Salidroside Ameliorates Renal Interstitial Fibrosis by Inhibiting the TLR4/NF-κB and MAPK Signaling Pathways


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Sal Alleviated Renal Function Parameters, Histopathology and Inflammation in Renal Fibrotic Mice
2.2. Sal Inhibited ECM Deposition and EMT in Renal Fibrotic Mice
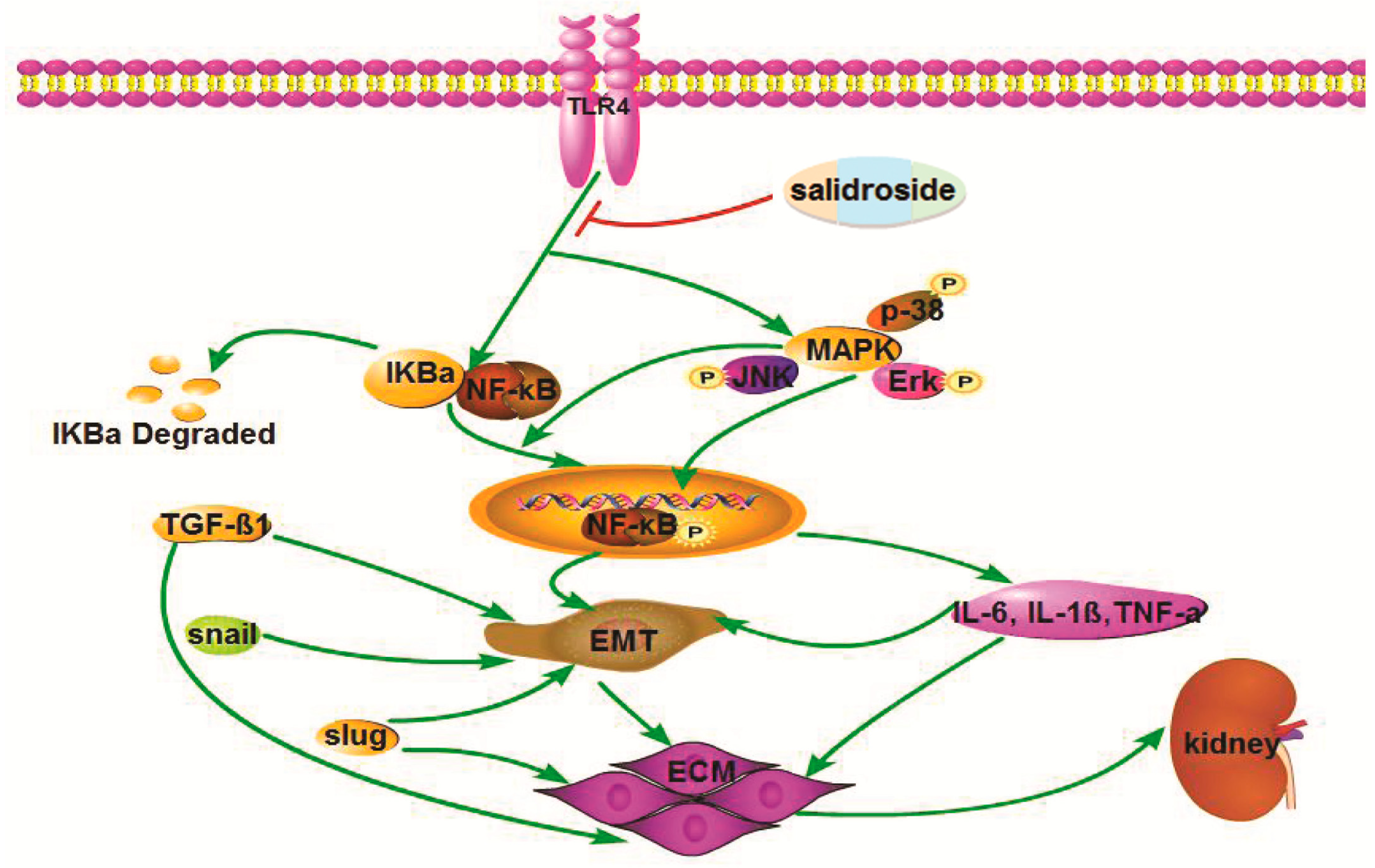
2.3. Sal Suppresses Renal Inflammation via the TLR4/NF-κB and MAPK Signaling Pathways in Renal Fibrotic Mice
2.4. Sal Inhibited TGF-β1-Induced HK-2 Cells ECM Deposition, EMT and Inflammatory Response via the TLR4/NF-κB and MAPK Signaling Pathways
2.5. Sal Suppressed the LPS-Induced Inflammatory Response by Inhibiting the TLR4/NF-κB and MAPK Signaling Pathways
3. Discussion
4. Materials and Methods
4.1. Main Reagents and Kits
4.2. Animals and Experimental Design
4.2.1. UUO Model
4.2.2. Folic Acid Model
4.3. H&E and Masson Staining
4.4. Biochemical Assays in Serum
4.5. Inflammatory Cytokines Levels in Serum
4.6. Immunohistochemistry Staining
4.7. Cell Culture
4.8. Western Blot
4.9. Cell Viability Assay
4.10. Immunofluorescence Staining
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jha, V.; Garciagarcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.; Yang, C.W. Chronic kidney disease: global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef]
- Kohan, D.E.; Barton, M. Endothelin and endothelin antagonists in chronic kidney disease. Kidney Int. 2014, 86, 896–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Declèves, A.E.; Sharma, K. Novel targets of antifibrotic and anti-inflammatory treatment in CKD. Nat. Rev. Nephrol. 2014, 10, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Klahr, S.; Morrissey, J. Obstructive nephropathy and renal fibrosis. Am. J. Physiol. Renal. Physiol. 2002, 283, 861–875. [Google Scholar] [CrossRef] [PubMed]
- Bohle, A.; Christ, H.; Grund, K.E.; Mackensen, S. The Role of the Interstitium of the Renal Cortex in Renal Disease. Contrib. Nephrol. 1979, 16, 109. [Google Scholar] [PubMed]
- Liu, Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J. Am. Soc. Nephrol. 2010, 21, 212. [Google Scholar] [CrossRef] [PubMed]
- Kriz, W.; Kaissling, B.; Hir, M.L. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: fact or fantasy? J. Clin. Investig. 2011, 121, 468–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sime, P.J.; O’reilly, K.M. Fibrosis of the lung and other tissues: new concepts in pathogenesis and treatment. Clin. Immunol. 2001, 99, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.; Ding, A. Nonresolving Inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, M.T.; Hile, K.L.; Zhang, H.; Asanuma, H.; Vanderbrink, B.A.; Rink, R.R.; Meldrum, K.K. Toll-Like Receptor 4: A Novel Signaling Pathway During Renal Fibrogenesis. J. Surg. Res. 2011, 168, e61–e69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinsey, G.R.; Li, L.; Okusa, M.D. Inflammation in acute kidney injury. Nephron Exp. Nephr. 2008, 109, e102–e107. [Google Scholar] [CrossRef] [PubMed]
- Fujihara, C.K.; Antunes, G.R.; Mattar, A.L.; Malheiros, D.M.; Vieira, J.M.; Zatz, R. Chronic inhibition of nuclear factor-κB attenuates renal injury in the 5/6 renal ablation model. Am. J. Physiol.-Renal 2007, 292, F92. [Google Scholar] [CrossRef] [PubMed]
- Gruden, G.; Zonca, S.; Hayward, A.; Thomas, S.; Maestrini, S.; Gnudi, L.; Viberti, G. Mechanical stretch-induced fibronectin and transforming growth factor-beta1 production in human mesangial cells is p38 mitogen-activated protein kinase-dependent. Diabetes 2000, 49, 655–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, D.L.; Chen, Y.; Xu, B.; Ren, K.; He, Z.; He, L.; Song, X. Development of lipid-shell and polymer core nanoparticles with water-soluble salidroside for anti-cancer therapy. Int. J. Mol. Sci. 2014, 15, 3373–3388. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Wei, T.; Gao, J.; Chang, X.; He, H.; Miao, M.; Yan, T. Salidroside attenuates lipopolysaccharide (LPS) induced serum cytokines and depressive-like behavior in mice. Neurosci. Lett. 2015, 606, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xiao, L.; Zhu, L.; Hu, M.; Wang, Q.; Yan, T. The Effect of Synthetic Salidroside on Cytokines and Airway Inflammation of Asthma Induced by Diisocyanate (TDI) in Mice by Regulating GATA3/T-bet. Inflammation 2015, 38, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; He, H.; Jiang, W.; Chang, X.; Zhu, L.; Luo, F.; Yan, T. Salidroside ameliorates cognitive impairment in a d-galactose-induced rat model of Alzheimer’s disease. Behav. Brain Res. 2015, 293, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Luo, F.; Jiang, W.; Zhu, L.; Gao, J.; He, H.; Yan, T. Protective activity of salidroside against ethanol-induced gastric ulcer via the MAPK/NF-κB pathway in vivo and in vitro. J. Int. Immunopharma 2015, 28, 604–615. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Chang, X.; Gao, J.; Zhu, L.; Miao, M.; Yan, T. Salidroside Mitigates Sepsis-Induced Myocarditis in Rats by Regulating IGF-1/PI3K/Akt/GSK-3β Signaling. Inflammation 2015, 38, 2178–2184. [Google Scholar] [CrossRef] [PubMed]
- Ota, I.; Li, X.Y.; Hu, Y.; Weiss, S.J. Induction of a MT1-MMP and MT2-MMP-dependent basement membrane transmigration program in cancer cells by Snail1. Proc. Natl. Acad. Sci. USA 2009, 106, 20318. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory processes in renal fibrosis. Nat. Rev. Nephrol. 2014, 10, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Thornhill, B.A.; Burt, L.E.; Chen, C.O.; Forbes, M.S.; Chevalier, R.L. Variable chronic partial ureteral obstruction in the neonatal rat: A new model of ureteropelvic junction obstruction1. Kidney Int. 2005, 67, 42–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevalier, R.L.; Forbes, M.S.; Thornhill, B.A. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int. 2009, 75, 1145–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevalier, R.L. Obstructive nephropathy: towards biomarker discovery and gene therapy. Nat. Clin. Pract. Neph 2006, 2, 157. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.C.; Zuo, Y.; Fogo, A.B. Models of chronic kidney disease. Drug Discov. Today Dis. Models 2010, 7, 13–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grande, M.T.; Sánchezlaorden, B.; Lópezblau, C.; De Frutos, C.A.; Boutet, A.; Arevalo, M.; Rowe, R.J.; Weiss, S.J.; López-Novoa, G.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuma, A.; Tamura, M.; Otsuji, Y. Mechanism of and Therapy for Kidney Fibrosis. J. Uoeh 2016, 38, 25. [Google Scholar] [CrossRef] [PubMed]
- Meran, S.; Steadman, R. Fibroblasts and myofibroblasts in renal fibrosis. Int J. Exp. Pathol. 2011, 92, 158–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grande, M.T.; López-Novoa, J.M. Fibroblast activation and myofibroblast generation in obstructive nephropathy. Nat. Rev. Nephrol. 2009, 5, 319. [Google Scholar] [CrossRef] [PubMed]
- Böttinger, E.P. TGF-beta in renal injury and disease. Semin. Nephrol. 2007, 27, 309. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Liu, S.; Shi, Y.; Hao, J.; Xing, L.; Cao, Y.; Duan, H. Suppressors of Cytokine Signaling Inhibit Tubular Epithelial Cell-Myofibroblast Transdifferentiation. Am. J. Nephrol. 2011, 34, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Letterio, J.J.; Roberts, A.B. Regulation of Immune Responses by TGF-β1. Annu. Rev. Immunol. 1998, 16, 137–161. [Google Scholar] [CrossRef] [PubMed]
- Holdsworth, S.R.; Summers, S.A. Role of Mast Cells in Progressive Renal Diseases. J. Am. Soc. Nephrol. JASN 2008, 19, 2254. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.J.; Banas, B.; Schlöndorff, D. Signaling danger: Toll-like receptors and their potential roles in kidney disease. J. Am. Soc. Nephrol. JASN 2004, 15, 854. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Henao-Mejia, J.; Flavell, R.A. Innate immune receptors: Key regulators of metabolic disease progression. Cell. Metab. 2013, 17, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, X.; Yu, G.; Chen, H.; Wang, L.; Wang, Z.; Weng, X. Ozone therapy ameliorates tubulointerstitial inflammation by regulating TLR4 in adenine-induced CKD rats. Ren. Fail. 2016, 38, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Liu, B.C.; Lv, L.L.; Ma, K.; Zhang, X.; Phillips, A.O. Monocytes induce proximal tubular epithelial–mesenchymal transition through NF-kappa B dependent upregulation of ICAM-1. J. Cell. Biochem. 2011, 112, 1585–1592. [Google Scholar] [CrossRef] [PubMed]
- Kanellis, J.; Ma, F.Y.; Kandane-Rathnayake, R.; Dowling, J.P.; Polkinghorne, K.R.; Bennett, B.L.; Nikolicpaterson, D.J. JNK signalling in human and experimental renal ischaemia/reperfusion injury. Nephrol. Dialysis Transplant. 2010, 25, 2898–2908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, K.; Han, J. The p38 signal transduction pathway: Activation and function. Cell. Signalling 2000, 12, 1–13. [Google Scholar] [CrossRef]
- Lim, A.K.H.; Ma, F.Y.; Nikolicpaterson, D.J.; Ozols, E.; Young, M.J.; Bennett, B.L.; Friedman, G.C.; Tesch, G.H. Evaluation of JNK Blockade as an Early Intervention Treatment for Type 1 Diabetic Nephropathy in Hypertensive Rats. Am. J. Nephrol. 2011, 34, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.Y.; Tesch, G.H.; Ozols, E.; Xie, M.; Schneider, M.D.; Nikolicpaterson, D.J. TGF-β1-activated kinase-1 regulates inflammation and fibrosis in the obstructed kidney. Am. J. Physiol. Renal. 2011, 300, F1410. [Google Scholar] [CrossRef] [PubMed]
- Workman, L.M.; Habelhah, H. TNFR1 signaling kinetics: Spatiotemporal control of three phases of IKK activation by posttranslational modification. Cell. Signal. 2013, 25, 1654–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luan, D.T.; Choi, Y.J.; Tsao, C.C.; Ayala, G.; Sheikhhamad, D.; Nassar, G.M.; Suki, W.N. Renal cell apoptosis in chronic obstructive uropathy: The roles of caspases. Kidney Int. 2001, 60, 924. [Google Scholar]
- Wu, Y.; Wang, L.; Deng, D.; Zhang, Q.; Liu, W. Renalase Protects against Renal Fibrosis by Inhibiting the Activation of the ERK Signaling Pathways. Int. J. Mol. Sci. 2017, 18, 855. [Google Scholar] [Green Version]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, R.; Guo, Y.; Zhang, Y.; Zhang, X.; Zhu, L.; Yan, T. Salidroside Ameliorates Renal Interstitial Fibrosis by Inhibiting the TLR4/NF-κB and MAPK Signaling Pathways. Int. J. Mol. Sci. 2019, 20, 1103. https://doi.org/10.3390/ijms20051103
Li R, Guo Y, Zhang Y, Zhang X, Zhu L, Yan T. Salidroside Ameliorates Renal Interstitial Fibrosis by Inhibiting the TLR4/NF-κB and MAPK Signaling Pathways. International Journal of Molecular Sciences. 2019; 20(5):1103. https://doi.org/10.3390/ijms20051103
Chicago/Turabian StyleLi, Rui, Yujuan Guo, Yiming Zhang, Xue Zhang, Lingpeng Zhu, and Tianhua Yan. 2019. "Salidroside Ameliorates Renal Interstitial Fibrosis by Inhibiting the TLR4/NF-κB and MAPK Signaling Pathways" International Journal of Molecular Sciences 20, no. 5: 1103. https://doi.org/10.3390/ijms20051103