Balancing mTOR Signaling and Autophagy in the Treatment of Parkinson’s Disease

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
Abstract
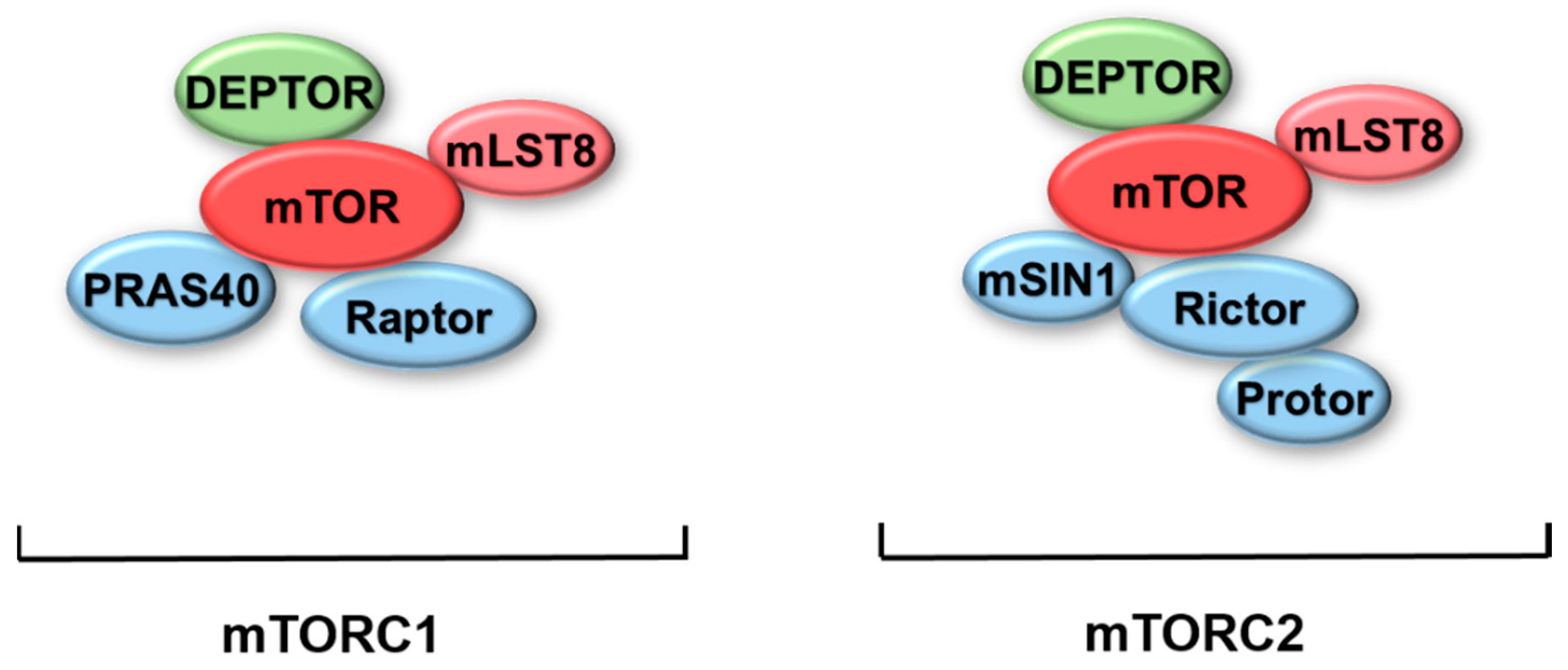
:1. Introduction of mTOR
2. Role of mTOR in Autophagy
2.1. mTOR/AMPK/ULK1 Signaling
2.2. mTOR/VPS34-ATG14 Complex Signaling
2.3. mTOR/TFEB/TFE3
2.4. mTOR in Autophagic Lysosome Reformation (ALR)
3. Role of mTOR in Parkinson’s Disease
4. Potential PD Treatment by Targeting mTOR
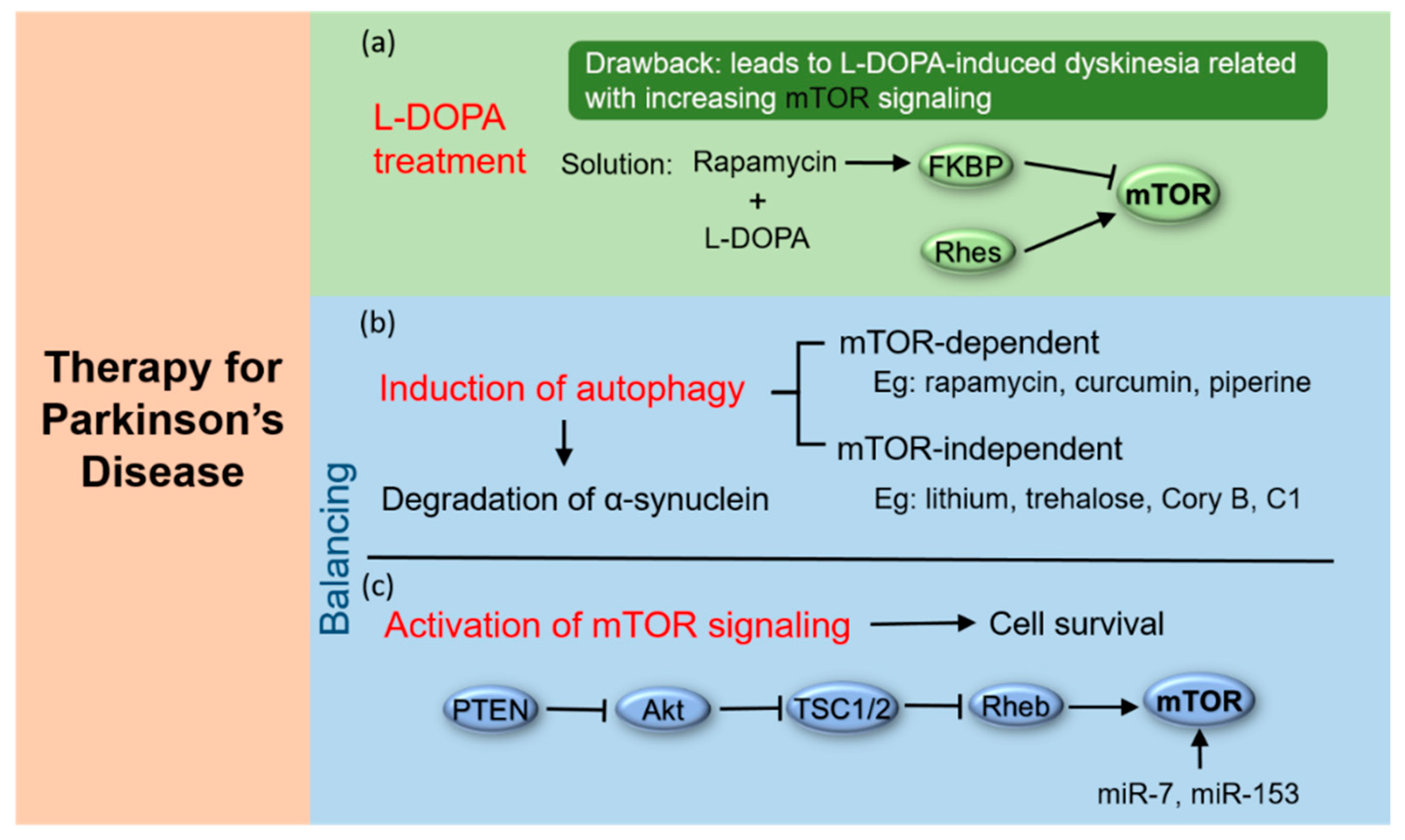
4.1. Treatment of PD by Combining L-DOPA with mTOR Inhibitors
4.2. Induction of Autophagy
4.3. Activation of mTOR Signaling
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALR | Autophagic lysosome reformation |
| AMPK | AMP-activated protein kinase |
| CLEAR | Coordinated lysosomal expression and regulation |
| DEPTOR | DEP-domain containing mTOR-interacting protein |
| ER | Endoplasmic reticulum |
| FIP200 | Focal adhesion kinase family interacting protein of 200 kD |
| FKBP | FK506-binding protein |
| FRAP | FKBP12-rapamycin complex-associated protein |
| GAP | GTPase-activating protein |
| GBA1 | Glucosidase beta acid 1 |
| IMPase | Inositol monophosphatase |
| IP3 | Inositol 1,4,5-trisphosphate |
| IRS | Insulin receptor substrate |
| LRRK2 | Leucine-rich repeat kinase 2 |
| miR | MicroRNA |
| mLST8 | Mammalian lethal with sec-13 protein 8 |
| MPTP | 1-methyl-4-phenyl-1,2,3,6 tetrahydropyridine |
| mSIN1 | Mammalian stress-activated map kinase-interacting protein 1 |
| mTOR | Mammalian targets of rapamycin |
| mTORC1 | mTOR complex 1 |
| mTORC2 | mTOR complex 2 |
| NRBF2 | Nuclear receptor binding factor 2 |
| PD | Parkinson’s disease |
| PDK1 | 3-Phosphoinositide dependent protein kinase-1 |
| PI | Phosphatidylinositol |
| PINK1 | PTEN-induced putative kinase 1 |
| PI3K | Phosphatidylinositol 3-kinase |
| PI3P | Phosphatidylinositol 3-phosphate |
| PKC | Protein kinase C |
| PRAS40 | Proline-rich Akt substrate of 40 kDa |
| Protor | Protein observed with Rictor |
| P70S6K | P70 ribosomal S6 kinase |
| Raptor | Regulatory associated protein of mammalian target of rapamycin |
| Rheb | Ras homolog enriched in brain |
| Rhes | Ras homolog enriched in striatum |
| Rictor | Rapamycin-insensitive companion of mTOR |
| SCARB2 | Scavenger receptor class B member 2 |
| SGK | Serum/glucocorticoid regulated kinase |
| SNpc | Substantia nigra pars compacta |
| TOR | Target of rapamycin |
| TSC | Tuberous sclerosis complex |
| ULK1 | Unc51-like kinase 1 |
| VPS34 | Vacuolar protein sorting 34 |
| VPS35 | Vacuolar protein sorting 35 |
| 4EBP1 | Eukaryotic initiation factor 4E (eIF4E) binding protein 1 |
| 6-OHDA | 6-hydroxydopamine |
References
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Sabers, C.J.; Martin, M.M.; Brunn, G.J.; Williams, J.M.; Dumont, F.J.; Wiederrecht, G.; Abraham, R.T. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J. Biol. Chem. 1995, 270, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, M.C.; Heitman, J. TOR mutations confer rapamycin resistance by preventing interaction with FKBP12-rapamycin. J. Biol. Chem. 1995, 270, 27531–27537. [Google Scholar] [CrossRef] [PubMed]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef]
- Zheng, X.F.; Florentino, D.; Chen, J.; Crabtree, G.R.; Schreiber, S.L. TOR kinase domains are required for two distinct functions, only one of which is inhibited by rapamycin. Cell 1995, 82, 121–130. [Google Scholar] [CrossRef]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002, 110, 177–189. [Google Scholar] [CrossRef]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; Latek, R.R.; Guntur, K.V.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol. Cell 2003, 11, 895–904. [Google Scholar] [CrossRef]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef]
- Di Malta, C.; Siciliano, D.; Calcagni, A.; Monfregola, J.; Punzi, S.; Pastore, N.; Eastes, A.N.; Davis, O.; De Cegli, R.; Zampelli, A.; et al. Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science 2017, 356, 1188–1192. [Google Scholar] [CrossRef] [PubMed]
- Beirowski, B.; Wong, K.M.; Babetto, E.; Milbrandt, J. mTORC1 promotes proliferation of immature Schwann cells and myelin growth of differentiated Schwann cells. Proc. Natl. Acad. Sci. USA 2017, 114, e4261–e4270. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, F.; Evangelisti, C.; McCubrey, J.A.; Martelli, A.M. Current treatment strategies for inhibiting mTOR in cancer. Trends Pharmacol. Sci. 2015, 36, 124–135. [Google Scholar] [CrossRef]
- Caron, A.; Richard, D.; Laplante, M. The Roles of mTOR Complexes in Lipid Metabolism. Annu. Rev. Nutr. 2015, 35, 321–348. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Jie, Z.; Joo, D.; Ordureau, A.; Liu, P.; Gan, W.; Guo, J.; Zhang, J.; North, B.J.; Dai, X.; et al. TRAF2 and OTUD7B govern a ubiquitin-dependent switch that regulates mTORC2 signalling. Nature 2017, 545, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Frias, M.A.; Thoreen, C.C.; Jaffe, J.D.; Schroder, W.; Sculley, T.; Carr, S.A.; Sabatini, D.M. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr. Biol. 2006, 16, 1865–1870. [Google Scholar] [CrossRef]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Ruegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef]
- Pearce, L.R.; Huang, X.; Boudeau, J.; Pawlowski, R.; Wullschleger, S.; Deak, M.; Ibrahim, A.F.; Gourlay, R.; Magnuson, M.A.; Alessi, D.R. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem. J. 2007, 405, 513–522. [Google Scholar] [CrossRef]
- Mizunuma, M.; Neumann-Haefelin, E.; Moroz, N.; Li, Y.J.; Blackwell, T.K. mTORC2-SGK-1 acts in two environmentally responsive pathways with opposing effects on longevity. Aging Cell 2014, 13, 869–878. [Google Scholar] [CrossRef]
- Chantaravisoot, N.; Wongkongkathep, P.; Loo, J.A.; Mischel, P.S.; Tamanoi, F. Significance of filamin A in mTORC2 function in glioblastoma. Mol. Cancer 2015, 14, 127. [Google Scholar] [CrossRef]
- Chen, B.W.; Chen, W.; Liang, H.; Liu, H.; Liang, C.; Zhi, X.; Hu, L.Q.; Yu, X.Z.; Wei, T.; Ma, T.; et al. Inhibition of mTORC2 Induces Cell-Cycle Arrest and Enhances the Cytotoxicity of Doxorubicin by Suppressing MDR1 Expression in HCC Cells. Mol. Cancer Ther. 2015, 14, 1805–1815. [Google Scholar] [CrossRef] [PubMed]
- Albert, V.; Svensson, K.; Shimobayashi, M.; Colombi, M.; Munoz, S.; Jimenez, V.; Handschin, C.; Bosch, F.; Hall, M.N. mTORC2 sustains thermogenesis via Akt-induced glucose uptake and glycolysis in brown adipose tissue. EMBO Mol. Med. 2016, 8, 232–246. [Google Scholar] [CrossRef]
- Liu, P.; Gan, W.; Chin, Y.R.; Ogura, K.; Guo, J.; Zhang, J.; Wang, B.; Blenis, J.; Cantley, L.C.; Toker, A.; et al. PtdIns(3,4,5)P3-Dependent Activation of the mTORC2 Kinase Complex. Cancer Discov. 2015, 5, 1194–1209. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Murashige, D.S.; Humphrey, S.J.; James, D.E. A Positive Feedback Loop between Akt and mTORC2 via SIN1 Phosphorylation. Cell Rep. 2015, 12, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, S.J.; Yang, G.; Yang, P.; Fazakerley, D.J.; Stockli, J.; Yang, J.Y.; James, D.E. Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab. 2013, 17, 1009–1020. [Google Scholar] [CrossRef]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef]
- Mabuchi, S.; Kuroda, H.; Takahashi, R.; Sasano, T. The PI3K/AKT/mTOR pathway as a therapeutic target in ovarian cancer. Gynecol. Oncol. 2015, 137, 173–179. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Brunn, G.J.; Hudson, C.C.; Sekulić, A.; Williams, J.M.; Hosoi, H.; Houghton, P.J.; Lawrence, J.C.; Abraham, R.T. Phosphorylation of the Translational Repressor PHAS-I by the Mammalian Target of Rapamycin. Science 1997, 277, 99–101. [Google Scholar] [CrossRef] [PubMed]
- Burnett, P.E.; Barrow, R.K.; Cohen, N.A.; Snyder, S.H.; Sabatini, D.M. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl. Acad. Sci. USA 1998, 95, 1432–1437. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Arias, E.; Cuervo, A.M. Chaperone-mediated autophagy in protein quality control. Curr. Opin. Cell Biol. 2011, 23, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Huber, L.A.; Teis, D. Lysosomal signaling in control of degradation pathways. Curr. Opin. Cell Biol. 2016, 39, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Sasaki, T.; Iemura, S.; Natsume, T.; Hara, T.; Mizushima, N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 2009, 5, 973–979. [Google Scholar] [CrossRef]
- Hurley, J.H.; Young, L.N. Mechanisms of Autophagy Initiation. Annu. Rev. Biochem. 2017, 86, 225–244. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Shang, L.; Chen, S.; Du, F.; Li, S.; Zhao, L.; Wang, X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc. Natl. Acad. Sci. USA 2011, 108, 4788–4793. [Google Scholar] [CrossRef]
- Lin, M.G.; Hurley, J.H. Structure and function of the ULK1 complex in autophagy. Curr. Opin. Cell Biol. 2016, 39, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Puente, C.; Hendrickson, R.C.; Jiang, X. Nutrient-regulated Phosphorylation of ATG13 Inhibits Starvation-induced Autophagy. J. Biol. Chem. 2016, 291, 6026–6035. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.F.; Chun, M.G.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.C.; Sheffler, D.J.; et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol. Cell 2015, 59, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Backer, J.M. The intricate regulation and complex functions of the Class III phosphoinositide 3-kinase Vps34. Biochem. J. 2016, 473, 2251–2271. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, K.; Morita, E.; Saitoh, T.; Akira, S.; Ktistakis, N.T.; Izumi, T.; Noda, T.; Yoshimori, T. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J. Cell Biol. 2010, 190, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Thapa, N.; Liao, Y.; Choi, S.; Anderson, R.A. PtdIns(4,5)P2 signaling regulates ATG14 and autophagy. Proc. Natl. Acad. Sci. USA 2016, 113, 10896–10901. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Cao, W.; Li, W.; Gao, C.; Qi, Z.; Zhao, Y.; Du, J.; Xue, H.; Peng, J.; Wen, J.; et al. Dapper1 promotes autophagy by enhancing the Beclin1-Vps34-Atg14L complex formation. Cell Res. 2014, 24, 912–924. [Google Scholar] [CrossRef]
- Byfield, M.P.; Murray, J.T.; Backer, J.M. hVps34 is a nutrient-regulated lipid kinase required for activation of p70 S6 kinase. J. Biol. Chem. 2005, 280, 33076–33082. [Google Scholar] [CrossRef]
- Nobukuni, T.; Joaquin, M.; Roccio, M.; Dann, S.G.; Kim, S.Y.; Gulati, P.; Byfield, M.P.; Backer, J.M.; Natt, F.; Bos, J.L.; et al. Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc. Natl. Acad. Sci. USA 2005, 102, 14238–14243. [Google Scholar] [CrossRef]
- Yuan, H.-X.; Russell, R.C.; Guan, K.-L. Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient stress-induced autophagy. Autophagy 2014, 9, 1983–1995. [Google Scholar] [CrossRef]
- Ohashi, Y.; Soler, N.; Garcia Ortegon, M.; Zhang, L.; Kirsten, M.L.; Perisic, O.; Masson, G.R.; Burke, J.E.; Jakobi, A.J.; Apostolakis, A.A.; et al. Characterization of Atg38 and NRBF2, a fifth subunit of the autophagic Vps34/PIK3C3 complex. Autophagy 2016, 12, 2129–2144. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Zhang, S.; He, L.; Rong, Y.; Brier, L.W.; Sun, Q.; Liu, R.; Fan, W.; Chen, S.; Yue, Z.; et al. MTORC1-mediated NRBF2 phosphorylation functions as a switch for the class III PtdIns3K and autophagy. Autophagy 2017, 13, 592–607. [Google Scholar] [CrossRef] [PubMed]
- Araki, Y.; Ku, W.C.; Akioka, M.; May, A.I.; Hayashi, Y.; Arisaka, F.; Ishihama, Y.; Ohsumi, Y. Atg38 is required for autophagy-specific phosphatidylinositol 3-kinase complex integrity. J. Cell Biol. 2013, 203, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; He, L.; Behrends, C.; Araki, M.; Araki, K.; Jun Wang, Q.; Catanzaro, J.M.; Friedman, S.L.; Zong, W.X.; Fiel, M.I.; et al. NRBF2 regulates autophagy and prevents liver injury by modulating Atg14L-linked phosphatidylinositol-3 kinase III activity. Nat. Commun. 2014, 5, 3920. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Cai, C.Z.; Song, J.X.; Tan, J.Q.; Durairajan, S.S.K.; Iyaswamy, A.; Wu, M.Y.; Chen, L.L.; Yue, Z.; Li, M.; et al. NRBF2 is involved in the autophagic degradation process of APP-CTFs in Alzheimer disease models. Autophagy 2017, 13, 2028–2040. [Google Scholar] [CrossRef] [PubMed]
- Puertollano, R.; Ferguson, S.M.; Brugarolas, J.; Ballabio, A. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef]
- Martina, J.A.; Diab, H.I.; Lishu, L.; Jeong, A.L.; Patange, S.; Raben, N.; Puertollano, R. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci. Signal. 2014, 7, ra9. [Google Scholar] [CrossRef]
- Raben, N.; Puertollano, R. TFEB and TFE3: Linking Lysosomes to Cellular Adaptation to Stress. Annu. Rev. Cell Dev. Biol. 2016, 32, 255–278. [Google Scholar] [CrossRef]
- Napolitano, G.; Esposito, A.; Choi, H.; Matarese, M.; Benedetti, V.; Di Malta, C.; Monfregola, J.; Medina, D.L.; Lippincott-Schwartz, J.; Ballabio, A. mTOR-dependent phosphorylation controls TFEB nuclear export. Nat. Commun. 2018, 9, 3312. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, L. Scissors for autolysosome tubules. EMBO J. 2015, 34, 2217–2218. [Google Scholar] [CrossRef] [PubMed]
- Munson, M.J.; Allen, G.F.; Toth, R.; Campbell, D.G.; Lucocq, J.M.; Ganley, I.G. mTOR activates the VPS34-UVRAG complex to regulate autolysosomal tubulation and cell survival. EMBO J. 2015, 34, 2272–2290. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhou, W.; Lin, J.; Wei, P.; Zhang, Y.; Jin, P.; Chen, M.; Man, N.; Wen, L. Autophagic lysosomal reformation depends on mTOR reactivation in H2O2-induced autophagy. Int. J. Biochem. Cell Biol. 2016, 70, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, W.; Lu, Y.; Tian, H.; Duan, C.; Lu, L.; Gao, G.; Wu, X.; Wang, X.; Yang, H. Piperlongumine restores the balance of autophagy and apoptosis by increasing BCL2 phosphorylation in rotenone-induced Parkinson disease models. Autophagy 2018, 14, 845–861. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [PubMed]
- Heikkila, R.E.; Manzino, L.; Cabbat, F.S.; Duvoisin, R.C. Protection against the dopaminergic neurotoxicity of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine by monoamine oxidase inhibitors. Nature 1984, 311, 467–469. [Google Scholar] [CrossRef]
- Recasens, A.; Dehay, B.; Bove, J.; Carballo-Carbajal, I.; Dovero, S.; Perez-Villalba, A.; Fernagut, P.O.; Blesa, J.; Parent, A.; Perier, C.; et al. Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann. Neurol. 2014, 75, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Krainc, D. alpha-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef]
- Crews, L.; Spencer, B.; Desplats, P.; Patrick, C.; Paulino, A.; Rockenstein, E.; Hansen, L.; Adame, A.; Galasko, D.; Masliah, E. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS ONE 2010, 5, e9313. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Duan, C.; Gao, G.; Wang, X.; Yang, H. Alpha-synuclein overexpression negatively regulates insulin receptor substrate 1 by activating mTORC1/S6K1 signaling. Int. J. Biochem. Cell Biol. 2015, 64, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.F.; Zhang, Y.J.; Zhou, H.Y.; Wang, H.M.; Tian, L.P.; Liu, J.; Ding, J.Q.; Chen, S.D. Curcumin ameliorates the neurodegenerative pathology in A53T alpha-synuclein cell model of Parkinson’s disease through the downregulation of mTOR/p70S6K signaling and the recovery of macroautophagy. J. Neuroimmune Pharmacol. 2013, 8, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Malagelada, C.; Ryu, E.J.; Biswas, S.C.; Jackson-Lewis, V.; Greene, L.A. RTP801 is elevated in Parkinson brain substantia nigral neurons and mediates death in cellular models of Parkinson’s disease by a mechanism involving mammalian target of rapamycin inactivation. J. Neurosci. 2006, 26, 9996–10005. [Google Scholar] [CrossRef] [PubMed]
- Corradetti, M.N.; Inoki, K.; Guan, K.L. The stress-inducted proteins RTP801 and RTP801L are negative regulators of the mammalian target of rapamycin pathway. J. Biol. Chem. 2005, 280, 9769–9772. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, S.; Sun, Y.; Watt, J.A.; Wang, S.; Lei, S.; Birnbaumer, L.; Singh, B.B. Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J. Clin. Investig. 2012, 122, 1354–1367. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, C.; Chen, S.; Ye, Y.; Guo, M.; Ren, Q.; Liu, L.; Zhang, H.; Xu, C.; Zhou, Q.; et al. Activation of AMPK and inactivation of Akt result in suppression of mTOR-mediated S6K1 and 4E-BP1 pathways leading to neuronal cell death in in vitro models of Parkinson’s disease. Cell Signal. 2014, 26, 1680–1689. [Google Scholar] [CrossRef]
- Rajput, A.H. Factors predictive of the development of levodopa-induced dyskinesia and Wearing-Off in Parkinson’s disease. Mov. Disord. 2014, 29, 429. [Google Scholar] [CrossRef]
- Huot, P.; Johnston, T.H.; Koprich, J.B.; Fox, S.H.; Brotchie, J.M. The pharmacology of L-DOPA-induced dyskinesia in Parkinson’s disease. Pharmacol. Rev. 2013, 65, 171–222. [Google Scholar] [CrossRef]
- Urs, N.M.; Bido, S.; Peterson, S.M.; Daigle, T.L.; Bass, C.E.; Gainetdinov, R.R.; Bezard, E.; Caron, M.G. Targeting beta-arrestin2 in the treatment of l-DOPA-induced dyskinesia in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2015, 112, E2517–E2526. [Google Scholar] [CrossRef]
- Martin-Flores, N.; Fernandez-Santiago, R.; Antonelli, F.; Cerquera, C.; Moreno, V.; Marti, M.J.; Ezquerra, M.; Malagelada, C. MTOR Pathway-Based Discovery of Genetic Susceptibility to L-DOPA-Induced Dyskinesia in Parkinson’s Disease Patients. Mol. Neurobiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Santini, E.; Heiman, M.; Greengard, P.; Valjent, E.; Fisone, G. Inhibition of mTOR signaling in Parkinson’s disease prevents L-DOPA-induced dyskinesia. Sci. Signal. 2009, 2, ra36. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.; Napolitano, F.; Mealer, R.G.; Kim, S.; Errico, F.; Barrow, R.; Shahani, N.; Tyagi, R.; Snyder, S.H.; Usiello, A. Rhes, a striatal-enriched small G protein, mediates mTOR signaling and L-DOPA-induced dyskinesia. Nat. Neurosci. 2011, 15, 191–193. [Google Scholar] [CrossRef] [PubMed]
- Brugnoli, A.; Napolitano, F.; Usiello, A.; Morari, M. Genetic deletion of Rhes or pharmacological blockade of mTORC1 prevent striato-nigral neurons activation in levodopa-induced dyskinesia. Neurobiol. Dis. 2016, 85, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Moors, T.E.; Hoozemans, J.J.; Ingrassia, A.; Beccari, T.; Parnetti, L.; Chartier-Harlin, M.C.; van de Berg, W.D. Therapeutic potential of autophagy-enhancing agents in Parkinson’s disease. Mol. Neurodegener. 2017, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Verstraeten, A.; Theuns, J.; Van Broeckhoven, C. Progress in unraveling the genetic etiology of Parkinson disease in a genomic era. Trends Genet. 2015, 31, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Przedborski, S. The two-century journey of Parkinson disease research. Nat. Rev. Neurosci. 2017, 18, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Gan-Or, Z.; Dion, P.A.; Rouleau, G.A. Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy 2015, 11, 1443–1457. [Google Scholar] [CrossRef]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Brown, E.; Hallett, P.; Isacson, O. Progressive decline of glucocerebrosidase in aging and Parkinson’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 433–438. [Google Scholar] [CrossRef]
- Decressac, M.; Mattsson, B.; Weikop, P.; Lundblad, M.; Jakobsson, J.; Björklund, A. TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc. Natl. Acad. Sci. USA 2013, E1817–E1826. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.; Yu, W.H.; Corti, O.; Mollereau, B.; Henriques, A.; Bezard, E.; Pastores, G.M.; Rubinsztein, D.C.; Nixon, R.A.; Duchen, M.R.; et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2018, 17, 660–688. [Google Scholar] [CrossRef]
- Dehay, B.; Bove, J.; Rodriguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef] [PubMed]
- Malagelada, C.; Jin, Z.H.; Jackson-Lewis, V.; Przedborski, S.; Greene, L.A. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J. Neurosci. 2010, 30, 1166–1175. [Google Scholar] [CrossRef] [PubMed]
- Bove, J.; Martinez-Vicente, M.; Vila, M. Fighting neurodegeneration with rapamycin: Mechanistic insights. Nat. Rev. Neurosci. 2011, 12, 437–452. [Google Scholar] [CrossRef]
- Liu, J.; Chen, M.; Wang, X.; Wang, Y.; Duan, C.; Gao, G.; Lu, L.; Wu, X.; Wang, X.; Yang, H. Piperine induces autophagy by enhancing protein phosphotase 2A activity in a rotenone-induced Parkinson’s disease model. Oncotarget 2016, 7, 60823–60843. [Google Scholar] [CrossRef] [PubMed]
- Bekinschtein, P.; Katche, C.; Slipczuk, L.N.; Igaz, L.M.; Cammarota, M.; Izquierdo, I.; Medina, J.H. mTOR signaling in the hippocampus is necessary for memory formation. Neurobiol. Learn. Mem. 2007, 87, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Hoeffer, C.A.; Klann, E. mTOR signaling: At the crossroads of plasticity, memory and disease. Trends Neurosci. 2010, 33, 67–75. [Google Scholar] [CrossRef]
- Sarkar, S.; Floto, R.A.; Berger, Z.; Imarisio, S.; Cordenier, A.; Pasco, M.; Cook, L.J.; Rubinsztein, D.C. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 2005, 170, 1101–1111. [Google Scholar] [CrossRef]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar] [CrossRef]
- Song, J.X.; Lu, J.H.; Liu, L.F.; Chen, L.L.; Durairajan, S.S.; Yue, Z.; Zhang, H.Q.; Li, M. HMGB1 is involved in autophagy inhibition caused by SNCA/alpha-synuclein overexpression: A process modulated by the natural autophagy inducer corynoxine B. Autophagy 2014, 10, 144–154. [Google Scholar] [CrossRef]
- Song, J.X.; Sun, Y.R.; Peluso, I.; Zeng, Y.; Yu, X.; Lu, J.H.; Xu, Z.; Wang, M.Z.; Liu, L.F.; Huang, Y.Y.; et al. A novel curcumin analog binds to and activates TFEB in vitro and in vivo independent of MTOR inhibition. Autophagy 2016, 12, 1372–1389. [Google Scholar] [CrossRef]
- Sarkar, S.; Ravikumar, B.; Floto, R.A.; Rubinsztein, D.C. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009, 16, 46–56. [Google Scholar] [CrossRef]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Domanskyi, A.; Geissler, C.; Vinnikov, I.A.; Alter, H.; Schober, A.; Vogt, M.A.; Gass, P.; Parlato, R.; Schutz, G. Pten ablation in adult dopaminergic neurons is neuroprotective in Parkinson’s disease models. FASEB J. 2011, 25, 2898–2910. [Google Scholar] [CrossRef]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef]
- Aspuria, P.J.; Tamanoi, F. The Rheb family of GTP-binding proteins. Cell Signal. 2004, 16, 1105–1112. [Google Scholar] [CrossRef]
- Potter, C.J.; Pedraza, L.G.; Xu, T. Akt regulates growth by directly phosphorylating Tsc2. Nat. Cell Biol. 2002, 4, 658. [Google Scholar] [CrossRef]
- Kim, S.R.; Chen, X.; Oo, T.F.; Kareva, T.; Yarygina, O.; Wang, C.; During, M.; Kholodilov, N.; Burke, R.E. Dopaminergic pathway reconstruction by Akt/Rheb-induced axon regeneration. Ann. Neurol. 2011, 70, 110–120. [Google Scholar] [CrossRef]
- Iwakawa, H.O.; Tomari, Y. The Functions of MicroRNAs: mRNA Decay and Translational Repression. Trends Cell Biol. 2015, 25, 651–665. [Google Scholar] [CrossRef]
- Wilczynska, A.; Bushell, M. The complexity of miRNA-mediated repression. Cell Death Differ. 2015, 22, 22–33. [Google Scholar] [CrossRef]
- Ma, L.; Wei, L.; Wu, F.; Hu, Z.; Liu, Z.; Yuan, W. Advances with microRNAs in Parkinson’s disease research. Drug Des. Dev.Ther. 2013, 7, 1103–1113. [Google Scholar] [CrossRef]
- Fragkouli, A.; Doxakis, E. miR-7 and miR-153 protect neurons against MPP(+)-induced cell death via upregulation of mTOR pathway. Front. Cell. Neurosci. 2014, 8, 182. [Google Scholar] [CrossRef]
- Bee, J.; Fuller, S.; Miller, S.; Johnson, S.R. Lung function response and side effects to rapamycin for lymphangioleiomyomatosis: A prospective national cohort study. Thorax 2018, 73, 369–375. [Google Scholar] [CrossRef]
- Lamming, D.W.; Ye, L.; Sabatini, D.M.; Baur, J.A. Rapalogs and mTOR inhibitors as anti-aging therapeutics. J. Clin. Investig. 2013, 123, 980–989. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Z.; Yang, C.; Iyaswamy, A.; Krishnamoorthi, S.; Sreenivasmurthy, S.G.; Liu, J.; Wang, Z.; Tong, B.C.-K.; Song, J.; Lu, J.; et al. Balancing mTOR Signaling and Autophagy in the Treatment of Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 728. https://doi.org/10.3390/ijms20030728
Zhu Z, Yang C, Iyaswamy A, Krishnamoorthi S, Sreenivasmurthy SG, Liu J, Wang Z, Tong BC-K, Song J, Lu J, et al. Balancing mTOR Signaling and Autophagy in the Treatment of Parkinson’s Disease. International Journal of Molecular Sciences. 2019; 20(3):728. https://doi.org/10.3390/ijms20030728
Chicago/Turabian StyleZhu, Zhou, Chuanbin Yang, Ashok Iyaswamy, Senthilkumar Krishnamoorthi, Sravan Gopalkrishnashetty Sreenivasmurthy, Jia Liu, Ziying Wang, Benjamin Chun-Kit Tong, Juxian Song, Jiahong Lu, and et al. 2019. "Balancing mTOR Signaling and Autophagy in the Treatment of Parkinson’s Disease" International Journal of Molecular Sciences 20, no. 3: 728. https://doi.org/10.3390/ijms20030728
APA StyleZhu, Z., Yang, C., Iyaswamy, A., Krishnamoorthi, S., Sreenivasmurthy, S. G., Liu, J., Wang, Z., Tong, B. C. -K., Song, J., Lu, J., Cheung, K. -H., & Li, M. (2019). Balancing mTOR Signaling and Autophagy in the Treatment of Parkinson’s Disease. International Journal of Molecular Sciences, 20(3), 728. https://doi.org/10.3390/ijms20030728