Integrated Proteomics and Metabolomic Analyses of Plasma Injury Biomarkers in a Serious Brain Trauma Model in Rats

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Evaluation of the DAI Rat Model
2.2. Plasma Metabolic Analysis Using UPLC-Q-TOF/MS Technology
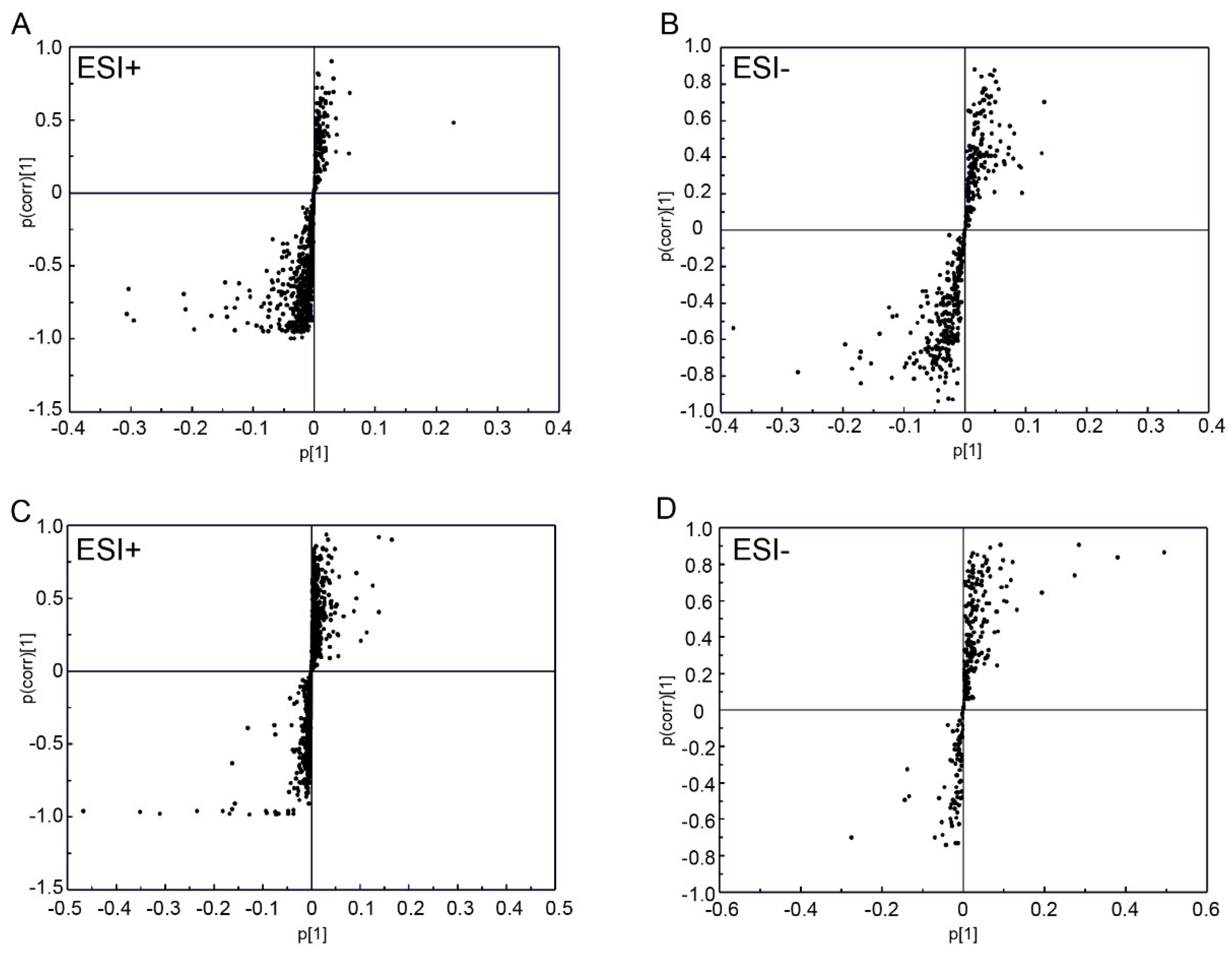
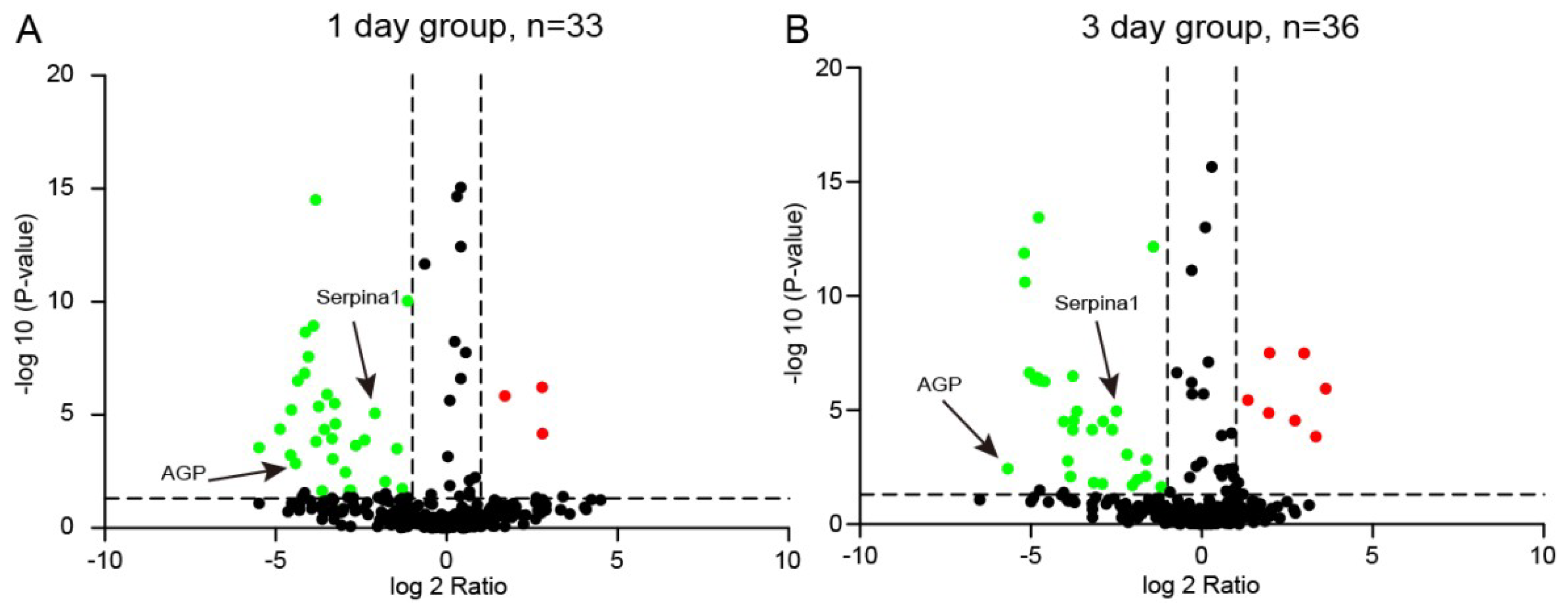
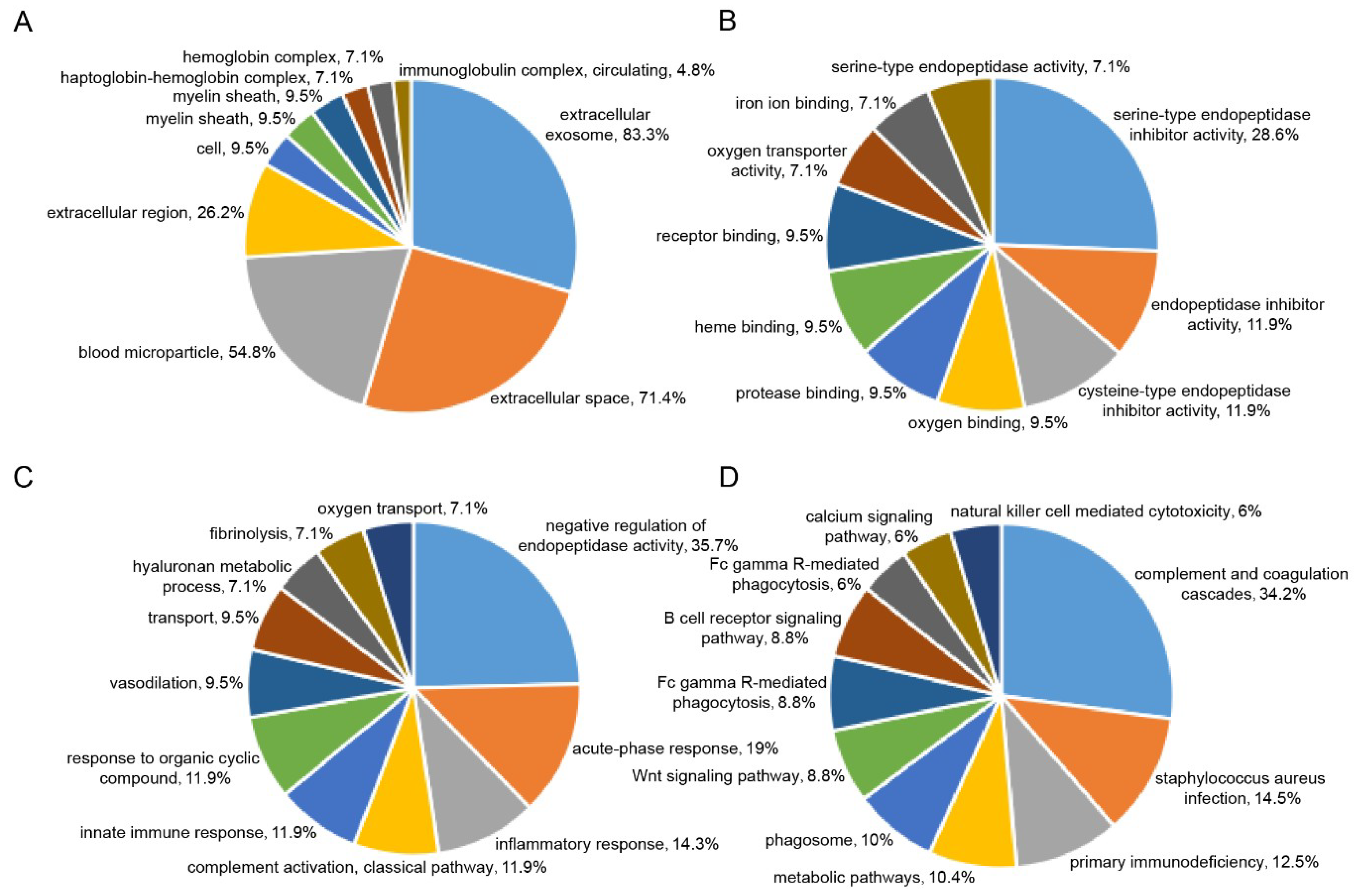
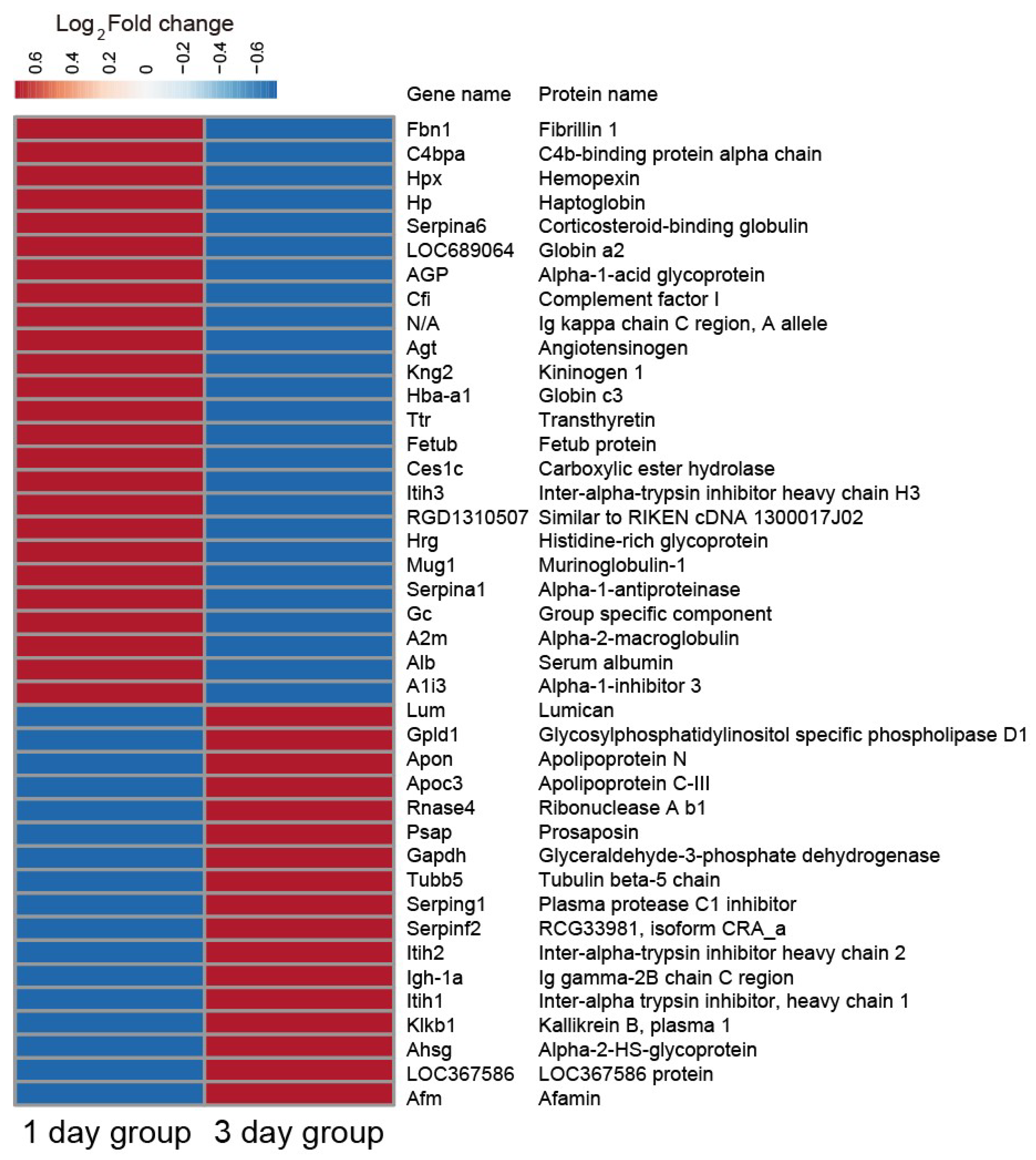
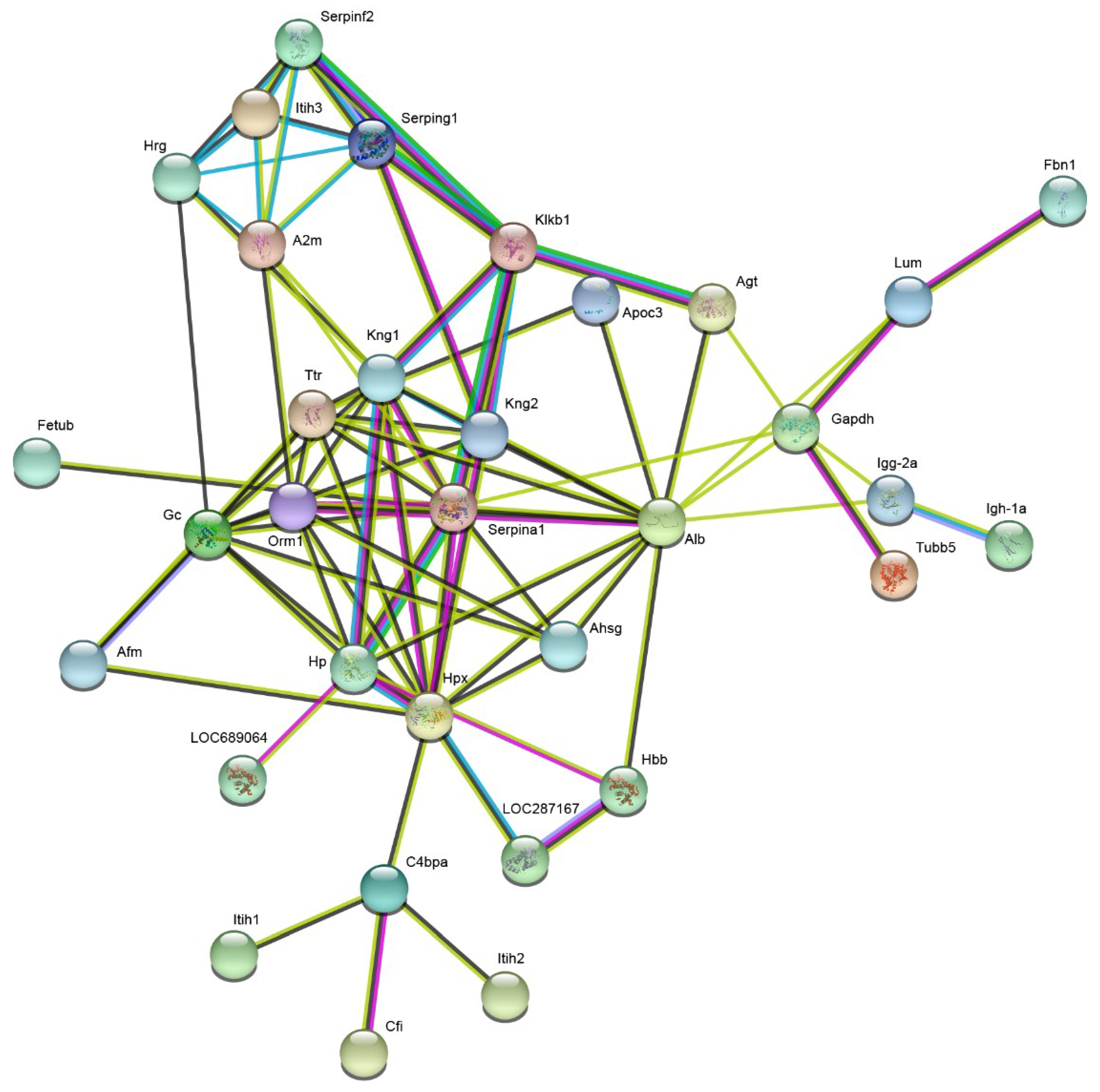
2.3. Plasma Proteomic Analysis Using iTRAQ
2.4. Validation of the iTRAQ-Based Proteomics Results via Western Blot Analysis
3. Discussion
4. Material and Methods
4.1. Chemicals and Reagents
4.2. Animals and Ethics Statement
4.3. Models and Sample Collection
4.3.1. Experimental Model of DAI
4.3.2. Groups
4.3.3. Sample Collection
4.4. Metabolomic Analysis
4.5. Proteomic Analysis
4.5.1. iTRAQ-Based Proteomic Analysis
4.5.2. Validation of Differentially Expressed Proteins
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Abu Hamdeh, S.; Marklund, N.; Lannsjö, M.; Howells, T.; Raininko, R.; Wikström, J.; Enblad, P. Extended Anatomical Grading in Diffuse Axonal Injury Using MRI: Hemorrhagic Lesions in the Substantia Nigra and Mesencephalic Tegmentum Indicate Poor Long-Term Outcome. J. Neurotrauma. 2017, 34, 341–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Wen, L. Inflammatory response following diffuse axonal injury. Int. J. Med. Sci. 2013, 10, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Vieira, R.C.; Paiva, W.S.; de Oliveira, D.V.; Teixeira, M.J.; de Andrade, A.F.; de Sousa, R.M. Diffuse axonal injury: Epidemiology, outcome and associated risk factors. Front. Neurol. 2016, 7, 178. [Google Scholar] [CrossRef]
- Kokkoz, Ç.; Irik, M.; Dayangaç, H.I.; Hayran, M.; Bilge, A.; Çavuş, M. Diagnosis of delayed diffuse axonal injury. Am. J. Emerg. Med. 2017, 35, 1788.e5–1788.e6. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhang, K.; Wang, Z.; Chen, G. Progress of research on diffuse axonal injury after traumatic brain injury. Neural. Plast. 2016, 2016, 9746313. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhu, S.; Zhao, M.; Dai, Y.; Zhang, L.; Ding, S.; Zhao, P.; Li, J. Integration of 1H NMR- and UPLC-Q-TOF/MS-based plasma metabonomics study to identify diffuse axonal injury biomarkers in rat. Brain. Res. Bull. 2018, 140, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhu, S.; Zhao, M.; Zhao, P.; Zhao, H.; Deng, J.; Li, J. Identification of plasma biomarkers for diffuse axonal injury in rats by iTRAQ-coupled LC-MS/MS and bioinformatics analysis. Brain. Res. Bull. 2018, 142, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, X.Y.; Feng, D.F.; Gu, L. Quantitative evaluation of microscopic injury with diffusion tensor imaging in a rat model of diffuse axonal injury. Eur. J. Neurosci. 2011, 33, 933–945. [Google Scholar] [CrossRef]
- Shenton, M.E.; Hamoda, H.M.; Schneiderman, J.S.; Bouix, S.; Pasternak, O.; Rathi, Y.; Vu, M.A.; Purohit, M.P.; Helmer, K.; Koerte, I.; et al. A review of magnetic resonance imaging and diffusion tensor imaging findings in mild traumatic brain injury. Brain. Imaging Behav. 2012, 6, 137–192. [Google Scholar] [CrossRef] [Green Version]
- Del Boccio, P.; Rossi, C.; di Ioia, M.; Cicalini, I.; Sacchetta, P.; Pieragostino, D. Integration of metabolomics and proteomics in multiple sclerosis: From biomarkers discovery to personalized medicine. Proteomics. Clin. Appl. 2015, 10, 470–484. [Google Scholar] [CrossRef]
- Tian, Y.; Jiang, F.; Li, Y.; Jiang, H.; Chu, Y.; Zhu, L.; Guo, W. Evaluation of the anti-hypertensive effect of Tengfu Jiangya tablet by combination of UPLC-Q-exactive-MS-based metabolomics and iTRAQ-based proteomics technology. Biomed. Pharmacother. 2018, 100, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yuan, S.; Pu, J.; Yang, L.; Zhou, X.; Liu, L.; Jiang, X.; Zhang, H.; Teng, T.; Tian, L.; et al. Integrated metabolomics and proteomics analysis of hippocampus in a rat model of depression. Neuroscience 2018, 371, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Cambiaghi, A.; Díaz, R.; Martinez, J.B.; Odena, A.; Brunelli, L.; Caironi, P.; Masson, S.; Baselli, G.; Ristagno, G.; Gattinoni, L.; et al. An Innovative Approach for the Integration of Proteomics and Metabolomics Data in Severe Septic Shock Patients Stratified for Mortality. Sci. Rep. 2018, 8, 6681. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, S.R.; Hwang, S.R.; Rolland, D.; Murga-Zamalloa, C.; Basrur, V.; Conlon, K.P.; Fermin, D.; Wolfe, T.; Raskind, A.; Ruan, C.; et al. Integrated phosphoproteomic and metabolomic profiling reveals NPM-ALK-mediated phosphorylation of PKM2 and metabolic reprogramming in anaplastic large cell lymphoma. Blood 2013, 122, 958–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Li, X.Y.; Feng, D.F.; Pan, D.C. Biomarkers associated with diffuse traumatic axonal injury: Exploring pathogenesis, early diagnosis, and prognosis. J. Trauma. 2010, 69, 1610–1618. [Google Scholar] [CrossRef] [PubMed]
- Cheow, E.S.H.; Cheng, W.C.; Yap, T.; Dutta, B.; Lee, C.N.; Kleijn, D.P.V.; Sorokin, V.; Sze, S.K. Myocardial injury is distinguished from stable angina by a set of candidate plasma biomarkers identified using iTRAQ/MRM-based approach. J. Proteome Res. 2018, 17, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.H.; Jeong, I.H.; Hyun, J.S.; Kong, B.S.; Kim, H.J.; Park, S.J. Metabolomic profiling of CSF in multiple sclerosis and neuromyelitis optica spectrum disorder by nuclear magnetic resonance. PLoS ONE 2017, 12, e0181758. [Google Scholar] [CrossRef] [PubMed]
- Nehlig, A. Brain uptake and metabolism of ketone bodies in animal models. Prostag. Leukot. Essent. Fatty Acids 2004, 70, 265–275. [Google Scholar] [CrossRef]
- Simeone, T.A.; Simeone, K.A.; Rho, J.M. Ketone Bodies as Anti-Seizure Agents. Neurochem. Res. 2017, 42, 2011–2018. [Google Scholar] [CrossRef] [PubMed]
- Reinke, S.N.; Walsh, B.H.; Boylan, G.B.; Sykes, B.D.; Kenny, L.C.; Murray, D.M.; Broadhurst, D.I. 1H NMR derived metabolomic profile of neonatal asphyxia in umbilical cord serum: Implications for hypoxic ischemic encephalopathy. J. Proteome Res. 2013, 12, 4230–4239. [Google Scholar] [CrossRef] [PubMed]
- Denihan, N.M.; Kirwan, J.A.; Walsh, B.H.; Dunn, W.B.; Broadhurst, D.I.; Boylan, G.B.; Murray, D.M. Untargeted metabolomic analysis and pathway discovery in perinatal asphyxia and hypoxic-ischaemic encephalopathy. J. Cereb. Blood Flow Metab. 2019, 39, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Li, M.; Xu, P.; Gu, T.; Ma, T.; Gu, S. (1)H NMR-based metabolomics exploring biomarkers in rat cerebrospinal fluid after cerebral ischemia/reperfusion. Mol. Biosyst. 2013, 9, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.H.; Lee, D.U.; Lee, J.T.; Kim, J.S.; Yong, C.S.; Kim, J.A.; Ha, J.S.; Huh, K. 4-Hydroxybenzaldehyde from Gastrodia elata B1. is active in the antioxidation and GABAergic neuromodulation of the rat brain. J. Ethnopharmacol. 2000, 73, 329–333. [Google Scholar] [CrossRef]
- Ookawa, S.; Wanibuchi, M.; Kataoka-Sasaki, Y.; Sasaki, M.; Oka, S.; Ohtaki, S.; Noshiro, S.; Komatsu, K.; Akiyama, Y.; Mikami, T.; et al. Digital polymerase chain reaction quantification of SERPINA1 predicts prognosis in high-grade glioma. World. Neurosurg. 2018, 111, e783–e789. [Google Scholar] [CrossRef]
- Ebbert, M.T.W.; Ross, C.A.; Pregent, L.J.; Lank, R.J.; Zhang, C.; Katzman, R.B.; Jansen-West, K.; Song, Y.; da Rocha, E.L.; Palmucci, C.; et al. Conserved DNA methylation combined with differential frontal cortex and cerebellar expression distinguishes C9orf72-associated and sporadic ALS, and implicates SERPINA1 in disease. Acta Neuropathol. 2017, 134, 715–728. [Google Scholar] [CrossRef]
- De Baumont, A.; Maschietto, M.; Lima, L.; Carraro, D.M.; Olivieri, E.H.; Fiorini, A.; Barreta, L.A.; Palha, J.A.; Belmonte-de-Abreu, P.; Moreira Filho, C.A.; et al. Innate immune response is differentially dysregulated between bipolar disease and schizophrenia. Schizophr. Res. 2015, 161, 215–221. [Google Scholar] [CrossRef]
- Mishra, A.; Ferrari, R.; Heutink, P.; Hardy, J.; Pijnenburg, Y.; Posthuma, D. Gene-based association studies report genetic links for clinical subtypes of frontotemporal dementia. Brain 2017, 140, 1437–1446. [Google Scholar] [CrossRef]
- Peng, S.; Xu, J.; Pelkey, K.A.; Chandra, G.; Zhang, Z.; Bagh, M.B.; Yuan, X.; Wu, L.G.; McBain, C.J.; Mukherjee, A.B. Suppression of agrin-22 production and synaptic dysfunction in Cln1 (-/-) mice. Ann. Clin. Transl. Neurol. 2015, 2, 1085–1104. [Google Scholar] [CrossRef]
- Zhang, S.; Mark, K.S. α1-Acid glycoprotein induced effects in rat brain microvessel endothelial cells. Microvasc. Res. 2012, 84, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Ceciliani, F.; Pocacqua, V.; Miranda-Ribera, A.; Bronzo, V.; Lecchi, C.; Sartorelli, P. α1-Acid glycoprotein modulates apoptosis in bovine monocytes. Vet. Immunol. Immunopathol. 2007, 116, 145–152. [Google Scholar] [CrossRef]
- Breslow, D.K.; Collins, S.R.; Bodenmiller, B.; Aebersold, R.; Simons, K.; Shevchenko, A.; Ejsing, C.S.; Weissman, J.S. Orm family proteins mediate sphingolipid homeostasis. Nature 2010, 463, 1048–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Z.; Lei, H.; Sun, Y.; Liu, X.; Su, D.F. Orosomucoid, an acute response protein with multiple modulating activities. J. Physiol. Biochem. 2015, 71, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Jiang, Y.; Zhu, J.; Wen, Z.; Xu, X.; Xu, X.; Xie, Y.; Yang, L.; Xu, L.; Lan, W.; et al. Orosomucoid1: Involved in vascular endothelial growth factor-induced blood-brain barrier leakage after ischemic stroke in mouse. Brain Res. Bull. 2014, 109, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhu, S.; Li, Y.; Zhao, M.; Liu, M.; Gao, J.; Ding, S.; Li, J. Quantitative proteomics analysis to identify diffuse axonal injury biomarkers in rats using iTRAQ coupled LC-MS/MS. J. Proteom. 2016, 133, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.M.; Feng, Y.F.; Liu, Y.T.; Chang, X.; Chen, L.; Zhang, H.W.; Ding, G.; Zou, Z.M. Integration of ¹H NMR and UPLC-Q-TOF/MS for a comprehensive urinary metabonomics study on a rat model of depression induced by chronic unpredictable mild stress. PLoS ONE 2013, 8, e63624. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Liu, Z.; Fan, S.H.; Yang, D.Y.; Zheng, P.; Shao, W.H.; Qi, Z.G.; Xu, X.J.; Li, Q.; Mu, J.; et al. Combined application of NMR- and GC-MS-based metabonomics yields a superior urinary biomarker panel for bipolar disorder. Sci. Rep. 2014, 4, 5855. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; He, J.; Deng, J.; Yang, X.; Cui, L.; Ran, R.; Du, G.; Jiang, X. Small molecule metabolite biomarkers for hepatocellular carcinoma with bile duct tumor thrombus diagnosis. Sci. Rep. 2018, 8, 3309. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, T.; Zhu, Y.; Zhang, P.; Zhao, M.; Zhao, D.; Ding, S.; Zhu, S.; Li, J. Integrated Proteomics and Metabolomic Analyses of Plasma Injury Biomarkers in a Serious Brain Trauma Model in Rats. Int. J. Mol. Sci. 2019, 20, 922. https://doi.org/10.3390/ijms20040922
Song T, Zhu Y, Zhang P, Zhao M, Zhao D, Ding S, Zhu S, Li J. Integrated Proteomics and Metabolomic Analyses of Plasma Injury Biomarkers in a Serious Brain Trauma Model in Rats. International Journal of Molecular Sciences. 2019; 20(4):922. https://doi.org/10.3390/ijms20040922
Chicago/Turabian StyleSong, Tao, Ying Zhu, Peng Zhang, Minzhu Zhao, Dezhang Zhao, Shijia Ding, Shisheng Zhu, and Jianbo Li. 2019. "Integrated Proteomics and Metabolomic Analyses of Plasma Injury Biomarkers in a Serious Brain Trauma Model in Rats" International Journal of Molecular Sciences 20, no. 4: 922. https://doi.org/10.3390/ijms20040922