The Effect of Single CpG Demethylation on the Pattern of DNA-Protein Binding


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
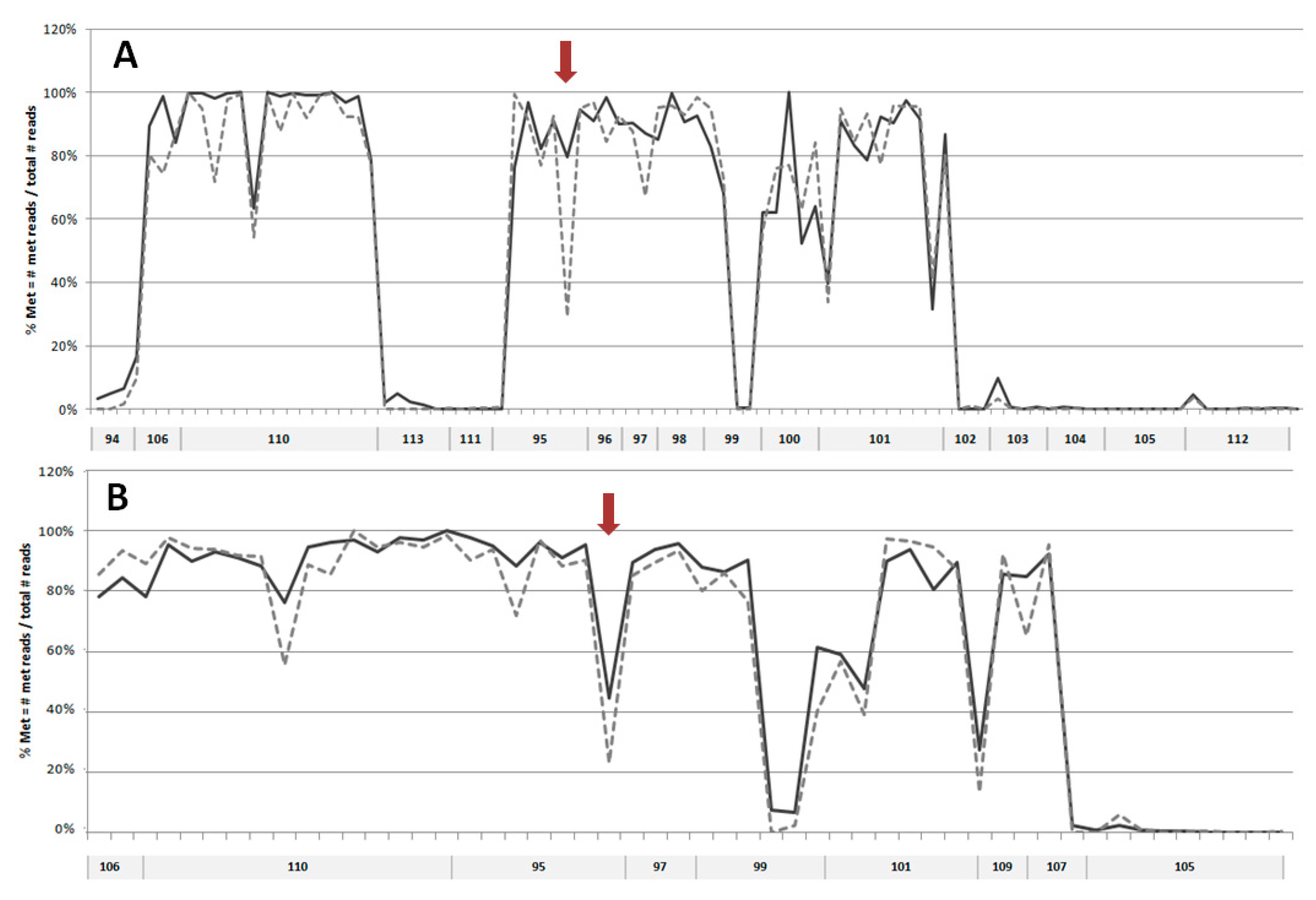
2.1. DNA Methylation Analysis
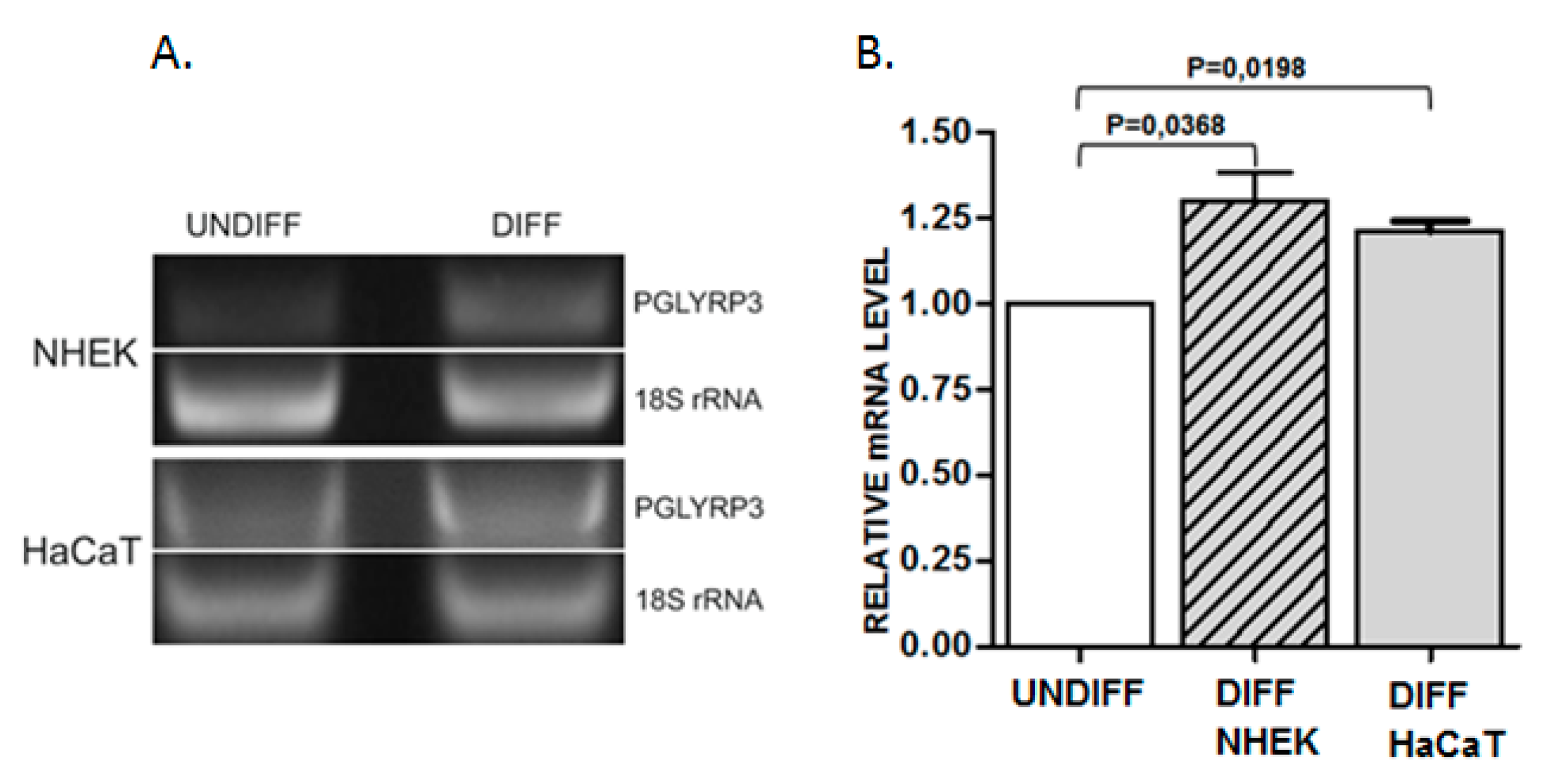
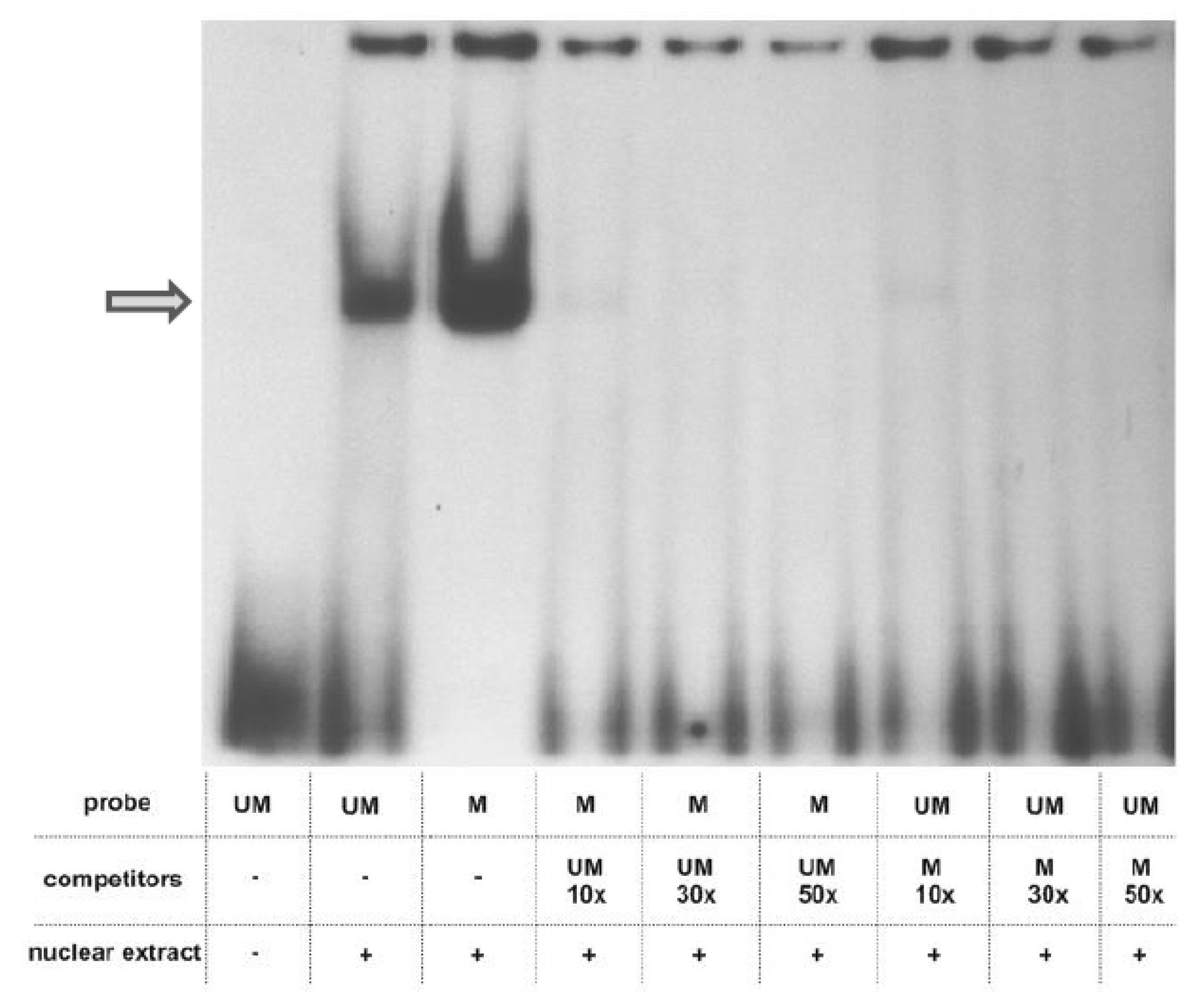
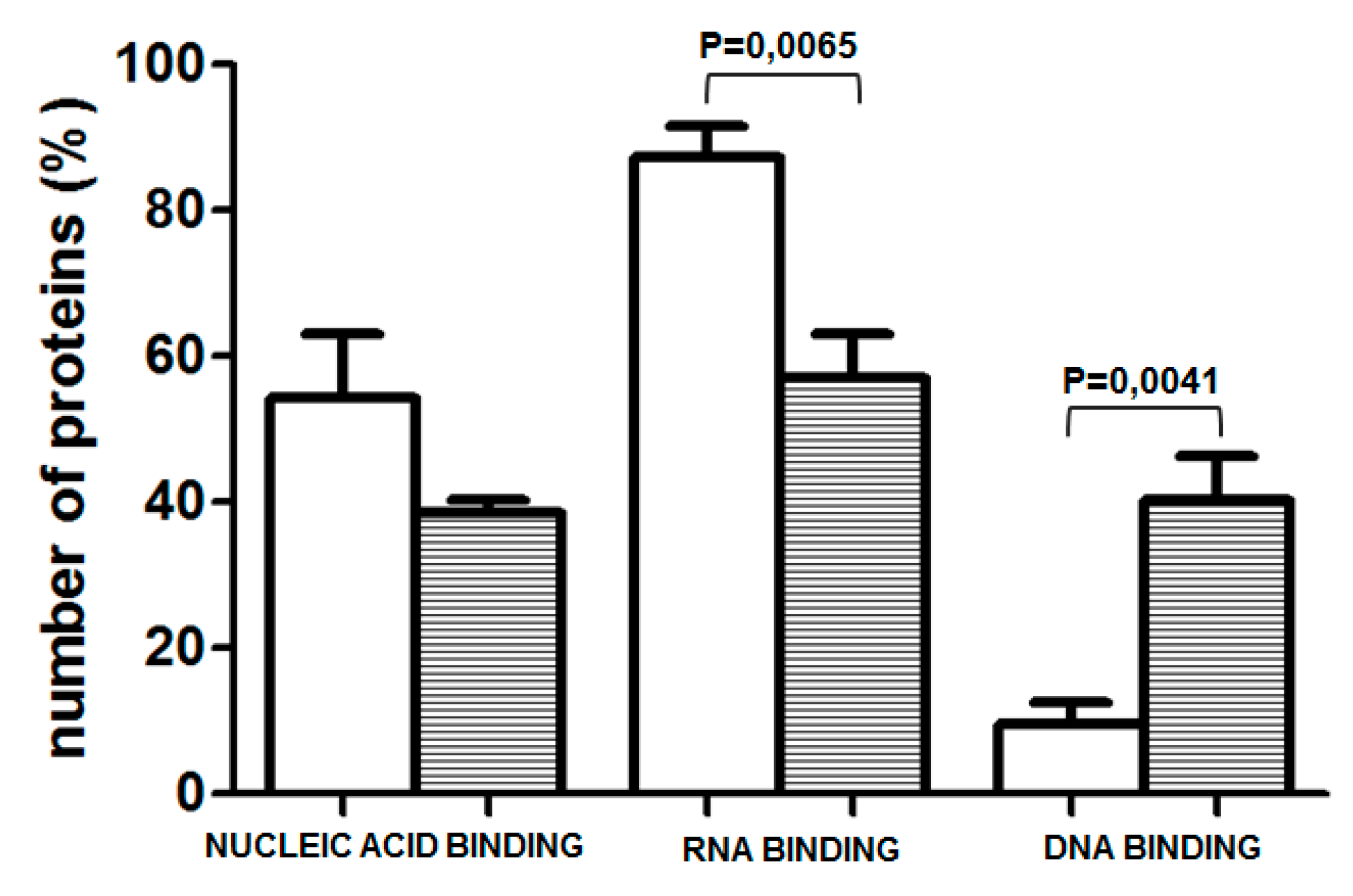
2.2. Analysis of Protein Binding to a Sequence Containing the Variably Methylated CpG Pair
3. Discussion
4. Methods
4.1. Cell Culture
4.2. DNA Methylation Analysis
4.3. Preparation of Nuclear Extracts
4.4. RNA Isolation and PCR
4.5. Electrophoretic Mobility Shift Assay (EMSA)
4.6. DNA Affinity Precipitation Assay (DAPA)
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Fuchs, E.; Raghavan, S. Getting under the skin of epidermal morphogenesis. Nat. Rev. Genet. 2002, 3, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Mischke, D.; Korge, B.P.; Marenholz, I.; Volz, A.; Ziegler, A. Genes encoding structural proteins of epidermal cornification and S100 calcium-binding proteins form a gene complex “epidermal differentiation complex” on human chromosome 1q21. J. Investig. Dermatol. 1996, 106, 989–992. [Google Scholar] [CrossRef] [PubMed]
- Cabral, A.; Voskamp, P.; Cleton-Jansen, A.M.; South, A.; Nizetic, D.; Backendorf, C. Structural organization and regulation of the small proline-rich family of cornified envelope precursors suggest a role in adaptive barrier function. J. Biol. Chem. 2001, 276, 19231–19237. [Google Scholar] [CrossRef] [PubMed]
- Kypriotou, M.; Huber, M.; Hohl, D. The human epidermal differentiation complex: Cornified envelope precursors, S100 proteins and the “fused genes” family. Exp. Dermatol. 2012, 21, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Leśniak, W.; Graczyk-Jarzynka, A. The S100 proteins in epidermis: Topology and function. Biochim. Biophys. Acta 2015, 1850, 2563–2572. [Google Scholar] [CrossRef] [PubMed]
- Bickenbach, J.R.; Greer, J.M.; Bundman, D.S.; Rothnagel, J.A.; Roop, D.R. Loricrin expression is coordinated with other epidermal proteins and the appearance of lipid lamellar granules in development. J. Investig. Dermatol. 1995, 104, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.P.; Wolffe, A.P. Methylation-induced repression-belts, braces and chromatin. Cell 1999, 99, 451–454. [Google Scholar] [CrossRef]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Das, P.M.; Singal, R. DNA Methylation and Cancer. J. Clin. Oncol. 2004, 22, 4632–4642. [Google Scholar] [CrossRef]
- Crider, K.S.; Yang, T.P.; Berry, R.J.; Bailey, L.B. Folate and DNA methylation: A review of molecular mechanisms and the evidence for folate’s role. Adv. Nutr. 2012, 3, 21–38. [Google Scholar] [CrossRef]
- Johnson, A.A.; Akman, K.; Calimport, S.R.G.; Wuttke, D.; Stolzing, A.; de Magalhaes, J.P. The role of DNA methylation in aging, rejuvenation, and age-related disease. Rejuv. Res. 2012, 15, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Laurent, L.; Wong, E.; Li, G.; Huynh, T.; Tsirigos, A.; Ong, C.T.; Low, H.M.; Kin Sung, K.W.; Rigoutsos, I.; Loring, J.; et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010, 20, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Elder, J.T.; Zhao, X. Evidence for local control of gene expression in the epidermal differentiation complex. Exp. Dermatol. 2002, 11, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Sen, G.L.; Reuter, J.A.; Webster, D.E.; Zhu, L.; Khavari, P.A. DNMT1 maintains progenitor function in self-renewing somatic tissue. Nature 2010, 463, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Roberson, E.D.O.; Liu, Y.; Ryan, C.; Joyce, C.E.; Duan, S.; Cao, L.; Martin, A.; Liao, W.; Menter, A.; Bowcock, A.M. A subset of methylated CpG sites differentiate psoriatic from normal skin. J. Investig. Dermatol. 2012, 132, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Sobiak, B.; Graczyk-Jarzynka, A.; Leśniak, W. Comparison of DNA methylation and expression pattern of S100 and other Epidermal Differentiation Complex genes in differentiating keratinocytes. J. Cell. Biochem. 2016, 11, 1092–1098. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, G.; Qian, J. Transcription factors as readers and effectors of DNA methylation. Nat. Rev. Genet. 2016, 17, 551–565. [Google Scholar] [CrossRef]
- Cavazza, A.; Miccio, A.; Romano, O.; Petiti, L.; Malagoli Tagliazucchi, G.; Peano, C.; Severgnini, M.; Rizzi, E.; De Bellis, G.; Bicciato, S.; et al. Dynamic transcriptional and epigenetic regulation of human epidermal keratinocyte differentiation. Stem Cell Rep. 2016, 6, 618–632. [Google Scholar] [CrossRef]
- Dziarski, R.; Gupta, D. The peptidoglycan recognition proteins (PGRPs). Genome Biol. 2006, 7, 232. [Google Scholar] [CrossRef]
- Strasser, B.; Mlitz, V.; Hermann, M.; Rice, R.H.; Eigenheer, R.A.; Alibardi, L.; Tschachler, E.; Eckhart, L. Evolutionary origin and diversification of epidermal barrier proteins in Amniotes. Mol. Biol. Evol. 2014, 31, 3194–3205. [Google Scholar] [CrossRef]
- Ma, P.; Wang, Z.; Pflugfelder, S.C.; Li, D.Q. Toll-like receptors mediate induction of peptidoglycan recognition proteins in human corneal epithelial cells. Exp. Eye Res. 2010, 90, 130–136. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Park, S.Y.; Gupta, D.; Kim, C.H.; Dziarski, R. Differential effects of peptidoglycan recognition proteins on experimental atopic and contact dermatitis mediated by Treg and Th17 cells. PLoS ONE 2011, 6, e24961. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Chen, M.; Liu, L.; Cheng, H.; Yan, Y.E.; Feng, Y.H.; Wang, H. Nicotine induced CpG methylation of Pax6 binding motif in StAR promoter reduces the gene expression and cortisol production. Toxicol. Appl. Pharmacol. 2011, 257, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, M.; Xiao, H.; Lee, M.T.; Levin, L.; Leung, Y.K.; Ho, S.M. Methylation of a single intronic CpG mediates expression silencing of the PMP24 gene in prostate cancer. Prostate 2010, 70, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Venza, I.; Visalli, M.; Fortunato, C.; Ruggeri, M.; Ratone, S.; Caffo, M.; Caruso, G.; Alafaci, C.; Tomasello, F.; Teti, D.; et al. PGE2 induces interleukin-8 derepression in human astrocytoma through coordinated DNA demethylation and histone hyperacetylation. Epigenetics 2012, 7, 1315–1330. [Google Scholar] [CrossRef]
- Ceccarelli, V.; Racanicchi, S.; Martelli, M.P.; Nocentini, G.; Fettucciari, K.; Riccardi, C.; Marconi, P.; Di Nardo, P.; Grignani, F.; Binaglia, L.; et al. Eicosapentaenoic acid demethylates a single CpG that mediates expression of tumor suppressor CCAAT/enhancer-binding protein delta in U937 leukemia cells. J. Biol. Chem. 2011, 286, 27092–27102. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.E.; Patel, Z.H.; Lu, X.; Lynch, A.T.; Weirauch, M.T.; Kottyan, L.C. Screening for functional non-coding genetic variants using electrophoretic mobility shift assay (EMSA) and DNA-affinity precipitation assay (DAPA). J. Vis. Exp. 2016, 114, 54093. [Google Scholar] [CrossRef]
- Iurlaro, M.; Ficz, G.; Oxley, D.; Raiber, E.A.; Bachman, M.; Booth, M.J.; Andrews, S.; Balasubramanian, S.; Reik, W. A screen for hydroxymethylcytosine and formylcytosine binding proteins suggests functions in transcription and chromatin regulation. Genome Biol. 2013, 14, R119. [Google Scholar] [CrossRef]
- Maor, G.L.; Yearim, A.; Ast, G. The alternative role of DNA methylation in splicing regulation. Trends Genet. 2015, 31, 274–280. [Google Scholar] [CrossRef]
- Graczyk, A.; Lesniak, W. S100A6 expression in keratinocytes and its impact on epidermal differentiation. Int. J. Biochem. Cell Biol. 2014, 57, 135–141. [Google Scholar] [CrossRef]
- Leśniak, W.; Jezierska, A.; Kuznicki, J. Upstream stimulatory factor is involved in the regulation of the human calcyclin (S100A6) gene. Biochim. Biophys. Acta 2000, 1517, 73–81. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sobiak, B.; Leśniak, W. The Effect of Single CpG Demethylation on the Pattern of DNA-Protein Binding. Int. J. Mol. Sci. 2019, 20, 914. https://doi.org/10.3390/ijms20040914
Sobiak B, Leśniak W. The Effect of Single CpG Demethylation on the Pattern of DNA-Protein Binding. International Journal of Molecular Sciences. 2019; 20(4):914. https://doi.org/10.3390/ijms20040914
Chicago/Turabian StyleSobiak, Barbara, and Wiesława Leśniak. 2019. "The Effect of Single CpG Demethylation on the Pattern of DNA-Protein Binding" International Journal of Molecular Sciences 20, no. 4: 914. https://doi.org/10.3390/ijms20040914
APA StyleSobiak, B., & Leśniak, W. (2019). The Effect of Single CpG Demethylation on the Pattern of DNA-Protein Binding. International Journal of Molecular Sciences, 20(4), 914. https://doi.org/10.3390/ijms20040914