadiaka, M.; Kanelopoulos, P.; Boulamatsis, D. Immune changes in patients with advanced breast cancer undergoing chemotherapy with taxanes. Br. J. Cancer 2002, 87, 21–27. [Google Scholar] [CrossRef]
- Perera, P.Y.; Mayadas, T.N.; Takeuchi, O.; Akira, S.; Zaks-Zilberman, M.; Goyert, S.M.; Vogel, S.N. Cd11b/cd18 acts in concert with cd14 and toll-like receptor (tlr) 4 to elicit full lipopolysaccharide and taxol-inducible gene expression. J. Immunol. 2001, 166, 574–581. [Google Scholar] [CrossRef] [PubMed]
- John, J.; Ismail, M.; Riley, C.; Askham, J.; Morgan, R.; Melcher, A.; Pandha, H. Differential effects of paclitaxel on dendritic cell function. BMC Immunol. 2010, 11, 14. [Google Scholar] [CrossRef]
- Wen, C.-C.; Chen, H.-M.; Chen, S.-S.; Huang, L.-T.; Chang, W.-T.; Wei, W.-C.; Chou, L.-C.; Arulselvan, P.; Wu, J.-B.; Kuo, S.-C.; et al. Specific microtubule-depolymerizing agents augment efficacy of dendritic cell-based cancer vaccines. J. Biomed. Sci. 2011, 18, 44. [Google Scholar] [CrossRef] [Green Version]
- Mizumoto, N.; Tanaka, H.; Matsushima, H.; Vishwanath, M.; Takashima, A. Colchicine promotes antigen cross-presentation by murine dendritic cells. J. Investig. Dermatol. 2007, 127, 1543–1546. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Matsushima, H.; Nishibu, A.; Clausen, B.E.; Takashima, A. Dual therapeutic efficacy of vinblastine as a unique chemotherapeutic agent capable of inducing dendritic cell maturation. Cancer Res. 2009, 69, 6987. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Matsushima, H.; Mizumoto, N.; Takashima, A. Classification of chemotherapeutic agents based on their differential in vitro effects on dendritic cells. Cancer Res. 2009, 69, 6978–6986. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.J.N. The benefits of immunotherapy combinations. Nature 2017, 552, S67. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; Sundar, R.; Lopez, J.J.B.J.O.C. Combining DNA damaging therapeutics with immunotherapy: More haste, less speed. Br. J. Cancer 2017. [CrossRef] [PubMed]
- Johnson, D.B.; Chandra, S.; Sosman, J.A. Immune checkpoint inhibitor toxicity in 2018. JAMA 2018, 320, 1702–1703. [Google Scholar] [CrossRef]
- George, B.; Kelly, K.; Ko, A.; Soliman, H.; Trunova, N.; Wainberg, Z.; Waterhouse, D.; O’dwyer, P.J.J.O.T.O. P1. 46: Phase i study of nivolumab+ nab-paclitaxel in solid tumors: Preliminary analysis of the non-small cell lung cancer cohort: Track: Advanced nsclc. J. Thorac. Oncol. 2016, 11, S211–S212. [Google Scholar] [CrossRef]
- Garon, E.B.; Ciuleanu, T.-E.; Arrieta, O.; Prabhash, K.; Syrigos, K.N.; Goksel, T.; Park, K.; Gorbunova, V.; Kowalyszyn, R.D.; Pikiel, J.J.T.L. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage iv non-small-cell lung cancer after disease progression on platinum-based therapy (revel): A multicentre, double-blind, randomised phase 3 trial. Lancet 2014, 384, 665–673. [Google Scholar] [CrossRef]
- Gadgeel, S.M.; Stevenson, J.; Langer, C.J.; Gandhi, L.; Borghaei, H.; Patnaik, A.; Villaruz, L.C.; Gubens, M.A.; Hauke, R.J.; Yang, J.C.-H. Pembrolizumab (Pembro) Plus Chemotherapy as Front-Line Therapy for Advanced Nsclc: Keynote-021 Cohorts AC; American Society of Clinical Oncology: Alexandria, VA, USA, 2016. [Google Scholar]

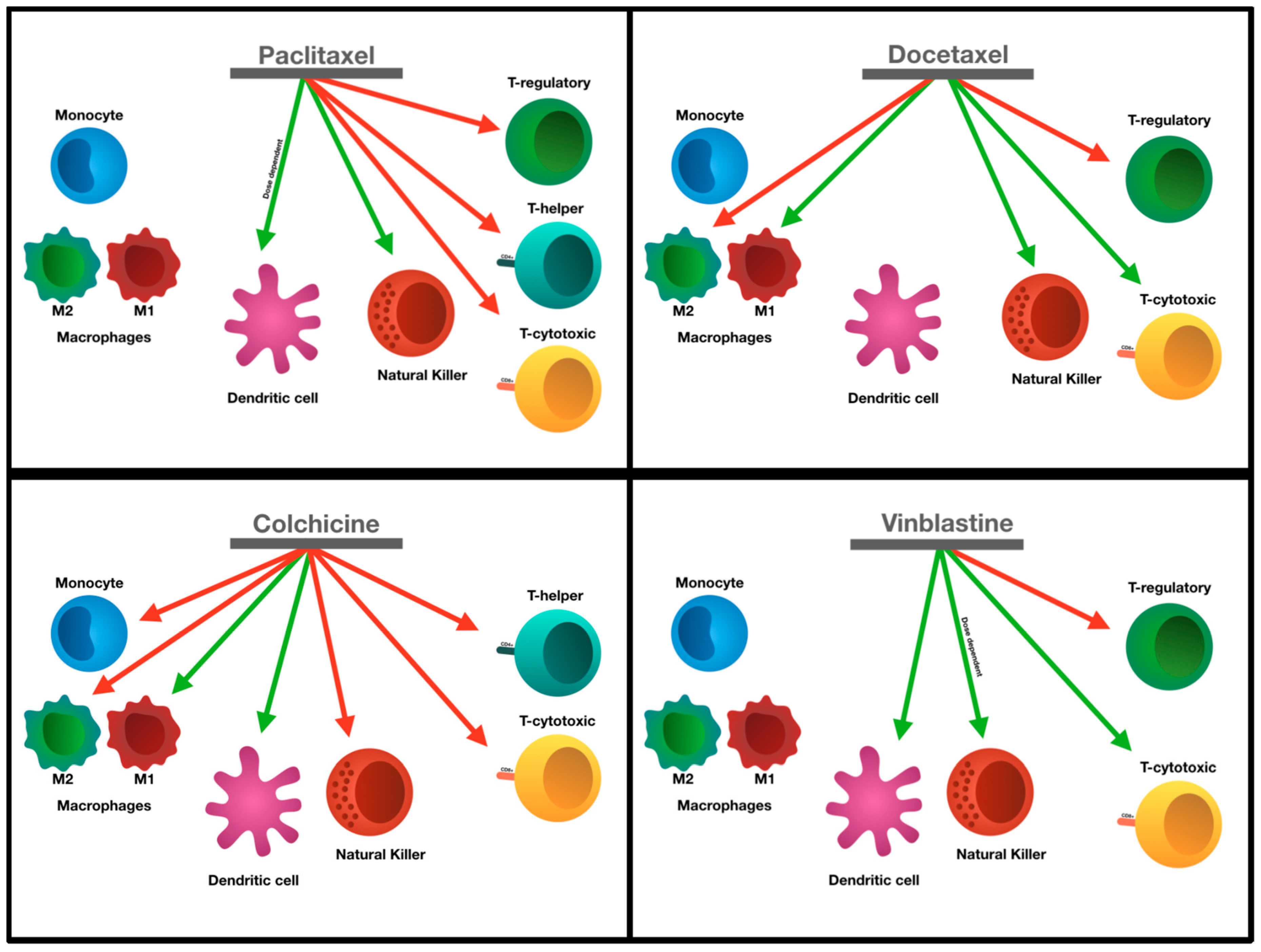

{kind=link}
{kind=link}
| Microtubule Inhibitors | Binding Domains | Cancer Treatments | Mode of Action | References |
|---|---|---|---|---|
| Paclitaxel (nab-paclitaxel) | Taxane-binding | Breast, ovarian, prostate, lung | Anti-microtubule depolymerization leading to mitotic arrest | [12,20] |
| Docetaxel | Taxane-binding | Breast, non-small cell lung, androgen-independent metastatic prostate cancer | Anti-microtubule depolymerization, and attenuation of bcl-2 and bcl-xL gene expression | [21,22] |
| Colchicine * | Colchicine-binding | Hepatocellular & prostate cancers | Anti-microtubule polymerization. Cell cycle arrest in metaphase | [19,23,24,25] |
| Vinblastine | Vinca-binding | Testicular, Hodgkins and non-Hodgkins lymphoma, breast, & germ cell cancers. | Induces wedge at tubulin interface causing tubulin self-association into spiral aggregates. Anti-microtubule polymerization, & cell cycle arrest in metaphase. | [17,26] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fong, A.; Durkin, A.; Lee, H. The Potential of Combining Tubulin-Targeting Anticancer Therapeutics and Immune Therapy. Int. J. Mol. Sci. 2019, 20, 586. https://doi.org/10.3390/ijms20030586
Fong A, Durkin A, Lee H. The Potential of Combining Tubulin-Targeting Anticancer Therapeutics and Immune Therapy. International Journal of Molecular Sciences. 2019; 20(3):586. https://doi.org/10.3390/ijms20030586
Chicago/Turabian StyleFong, Alexis, Amanda Durkin, and Hoyun Lee. 2019. "The Potential of Combining Tubulin-Targeting Anticancer Therapeutics and Immune Therapy" International Journal of Molecular Sciences 20, no. 3: 586. https://doi.org/10.3390/ijms20030586
APA StyleFong, A., Durkin, A., & Lee, H. (2019). The Potential of Combining Tubulin-Targeting Anticancer Therapeutics and Immune Therapy. International Journal of Molecular Sciences, 20(3), 586. https://doi.org/10.3390/ijms20030586