Improving the Performance of Horseradish Peroxidase by Site-Directed Mutagenesis


Abstract
1. Introduction
2. Results and Discussion
2.1. Protein Production
2.2. Biochemical Characterization
2.2.1. Biochemical Characterization of Benchmark rHRP and Seven rHRP Mutants
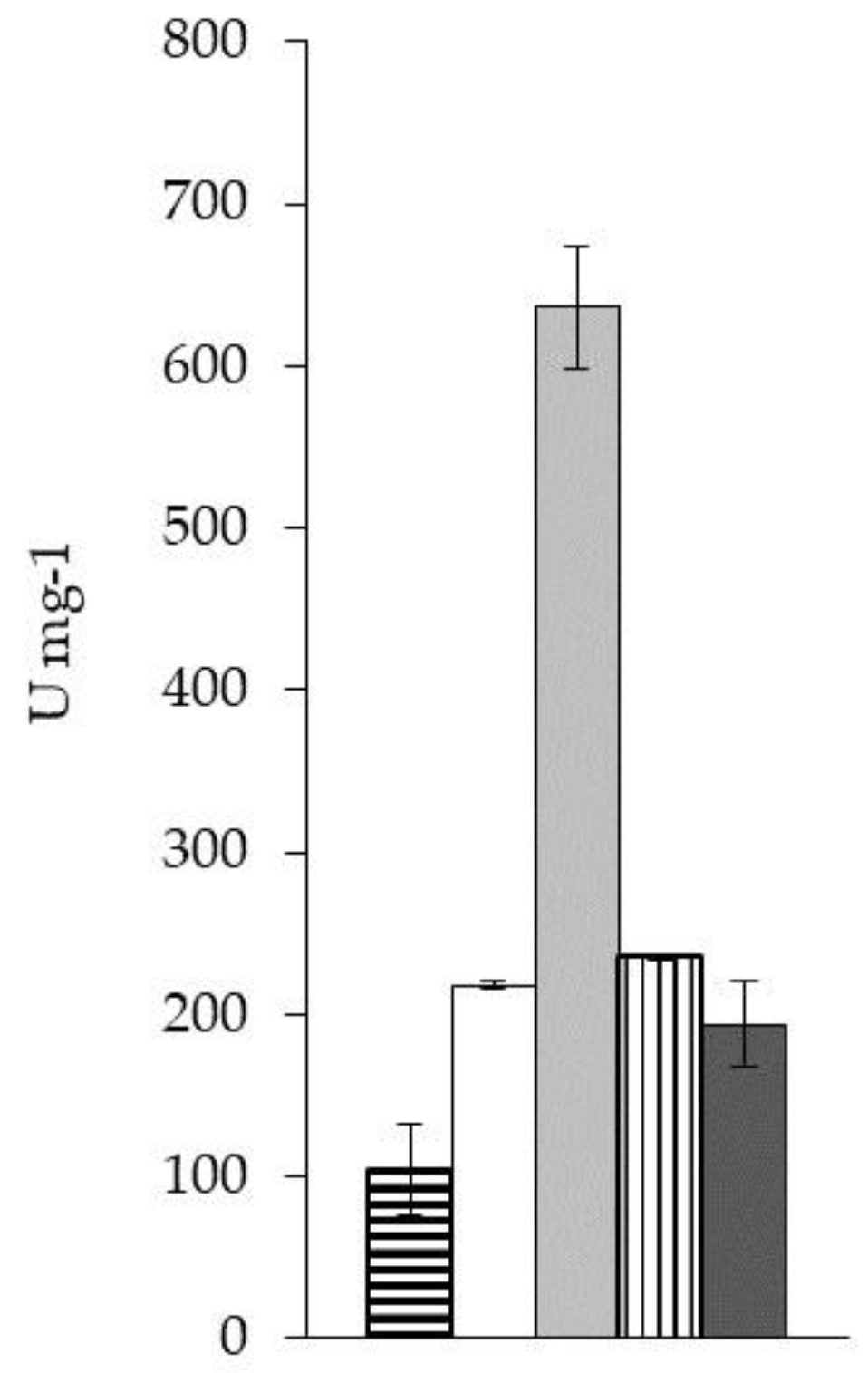
2.2.1.1. Enzyme Kinetics
2.2.1.2. Thermal Stability
2.2.2. Catalytic Activity of Plant HRP under Different Conditions
2.2.3. Optimized Biochemical Characterization of Benchmark rHRP and Mutant N13D/N57S/N255D/N268D
2.2.3.1. Enzyme Activity
2.2.3.2. Thermal Stability
3. Materials and Methods
3.1. Chemicals
3.2. Strains and Plasmids
3.3. Strain Generation by Site-Directed Mutagenesis
3.4. Growth Conditions and Protein Production
3.5. Protein Purification
3.6. SDS PAGE
3.7. Biochemical Enzyme Characterization
3.7.1. Assay Development
3.7.1.1. Pathlength Correction for Microplates
3.7.1.2. Determination of Extinction Coefficient for ABTS
3.7.1.3. Enzyme Activity Calculation
- d = dilution factor
- p = pathlength in cm, which is 1 for cuvettes and 0.58 in a 96-well plate when the reaction volume is 200 µL (see Section 3.7.1.1).
- ε = 27 mM−1 cm−1
3.7.2. Catalytic Activity of Plant HRP under Different Conditions
3.7.3. Enzyme Kinetics
3.7.3.1. Hitachi U-2900 Spectrophotometer
3.7.3.2. Tecan Infinite M200 PRO
3.7.4. Thermal Stability
- t1/2 = half life
- kin = slope of the logarithmic residual activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABTS | 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) |
| BisTris | Bis(2-hydroxyethyl)amino-tris(hydroxymethyl)methane |
| CV | Column volumes |
| DCW | Dry cell weight [g L−1] |
| ELISA | Enzyme-linked Immunosorbent Assay |
| EMSA | Electrophoretic Mobility Shift Assay |
| ER | Endoplasmatic reticulum |
| FDU | FastDigest unit, 1 µL of enzyme (1 FDU) cleaves 1 µg of DNA substrate in 5–15 min at 37 °C in 20 µL of 1× FastDigest buffer |
| HRP | Plant horseradish peroxidase |
| IAA | Indole-3-acetic acid |
| IMAC | Immobilized Metal Affinity Chromatography |
| IPTG | Isopropyl β-d-1-thiogalactopyranoside |
| rHRP | Recombinant Horseradish Peroxidase |
| SDS | Sodium dodecyl sulfate |
| UYF | Ultra Yield™ |
References
- Gholami-Borujeni, F.; Mahvi, A.H.; Naseri, S.; Faramarzi, M.A.; Nabizadeh, R.; Alimohammadi, M. Application of immobilized horseradish peroxidase for removal and detoxification of azo dye from aqueous solution. Res. J. Chem. Environ. 2011, 15, 217–222. [Google Scholar]
- Krieg, R.; Halbhuber, K. Recent advances in catalytic peroxidase histochemistry. Cell. Mol. Biol. (Noisy-le-Grand) 2003, 49, 547–563. [Google Scholar]
- Litescu, S.C.; Eremia, S.; Radu, G.L. Biosensors for the determination of phenolic metabolites. In Bio-Farms for Nutraceuticals; Springer: Boston, MA, USA, 2010; pp. 234–240. [Google Scholar] [CrossRef]
- Marquette, C.A.; Blum, L.J. Chemiluminescent enzyme immunoassays: A review of bioanalytical applications. Bioanalysis 2009, 1, 1259–1269. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.J.; Carolan, N.; O’Fagain, C. Horseradish and soybean peroxidases: Comparable tools for alternative niches? Trends Biotechnol. 2006, 24, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Vasileva, N.; Godjevargova, T.; Ivanova, D.; Gabrovska, K. Application of immobilized horseradish peroxidase onto modified acrylonitrile copolymer membrane in removing of phenol from water. Int. J. Biol. Macromol. 2009, 44, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Yang, H. Enzyme-based ultrasensitive electrochemical biosensors. Curr. Opin. Chem. Biol. 2012, 16, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Welinder, K.G. Covalent structure of the glycoprotein horseradish peroxidase (EC 1.11. 1.7). FEBS Lett. 1976, 72, 19–23. [Google Scholar] [CrossRef]
- Veitch, N.C. Horseradish peroxidase: A modern view of a classic enzyme. Phytochemistry 2004, 65, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Huddy, S.M.; Hitzeroth, I.I.; Meyers, A.E.; Weber, B.; Rybicki, E.P. Transient Expression and Purification of Horseradish Peroxidase C in Nicotiana benthamiana. Int. J. Mol. Sci. 2018, 19, 115. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Tabayashi, A.; Iwano, M.; Shinmyo, A.; Kato, K.; Nakayama, H. Activity of the C-terminal-dependent vacuolar sorting signal of horseradish peroxidase C1a is enhanced by its secondary structure. Plant Cell Physiol. 2011, 52, 413–420. [Google Scholar] [CrossRef]
- Matsui, T.; Nakayama, H.; Yoshida, K.; Shinmyo, A. Vesicular transport route of horseradish C1a peroxidase is regulated by N-and C-terminal propeptides in tobacco cells. Appl. Microbiol. Biotechnol. 2003, 62, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Wuhrer, M.; Hokke, C.H.; Deelder, A.M. Glycopeptide analysis by matrix-assisted laser desorption/ionization tandem time-of-flight mass spectrometry reveals novel features of horseradish peroxidase glycosylation. Rapid Commun. Mass Spectrom. 2004, 18, 1741–1748. [Google Scholar] [CrossRef] [PubMed]
- Wuhrer, M.; Balog, C.I.; Koeleman, C.A.; Deelder, A.M.; Hokke, C.H. New features of site-specific horseradish peroxidase (HRP) glycosylation uncovered by nano-LC-MS with repeated ion-isolation/fragmentation cycles. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2005, 1723, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Krainer, F.W.; Glieder, A. An updated view on horseradish peroxidases: Recombinant production and biotechnological applications. Appl. Microbiol. Biotechnol. 2015, 99, 1611–1625. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.T.; Santama, N.; Dacey, S.; Edwards, M.; Bray, R.C.; Thorneley, R.; Burke, J.F. Expression of a synthetic gene for horseradish peroxidase C in Escherichia coli and folding and activation of the recombinant enzyme with Ca2+ and heme. J. Biol. Chem. 1990, 265, 13335–13343. [Google Scholar] [PubMed]
- Gundinger, T.; Spadiut, O. A comparative approach to recombinantly produce the plant enzyme horseradish peroxidase in Escherichia coli. J. Biotechnol. 2017, 248, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Asad, S.; Khajeh, K.; Ghaemi, N. Investigating the structural and functional effects of mutating Asn glycosylation sites of horseradish peroxidase to Asp. Appl. Biochem. Biotechnol. 2011, 164, 454–463. [Google Scholar] [CrossRef]
- Capone, S.; Pletzenauer, R.; Maresch, D.; Metzger, K.; Altmann, F.; Herwig, C.; Spadiut, O. Glyco-variant library of the versatile enzyme horseradish peroxidase. Glycobiology 2014, 24, 852–863. [Google Scholar] [CrossRef]
- Asad, S.; Dastgheib, S.M.; Khajeh, K. Construction of a horseradish peroxidase resistant toward hydrogen peroxide by saturation mutagenesis. Biotechnol. Appl. Biochem. 2016, 63, 789–794. [Google Scholar] [CrossRef]
- Lin, Z.; Thorsen, T.; Arnold, F.H. Functional expression of horseradish peroxidase in E. coli by directed evolution. Biotechnol. Prog. 1999, 15, 467–471. [Google Scholar] [CrossRef]
- Ryan, B.J.; Ó’Fágáin, C. Effects of mutations in the helix G region of horseradish peroxidase. Biochimie 2008, 90, 1414–1421. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.J.; Ó’Fágáin, C. Effects of single mutations on the stability of horseradish peroxidase to hydrogen peroxide. Biochimie 2007, 89, 1029–1032. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.J.; O’Connell, M.J.; Ó’Fágáin, C. Consensus mutagenesis reveals that non-helical regions influence thermal stability of horseradish peroxidase. Biochimie 2008, 90, 1389–1396. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.J.; Ó’Fágáin, C. Arginine-to-lysine substitutions influence recombinant horseradish peroxidase stability and immobilisation effectiveness. BMC Biotechnol. 2007, 7, 86. [Google Scholar] [CrossRef] [PubMed]
- Morawski, B.; Quan, S.; Arnold, F.H. Functional expression and stabilization of horseradish peroxidase by directed evolution in Saccharomyces cerevisiae. Biotechnol. Bioeng. 2001, 76, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Morawski, B.; Lin, Z.; Cirino, P.; Joo, H.; Bandara, G.; Arnold, F.H. Functional expression of horseradish peroxidase in Saccharomyces cerevisiae and Pichia pastoris. Protein Eng. 2000, 13, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.A. Appropriate glycosylation of recombinant proteins for human use. Mol. Biotechnol. 2004, 28, 241–255. [Google Scholar] [CrossRef]
- Tupper, J.; Tozer, G.M.; Dachs, G.U. Use of horseradish peroxidase for gene-directed enzyme prodrug therapy with paracetamol. Br. J. Cancer 2004, 90, 1858–1862. [Google Scholar] [CrossRef]
- Dai, M.; Liu, J.; Chen, D.E.; Rao, Y.; Tang, Z.J.; Ho, W.Z.; Dong, C.Y. Tumor-targeted gene therapy using Adv-AFP-HRPC/IAA prodrug system suppresses growth of hepatoma xenografted in mice. Cancer Gene Ther. 2012, 19, 77–83. [Google Scholar] [CrossRef][Green Version]
- Folkes, L.; Candeias, L.; Wardman, P. Toward targeted “oxidation therapy” of cancer: Peroxidase-catalysed cytotoxicity of indole-3-acetic acids. Int. J. Radiat. Oncol. Biol. Phys. 1998, 42, 917–920. [Google Scholar] [CrossRef]
- Folkes, L.K.; Wardman, P. Oxidative activation of indole-3-acetic acids to cytotoxic species—A potential new role for plant auxins in cancer therapy. Biochem. Pharmacol. 2001, 61, 129–136. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Grigorenko, V.; Chubar, T.; Kapeliuch, Y.; Börchers, T.; Spener, F.; Egorova, A. New approaches for functional expression of recombinant horseradish peroxidase C in Escherichia coli. Biocatal. Biotransform. 1999, 17, 359–379. [Google Scholar] [CrossRef]
- Gilfoyle, D.J.; Rodriguez-Lopez, J.N.; Smith, A.T. Probing the aromatic-donor-binding site of horseradish peroxidase using site-directed mutagenesis and the suicide substrate phenylhydrazine. Eur. J. Biochem. 1996, 236, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Asad, S.; Torabi, S.-F.; Fathi-Roudsari, M.; Ghaemi, N.; Khajeh, K. Phosphate buffer effects on thermal stability and H2O2-resistance of horseradish peroxidase. Int. J. Biol. Macromol. 2011, 48, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Haifeng, L.; Yuwen, L.; Xiaomin, C.; Zhiyong, W.; Cunxin, W. Effects of sodium phosphate buffer on horseradish peroxidase thermal stability. J. Therm. Anal. Calorim. 2008, 93, 569–574. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]


{kind=link}
| Mutation | Effect | Reference |
|---|---|---|
| N13D | Increased stability towards H2O2 Increased thermal stability | Asad et al. [18] Capone et al. [19] |
| N268D | Increased stability towards H2O2 Increased thermal stability Increased substrate specificity for phenol/4-aminoantipyrine Increased activity with phenol/4-aminoantipyrine | Asad et al. [18] Asad et al. [20] Capone et al. [19] |
| N268G | Increased stability towards H2O2 Increased thermal stability Increased substrate specificity for phenol/4-aminoantipyrine | Asad et al. [20] |
| N57S | Increased activity with ABTS Increased activity with H2O2 Increased thermal stability | Capone et al. [19] |
| N186D | Increased activity with ABTS | Capone et al. [19] |
| N198D | Increased substrate specificity for ABTS | Capone et al. [19] |
| N255D | Better folding in E. coli Increased activity with ABTS Increased activity with H2O2 | Lin et al. [21] Capone et al. [19] |
| N158D | Increased activity with H2O2 | Capone et al. [19] |
| K232N | Increased activity with ABTS Increased thermal stability Increased solvent stability Increased stability towards H2O2 | Ryan et al. [22] Ryan et al. [23] |
| K232F | Increased activity with ABTS Increased thermal stability Increased solvent stability | Ryan et al. [22] |
| E238Q | Increased substrate specificity for ABTS | Ryan et al. [22] |
| K241N | Increased activity with ABTS | Ryan et al. [22] |
| K241E | Increased substrate specificity for ABTS Increased activity with ABTS | Ryan et al. [22] |
| K241A | Increased activity with ABTS Increased stability towards H2O2 | Ryan et al. [22] Ryan et al. [23] |
| K232N/K241N | Increased thermal stability Increased stability towards H2O2 | Ryan et al. [22] Ryan et al. [23] |
| K232F/K241N | Increased activity with ABTS Increased thermal stability Increased solvent stability Increased stability towards H2O2 | Ryan et al. [22] Ryan et al. [23] |
| K232Q/K241Q | Increased activity with ABTS | Ryan et al. [22] |
| T110V | Increased stability towards H2O2 Increased thermal stability | Ryan et al. [24] Ryan et al. [23] |
| T102A | Increased activity with ABTS | Ryan et al. [24] |
| K232E | Increased stability towards H2O2 | Ryan et al. [23] |
| K241F | Increased stability towards H2O2 | Ryan et al. [23] |
| R118K/R159K/K232N/K241F | Increased thermal stability Increased stability towards H2O2 | Ryan et al. [25] |
| 13A7 * | Increased activity with guaiacol | Morawski et al. [26] |
| H2-10G5 * | Increased activity with guaiacol Increased activity with ABTS Increased pH stability Increased thermal stability Increased stability towards SDS/urea/sodium chloride | Morawski et al. [26] |
| 13A7-N175S * | Increased activity with guaiacol Increased activity with ABTS Increased pH stability Increased thermal stability Increased stability towards SDS/urea/sodium chloride | Morawski et al. [26] |
| 13A10 * | Increased activity with guaiacol Increased activity with ABTS | Morawski et al. [27] |
| 17E12 * | Increased activity with guaiacol Increased activity with ABTS | Morawski et al. [27] |
| HRP variant | Km [mM] | Vmax [mol−1 L−1 × s] | Kcat [s−1] | Kcat/Km [mM−1 s−1] |
|---|---|---|---|---|
| Benchmark rHRP | 2.82 ± 1.52 | 1.7 × 10−6 ± 4.3 × 10−7 | 1.52 ± 0.38 | 0.54 ± 0.32 |
| N13D | 3.29 ± 0.33 | 8.8 × 10−7 ± 4.0 × 10−8 | 1.04 ± 0.05 | 0.32 ± 0.04 |
| N57S | 3.22 ± 0.59 | 2.7 × 10−6 ± 2.6 × 10−7 | 2.10 ± 0.19 | 0.64 ± 0.13 |
| N255D | 4.37 ± 0.86 | 8.9 × 10−7 ± 1.0 × 10−7 | 1.10 ± 0.12 | 0.24 ± 0.05 |
| N268D | 7.85 ± 4.98 | 2.4 × 10−6 ± 1.0 × 10−6 | 3.00 ± 1.25 | 0.38 ± 0.29 |
| N57S/N268D | 4.18 ± 3.55 | 1.0 × 10−6 ± 3.9 × 10−7 | 1.50 ± 0.58 | 0.36 ± 0.34 |
| N57S/N255D/N268D | 9.52 ± 6.89 | 4.0 × 10−6 ± 2.0 × 10−6 | 4.81 ± 2.37 | 0.51 ± 0.44 |
| N13D/N57S/N255D/N268D | 13.6 ± 6.63 | 1.7 × 10−5 ± 5.6 × 10−6 | 15.1 ± 5.00 | 1.11 ± 0.65 |
| HRP Type VI−A | 9.46 ± 5.18 | 5.7 × 10−3 ± 1.8 × 10−3 | 271 ± 87.4 | 28.7 ± 18.3 |
| HRP Variant | t1/2 at 60 °C |
|---|---|
| Benchmark rHRP | 2 min 39 s ± 16 s |
| N13D | 3 min 53 s ± 16 s |
| N57S | 2 min 46 s ± 14 s |
| N255D | 2 min 48 s ± 9 s |
| N268D | 9 min 32 s ± 2 min 18 s |
| N57S/N268D | 2 min 14 s ± 56 s |
| N57S/N255D/N268D | 5 min 51 s ± 18 s |
| N13D/N57S/N255D/N268D | 6 min 19 s ± 12 s |
| HRP Type VI-A | 117 min ± 9 min 55 s |
| Buffer | Km [mM] | Vmax [mol−1 L−1 × s] | Kcat [s−1] | Kcat/Km [mM−1 s−1] |
|---|---|---|---|---|
| Buffer 1 | 9.46 ± 5.18 | 5.7 × 10-3 ± 1.8×10-3 | 272 ± 87.4 | 28.7 ± 18.3 |
| Buffer 2 | 1.51 ± 0.15 | 7.9 × 10-3 ± 6.3 × 10-4 | 378 ± 30.0 | 251 ± 32.2 |
| HRP variant | Km [mM] | Vmax [mol−1 L−1 × s] | Kcat [s−1] | Kcat/Km [mM−1 s−1] |
|---|---|---|---|---|
| Benchmark rHRP | 0.44 ± 0.10 | 2.0 × 10−6 ± 9.8 × 10−8 | 2.24 ± 0.11 | 5.07 ± 1.16 |
| N13D/N57S/N255D/N268D | 0.45 ± 0.12 | 1.8 × 10−5 ± 1.1 × 10−6 | 17.4 ± 1.01 | 39.1 ± 10.5 |
| HRP Type VI-A | 0.27 ± 0.05 | 8.8 × 10−3 ± 6.0 × 10−4 | 422 ± 28.9 | 1,572 ± 306 |
| HRP Variant | t1/2 at 60 °C |
|---|---|
| Benchmark rHRP | 3 min 29 s ± 1 s |
| N13D/N57S/N255D/N268D | 7 min 41 s ± 31 s |
| HRP Type VI-A | 133 min ± 1 min 20 s |
| N-site | Name | Sequence (5′→3′ Direction) |
|---|---|---|
| Benchmark rHRP | pET39b+_hrp_fwd | GCGAATGCCCATGGATATGCAACTG |
| Benchmark rHRP | pET39b+_hrp_rev | CCCGGGACTCGAGTTACGAGTT |
| N13 | N13D_fwd2 | CTGCCCGGATGTGAGCAACA |
| N13 | N13D_rev2 | CGGGCAGCTATTATCATAGAAGG |
| N57 | N57S fwd | CTGCTGGACAGCACCACGTCC |
| N57 | N57S rev | GTCCAGCAGGATACTTGCATCACAGCC |
| N255 | N255D_fwd2 | TTAGTTCCCCGGATGC |
| N255 | N255D_rev2 | CGGGGAACTAAACAGTTCT |
| N268 | N268D fwd | GTTCGTTCATTTGCCGATTCGACCCAGA |
| N268 | N268D rev | GGCAAATGAACGAACCAGCGGAATCG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Humer, D.; Spadiut, O. Improving the Performance of Horseradish Peroxidase by Site-Directed Mutagenesis. Int. J. Mol. Sci. 2019, 20, 916. https://doi.org/10.3390/ijms20040916
Humer D, Spadiut O. Improving the Performance of Horseradish Peroxidase by Site-Directed Mutagenesis. International Journal of Molecular Sciences. 2019; 20(4):916. https://doi.org/10.3390/ijms20040916
Chicago/Turabian StyleHumer, Diana, and Oliver Spadiut. 2019. "Improving the Performance of Horseradish Peroxidase by Site-Directed Mutagenesis" International Journal of Molecular Sciences 20, no. 4: 916. https://doi.org/10.3390/ijms20040916
APA StyleHumer, D., & Spadiut, O. (2019). Improving the Performance of Horseradish Peroxidase by Site-Directed Mutagenesis. International Journal of Molecular Sciences, 20(4), 916. https://doi.org/10.3390/ijms20040916