1. Introduction
Cellular protrusions are involved in numerous biological functions, including cell migration, neurite outgrowth, phagocytosis, and cell–cell communication. Many are also implicated in pathological processes such as cancer–cell invasion [
1] and the intercellular transmission of misfolded proteins [
2] and infectious pathogens [
3]. These physiological and pathological functions are controlled by different subtypes of protrusions, such as pseudopodia [
1], filopodia [
4], growth cones (GCs) [
5,
6], or tunneling nanotubes (TNTs) [
2,
7,
8]. Interestingly, these different types of subcellular protrusions can be distinguished by light microscopy. For instance, GCs, which play an important role in axonal guidance, are specialized structures at the tip of axons, which comprise a core region with thin filopodia and lamellipodia at the periphery [
5,
6]. On the other hand, filopodia and TNTs are actin-rich “finger-like” structures that can only be distinguished by light microscopy from their differences in adhesion to the substratum. Indeed, filopodia, which are important sensory organelles, adhere to the substratum [
9], while TNTs, which transport intercellular material [
2,
7,
8], “hover” above the substratum [
10].
Unfortunately, while some functions have been characterized, many unanswered questions about their distinct mechanisms of formation, composition or signaling pathways remain, making it difficult to reach a consensus on their nomenclature and function [
11].
The first step needed to characterize their formation, function, and regulation, is to identify and compare their proteomes. Regrettably, no method currently exists to isolate disparate subtypes of protrusions. Subcellular fractionation has been used to isolate GCs [
12,
13]; however, the selectivity and purity of this method remains questionable, and its use with other protrusions is not possible. Other studies employ Boyden chambers [
14] or excimer laser-assisted etching [
15]; however, as with fractionation, these methods cannot isolate specific subsets of protrusions and are therefore limited and only appropriate for the study of protrusions in general. Thus, no technique has been described that can specifically isolate distinct subtypes of protrusions.
Here, we describe a novel method that combines Laser Capture Microdissection (LCM) and mass spectrometry (MS) to study specific subtypes of protrusions (
Figure S1). LCM is a way to microscopically visualize, segregate, and enrich specific cells of interest in a heterogeneous sample for the subsequent study of their DNA, RNA, and protein without altering downstream analyses [
16]. LCM has been successfully applied to isolate tumor cells [
17], neurons affected by Parkinson [
18] or Alzheimer disease [
19], virus-infected cells [
20], organelles [
21], and GCs [
22]. For genomic and transcriptomic studies, amplification methods exist, which lessen the need for large sample sizes. Unfortunately, for proteomic studies, no analogous amplification method currently exists. Thus, the use of LCM for proteomic studies has been limited by large sample size demands. However, recent advances in the sensitivity, detection limits, and acquisition rates of mass spectrometers based on quadrupole, ion-trap, and ultra-high field Orbitrap technologies have allowed for an unprecedented depth of analysis of low-abundance and high-complexity samples [
23]—due to their high mass accuracy and specificity—giving rise to what has been termed microproteomics (i.e., the identification of proteins from complex, yet microscale samples) [
23,
24,
25]. Using such technology, we recently described an LCM/MS method that efficiently identified the proteome of 1000 cells by improving both the fixation and protein extraction protocols [
26].
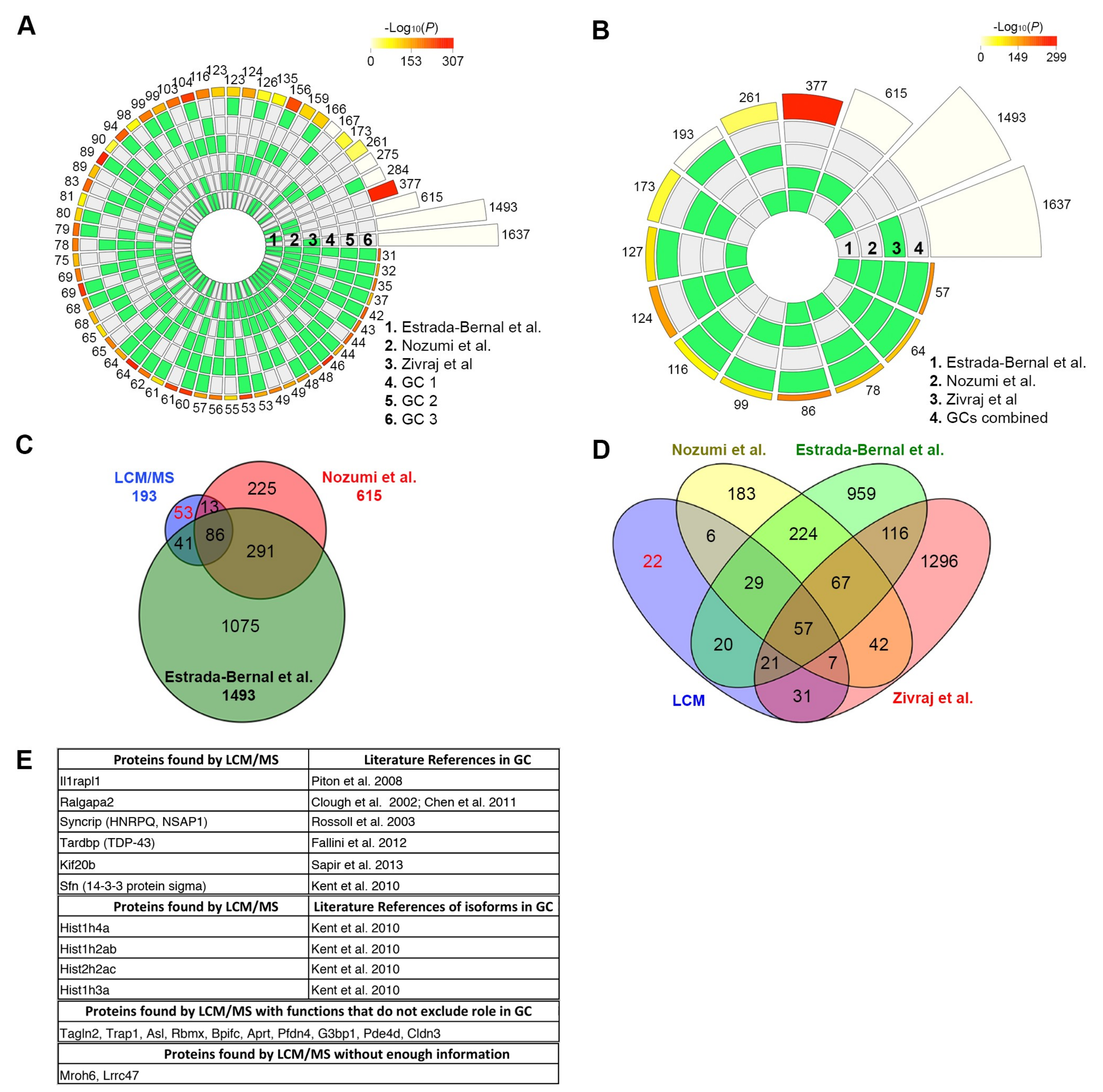
In this study, we successfully pushed the limits of LCM/microproteomics to the subcellular level and compiled the proteomes of distinct subtypes of protrusions. Since the proteome [
12,
13] and transcriptome [
22] of GCs have been characterized, we used these published studies to validate our method. Our LCM samples showed an extremely high overlap with those studies, demonstrating that microscale samples can accurately match the results of high-throughput samples, albeit at lower coverage rates depending upon the number of microscale samples analyzed.
To further validate this method, we isolated different categories of protrusions such as axons/dendrites/GCs from serum-deprived, differentiated neuronal CADs (dCADs), identified their proteomes, and compared them to filopodia/TNTs/GCs from neuronal CADs. We showed that these subtypes of protrusions possess unique proteomes, which is underscored by differences in their functional annotations and localization. Finally, we showed that the additive—“in two sample”—approach currently used in high-throughput proteomic studies, arbitrarily eliminates critical proteins from consideration and should be used cautiously in microproteomic studies. We demonstrate that this type of analysis leads to the rejection of relevant proteins, and obscures information regarding low abundant proteins in such samples. Instead, we propose a subtractive approach that dramatically improves the identification of low abundant proteins in small samples (
Figure S1). Overall, this method will significantly advance our knowledge of these specialized structures and will help to answer questions about their distinct formation and function that had thus far been out of reach.
3. Discussion
Cellular protrusions play a significant role in multicellular processes and have distinct functions. To date, it has not been possible to isolate and separate these various structures, which has hindered research regarding their protein composition, formation, and function, as well as the identification of biomarkers for their characterization in tissue samples or in vivo. Here, we described a novel method that allows for the specific isolation of individual protrusions, based on morphology. Using LCM/MS, we identified the proteome of GCs, which overlapped strongly with published data [
12,
13,
22].
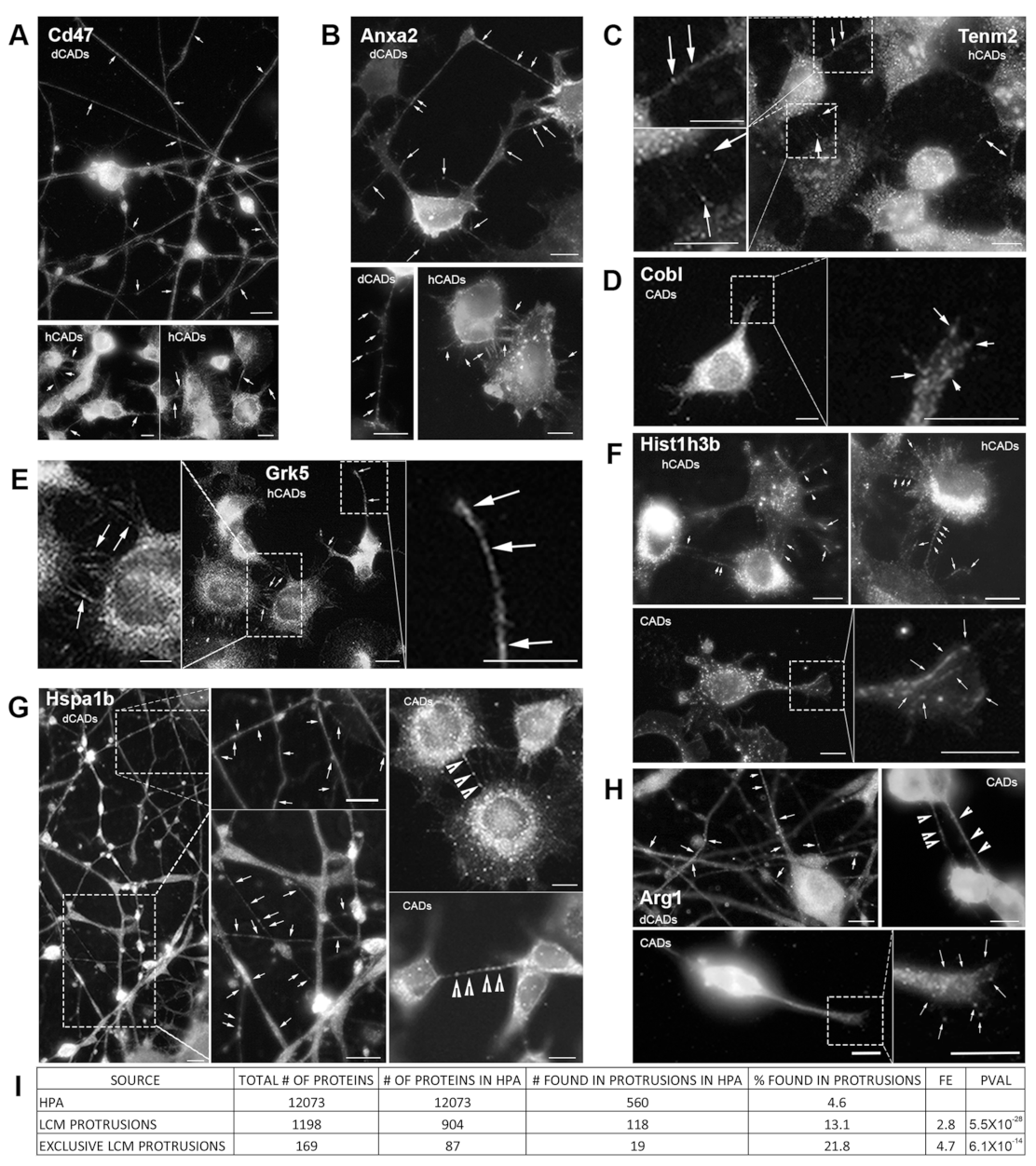
In order to further validate our LCM/MS method, we identify four “expected proteins” that were known to be associated with membranes and/or cellular protrusions and four “unexpected proteins” based on their known function and/or cellular localization (
Figure 5). In our LCM/MS data, the membrane protein Cd47, known to localize and induce filopodia formation [
50], and the phospholipid-binding protein Anxa2 [
51] were identified in hCAD/dCAD protrusions. IF experiments showed that both proteins localize throughout axons/dendrites in dCADs and in hCAD protrusions (
Figure 5A). In addition, Tenm2, another protein shown to induce filopodia formation and to localize to filopodia [
43], was found by LCM-MS in hCAD protrusions. In agreement, numerous punctates of Tenm2 were found within hCAD protrusions by IF (
Figure 5C). Finally, the actin nucleator Cobl identified by LCM/MS within GCs was observed within these structures by IF (
Figure 5D), in agreement with recent findings [
44].
Next, to specifically determine the purity of the LCM isolated protrusions, we analyzed the proteomes of the different subtypes of cellular protrusions, to specifically look for proteins that would appear “out of place” based on their known functions and/or cellular localization. We decided to look by IF, whether or not these proteins could be found within the cellular protrusions from which they were isolated and identified by LCM/MS, or whether this might be from cell contamination. The four “unexpected” proteins we looked at were Grk5, which regulates microtubule nucleation and normal cell cycle progression [
45], the nuclear histone protein, Hist1h3b, the molecular chaperone Hspa1b, and Arg1, a urea cycle enzyme essential for mouse embryonic development [
46]. As shown, in
Figure 5E–H, the presence of all four “unexpected” proteins was confirmed by IF within the isolated protrusions identified by LCM/MS. Overall, these IF experiments further demonstrate the purity of our LCM isolation method and demonstrate that this method can be used to identify new candidate protein in specific subtypes of cellular protrusions.
To further demonstrate that our LCM isolation method significantly enriches protrusion-localized proteins, we used the Human Protein Atlas (HPA) Subcell database [
39,
40]. The fact that HPA images are limited to only 12,073 proteins, with just a few images per antibody, and that those images are not optimized for the visualization of protrusions, suggests that this HPA enrichment analysis likely understates the actual enrichment. In fact, of the eight proteins that we tested, only six were in the HPA database, and of those, merely four could be clearly identified in protrusions (
Figure S3B). Thus, anecdotally, this enrichment may be significantly understated, possibly by as high as 33%.
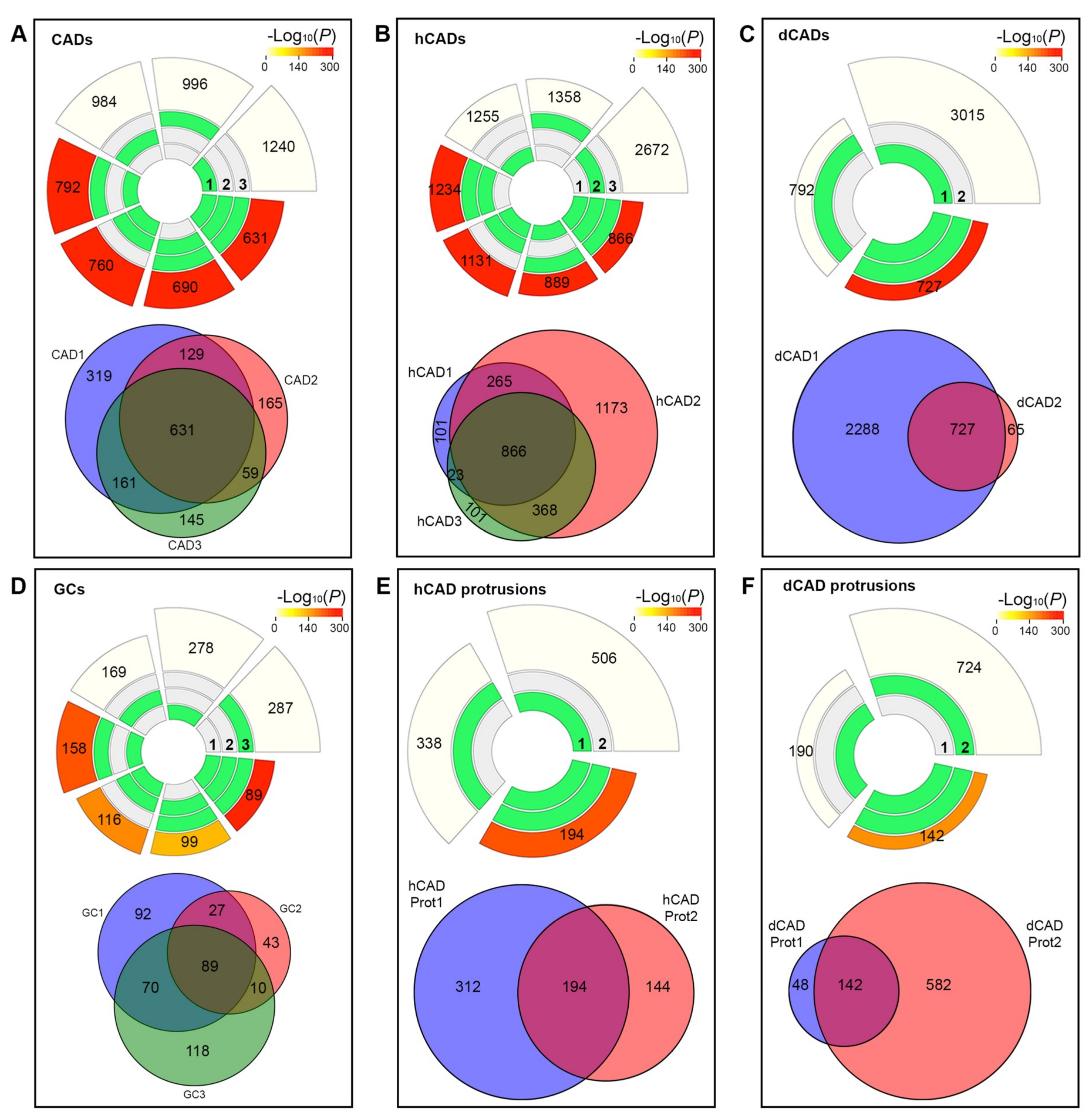
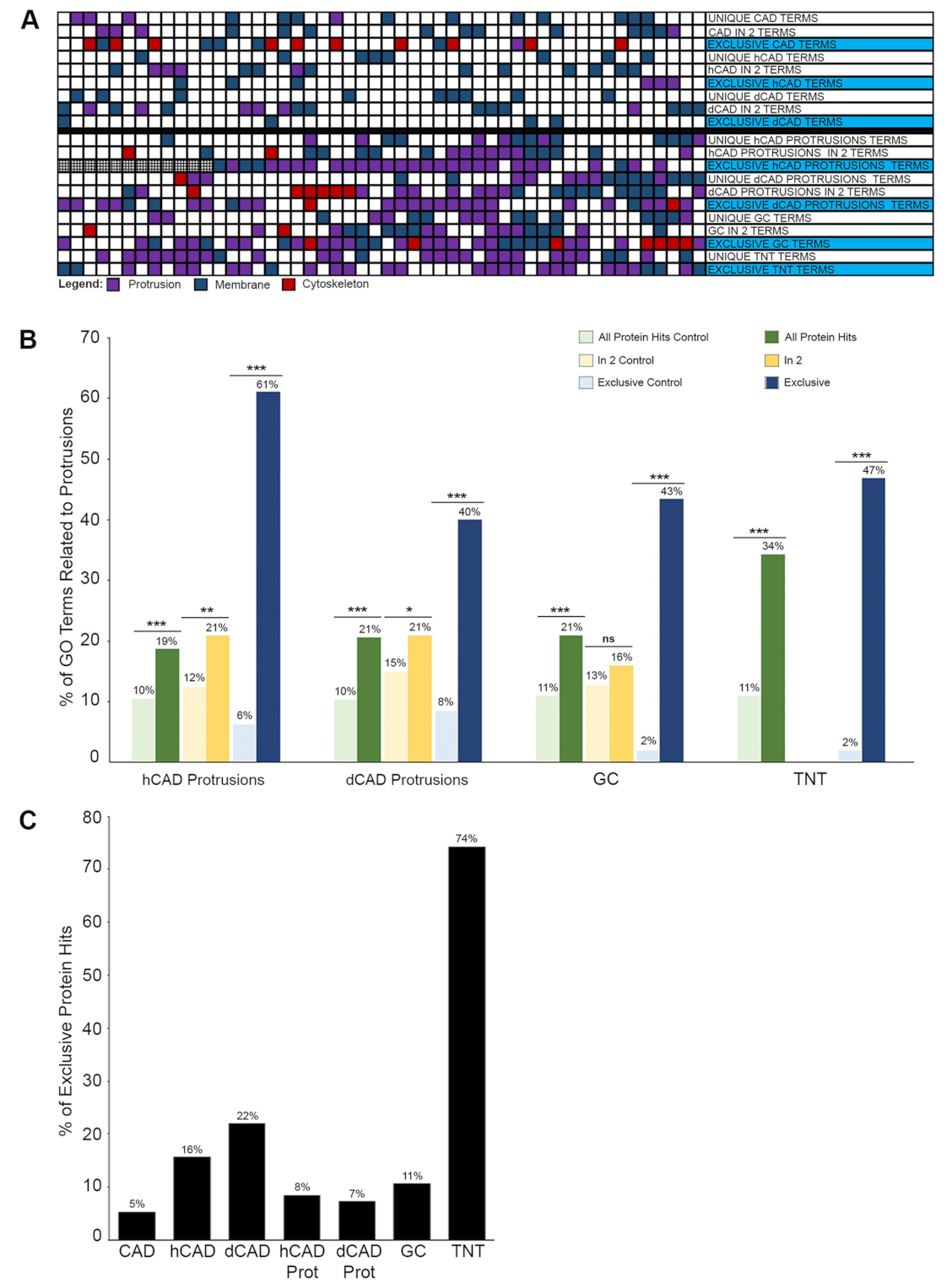
Next, we turned our attention to the current methods of proteomics analysis. We demonstrated that approaches used in high-throughput proteomics studies, where large datasets are acquired, were not appropriate to analyze extremely small samples such as cellular protrusions. The goals of proteomics and microproteomics studies are often opposed. Most high-throughput proteomics studies start with large datasets and try to reduce the number of protein hits by looking at the proteins identified in multiple replicates. In these studies, the additive approach helps to identify the most abundant proteins. However, when looking at microproteomic datasets, such as cellular protrusions, uncovering common, yet highly abundant proteins may not yield a list of functional proteins. In fact, many common, highly abundant proteins may be found in such structures due to random Brownian motion from the cytosol, as part of the transport/cargo machinery, or as part of the local protein synthesis machinery. As such, by eliminating the highly abundant, common proteins found in whole cell samples and other protrusions, we were able to identify proteins that were found exclusively in each protrusion subtype which our analysis suggested yields a far more interesting list of proteins.
For instance, since GCs are well characterized, we further analyzed their Proteomap (
Figure 7B). The most prominent annotation is “MAPK Signaling pathway.” Interestingly, this pathway plays an important role in GC regulation and navigation [
47]. Another major annotation is “Ion Channels.” Again, sodium/potassium channels are present in GCs [
48], along with calcium channels, which help regulate GC motility and axonal guidance [
49]. “Cofactor biosynthesis/regulation of autophagy/translation factors” are other important annotations, corroborated by the know functions of GCs. Indeed, transcriptional factors and local translation in GCs have been studied [
52] and autophagosome biogenesis in GCs has been observed [
53]. Thus, these Proteomap annotations from the “exclusive” GC list were remarkably accurate.
Furthermore, it is interesting to note that TNTs, whose primary function is to transport materials from one cell to another [
54]—including electrical signals [
55]—is the only Proteomap with “transport” as an annotation. It also contains “ion channels/cell adhesion molecules and their ligands/GTP binding signaling proteins,” all of which are consistent with TNT formation and function [
11]. This demonstrates that GCs and TNTs have a distinct set of proteins with specialized functions and attests to the necessity of changing from the additive to the subtractive approach. Indeed, this method of analysis is best suited for microproteomic studies from microscale sample sizes.
Similarly, using COMPleat, we found that all protrusions were depleted in “nucleolus/nuclear" localization (
Figure 7C). As expected, we observed an enrichment of “axon/plasma membrane/cell projection" localization in hCAD protrusions; “axon” localization in dCAD protrusions; “axon/plasma membrane/cytoskeleton” localization in GCs; and “plasma membrane” localization in TNTs (
Figure 7C). Once again, the fact that “axon” localization was not present in TNTs is a testimony to the accuracy of our method, and the fact that we can identify and isolate small cellular protrusions amongst larger protrusions.
In addition, since the isolation of small protrusions such as TNTs can be tedious, and technical replicates are not feasible, we wanted to test whether data could be obtained from a single experiment. Interestingly, we demonstrate that this method can be used with a single dataset—in addition to multiple control samples—as an initial screening method to identify protein targets. Such protein hits can then be independently confirmed and categorized using microscopy, biochemical experiments, or functional annotations (
Figure 5 and
Figure 7).
Overall, using LCM/MS, we also demonstrated that the proteomes of different subtypes of protrusions are unique and corroborate with their proposed functional roles (
Figure 6 and
Figure 7). Finally, using this approach, we identified proteins never determined to be part of protrusions (
Figure 5E–H), further highlighting the usefulness of such a method.
Up to now, researchers interested in specific subtypes of protrusions, had to rely on candidate-based approaches. Our methodology can now directly identify proteins contained within these structures and may uncover the unique proteins involved in the formation or function of each subtypes of protrusions.
For instance, since several cancers and diseases [
11,
56] appear to use TNTs for cell-to-cell transmission, there is a need to better characterize these unique structures. Interestingly, a recent paper using correlative cryo-EM has demonstrated that while filopodia and TNTs look similar by light microscopy, they are structurally different [
57]. Using our LCM/MS method, we are currently in the process of gathering new TNT samples along with filopodia only samples, which should finally bring to light some of the key structural and functional differences between these two types of protrusions. The identification of TNT-specific proteins could help distinguish the different transport mechanisms involved in specific diseases and lead to specific therapeutics [
58]. Beyond that, it may also lead to the discovery of specific biomarkers for in vivo studies, an important limitation that has slowed down research in the TNT field [
11].
Another advantage of this approach is its versatility. Indeed, isolation with LCM is not restricted to cell morphology, as structures of interest can also be identified using fluorescence labeling. For instance, Myosin-X (Myo10) plays a role in both filopodia and TNT formation [
28]. Using LCM combined with fluorescence microscopy we are currently isolating Myo10-dependent filopodia (
Figure 8), to identify their key components by MS.
4. Materials and Methods
4.1. Cell Culture
Cath.A differentiated (CAD) cells, derived from a catecholaminergic neuronal tumor, were obtained from mouse B6/D2 F1 hybrid; Sigma-Aldrich (St Louis, MO, USA) under the control of the European Collection of Authenticated Cell Cultures (ECACC), which assures both the authentication of the cell line and that it is mycoplasma free) and cultured with Opti-MEM Reduced Serum Medium, GlutaMAX Supplement (Gibco Life Technologies, Carlsbad, CA, USA) and 10% fetal bovine serum (FBS; Biowest, Riverside, CA, USA). To differentiate CAD cells into neurons, cells were grown in Opti-MEM without FBS for 10 days.
4.2. Fixation Protocol
Cellular protrusions are fragile and require strong fixative solutions to preserve their structures. We have previously demonstrated that the optimal fixation conditions for immunofluorescence of TNTs, one of the most delicate type of cellular structures to preserve due to their nature (i.e., not touching the substratum), was using a fixative containing glutaraldehyde (0.05%; Sigma-Aldrich), 2% of paraformaldehyde (PFA; Electron Microscopy Sciences, Hatfield, PA, USA) and 0.2 M HEPES (Life Technology, Carlsbad, CA, USA) in phosphate-buffered saline (PBS) for 15 min, followed by a second fixative containing 4% PFA and 0.2 M HEPES in PBS for another 15 min [
28]. Unfortunately, while this fixation is ideal for maintaining these structures, the MS results showed lower quantity and quality of the identified proteins, with mostly single peptide hits. For this reason, a novel fixative protocol based on dithiobispropionimidate (DTBP; ThermoFisher, Carlsbad, CA, USA), a chemical that can be de-crosslinked, was developed in order to optimize both the maintenance of the structural integrity of TNTs, as well as for the MS recovery. This new method greatly improved the quantity and quality of the proteins identified by MS [
26]. Briefly, CAD cells were fixed using 4% PFA and 0.2 M HEPES in PBS for 15 min, followed by a fixative containing 5mM DTBP and 25mM HEPES in PBS for another 15 min.
For immunofluorescence experiments where samples were not analyzed by MS, the glutaraldehyde-based protocol was used.
4.3. Laser Capture Microdissection
LCM was performed using a laser microdissection system from Molecular Machines and Industries (MMI CellCut Laser Microdissection, Eching, Germany) controlled by the MMI Cell Tools Software from the same company.
4.3.1. LCM Dish Preparation
The MMI Live cell chambers with membrane and petri dish (MMI, Haslett, MI, USA) were used to isolate all types of cellular protrusion by laser capture microdissection (LCM). The attachment of the cells to the membrane was facilitated by treating the membrane with 6 mM of fibronectin (Sigma-Aldrich) in PBS for 20 min at 37 °C. The solution was then removed and the membrane was washed twice with filtered PBS and once with Opti-MEM. Next, CAD cells were cultured in the dish and incubated at 37 °C for 3 h to allow the cells to attach to the surface and make protrusions. It should be taken into account that the cell density is a critical step and will depend on the cell line and the type of protrusion of interest (i.e., 60,000 or 180,000 CAD cells were plated to isolate growth cones or TNTs, respectively). In addition, cellular protrusions can be increased by different approaches, in the current work we used an exposure to 100 µM H
2O
2 (Fisher Scientific, Hampton, VA, USA) in Opti-MEM for 5 min [
28].
4.3.2. Laser Calibrations
The MMI system uses a fixed UV-laser with high pulse rate and low power, allowing for sharp and precise cutting line of 0.3 to 0.5 microns.
Laser cut speed: the laser cut speed was set low in order to avoid removing cells and/or the fragile cellular projections of interest during the cutting process. The speeds used in this study were in the range of 10 to 35 µm/s depending on the type of cellular protrusion cut. TNTs, which were the most fragile types of protrusions, were cut at 10 µm/s, while other adhering protrusions were cut at 20 or 35 µm/s, with three cutting repetitions (i.e., the laser cut the same area three times in order to ensure that the membrane and cellular protrusions were entirely cut from the rest of the dish).
Laser focus: the Laser focus (i.e., the position of the laser beam in the Z-direction) was adjusted before each experiment to ensure optimal and precise cutting. This was ensured by calibrating the laser focus according to the plane tilt of the dish. The laser focus used in this study was in the range of 2500 µm to 3400 µm.
Laser power: the power needed to samples is normally proportional to the sample thickness. The laser power was always set to get the smallest, precise laser cut through the membrane and was calibrating along with the Laser focus before each experiment. The power used in this study was in the range of 60% to 75%.
4.3.3. Objective Used
To improve the enrichment of different cellular protrusion from the cells of interest, the settings for LCM were adjusted for the growth cones, hCAD protrusions or TNTs with a 40× dry objective. When protrusions of dCADs were longer than 250 µm, the 20× objective was used.
4.3.5. Post-Cutting
To ensure that the LCM membranes, cellular debris, proteins from exosomal origin, secreted by the cells or from the FBS in the media did not contaminate our samples, negative controls were also isolated. Briefly, cells were plated similar to all other experiments, but instead of isolating specific types of protrusions, ROIs were obtained from “empty” regions in the LCM dishes, between groups of cells. These negative control samples were extracted and analyzed by MS, in an identical manner to all other samples. After isolating the different cell protrusions, the membrane rings were gently lifted from the microdissection chamber dishes, leaving the cut protrusions on the attached membrane. This step was carried out under the microscope at 4× magnification to check for any loss of cuts from the membrane while separating it from the microdissection chamber. The 40x magnification was used to verify that there was no contamination of cell bodies.
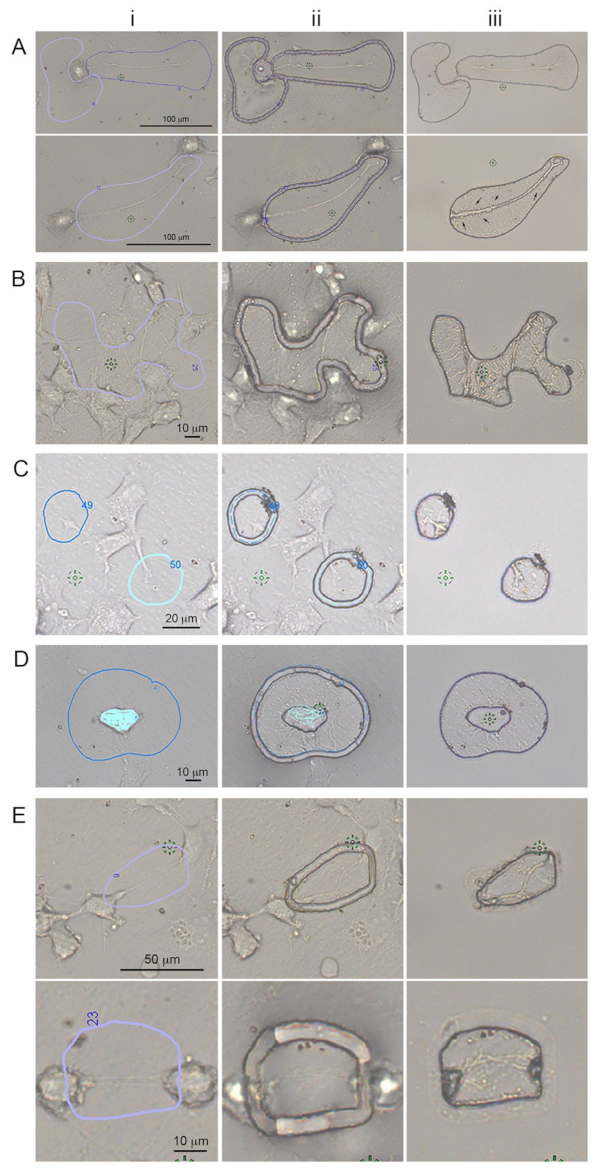
In order to gather enough proteins for each subcellular protrusion types, LCM-isolated protrusions had to be pooled together from different dishes for each independent experiment. For all experiment subtypes of protrusions, and/or whole cells, samples were cut within five days post fixation. On day five, the cellular protrusions were removed from the LCM dishes, recovered by pipetting with modified RIPA buffer and stored in a centrifuge tube at −80 °C. For growth cones, ~5000–6000 cuts from three dishes were pooled together for all three independent experiments. For hCAD protrusions, ~1000 cuts were obtained from three LCM dishes and for dCAD protrusions, ~1000–2000 cuts from two LCM dishes were pooled together for both duplicate experiments and stored at −80 °C until extraction. The cuts obtained for the different subcellular were similar to the example shown in
Figure 1. For each independent experiment, the growth cones and hCAD protrusions took ~2–3 weeks to isolate and ~2 weeks for the dCAD protrusions.
In the case of whole cell samples, the complete membrane was separated from the ring and immersed in modified RIPA buffer. All samples were stored at −80 °C until protein extraction.
For TNT isolation from cells fixed with glutaraldehyde/PFA fixation (see above), ~12,000 cuts were isolated from ~15 dishes in RIPA buffer. All of the samples from the 15 dishes were pooled into a single tube and stored at −80 °C until protein extraction. It took ~3–4 months to gather enough samples for one independent MS experiment.
4.4. Protein Extraction
As recently described [
26], the samples in modified RIPA buffer [10 mM Tris-HCl (pH 8.0), 1mM EDTA, 0.5 mM EGTA, 1% Triton X-100, 0.1% Sodium deoxycholate, and 140mM Sodium chloride], along with 1:100 Halt protease inhibitor cocktail (ThermoFisher)] containing 2% sodium dodecyl sulfate (SDS) and 100 mM DTT (Sigma-Aldrich) were sonicated for 5 min. Next, samples were incubated on ice for 20 min, followed by incubations at 37 °C in a water bath for 30 min, at 100 °C (dry bath) for 20 min, and at 60 °C (dry bath) for 2 h. Finally, samples were again sonicated for 5 min and centrifuged for 2 min at max speed. Protein concentration was assessed using RCDC (Biorad, Hercules, CA, USA), a detergent- and reducing agent-compatible assay.
4.5. Mass Spectrometry Sample Preparation
Five or three micrograms of protein lysate from pooled samples of isolated cellular protrusions or from whole cells/negative controls, respectively, were denatured using laemmli buffer containing 100 mM DTT and boiled for 5 min. Next, samples were run on 7.5% Mini-Protean TGX gels (BioRad) at 30 mAmp, for about 5–10 min, down to ~1 cm inside the gel (enough to visualize the MW marker ladder separation). This “limited gel” allows for the separation of the proteins by size in preparation for MS. The limited gel was stopped and fixed for 1h in fixing solution (Water:Methanol:Acetic acid = 40:40:8). Inside a biosafety cabinet, the samples were excised carefully from the gel and stored in 1% acetic acid water solution until analyzed by MS.
4.6. Mass Spectrometry—Sample Preparation for LC-MS
All mass spectrometry data collection and protein identification were contracted and performed by the Vincent Coates Foundation Mass Spectrometry Laboratory at Stanford University to eliminate bias. The analysis of the protein identification data was performed in-house.
The samples were diced into 1 mm × 1 mm squares, rinsed multiple times with 50 mM ammonium bicarbonate and reduced with 5 mM DTT, 50 mM ammonium bicarbonate at 55 °C for 30 min. Residual solvent was removed and alkylation was performed using 10 mM propionamide in 50 mM ammonium bicarbonate for 30 min at room temperature (RT). The gel pieces were rinsed with 50% acetonitrile 50 mM ammonium bicarbonate and place in a speed vacuum for 5 min. Digestion was performed with trypsin/LysC (Promega, Madison, WI, USA) overnight digest at 37 °C. Tubes were spun and the solvent including peptides were collected, further peptide extraction was performed by the addition of 60% acetonitrile, 39.9% water, 0.1% formic acid and incubated for 10–15 min. The peptide pools were dried in a speed vacuum.
4.7. Experimental Design and Statistical Rationale
Digested peptide pools were reconstituted and injected onto a 100 micron I.D. C18 reversed phase analytical column (Dr. Maisch, Ammerbuch-Entringen, Germany; 2.4 μM Reprosil-Pur) 25–50 cm in length. The UPLC was a Waters M class, operated at 300 nL/min using a linear gradient from 4% mobile phase B to 35% B. Mobile phase A consisted of 0.2% formic acid, 5% DMSO and water; mobile phase B was 0.2% formic acid, 5% DMSO, acetonitrile. All data were collected using an Orbitrap Fusion mass spectrometer set to acquire data in a data dependent fashion selecting and fragmenting by collision-induced dissociation the most intense precursor ions optimized to maximize duty cycle. An exclusion window of 60 seconds was used to improve proteomic depth and multiple charge states of the same ion were not sampled. A sample of hCAD and dCAD protrusions were run on a TIMS TOF, which gave comparable results. All raw MS/MS data were analyzed using Preview and Byonic v2.6.49 (Protein Metrics) as well as custom tools for data analysis developed in MatLab at Stanford University. Peak selection was handled automatically within Byonic.
MS/MS data were searched against a UniProtKB FASTA database containing 16,972 reviewed Mus musculus entries (various dates). Propionamidation (+71.037114 @ C) was set as a fixed modification, deamination (+0.984016 @ N) and Acetylation (+42.010565 @ K) were set as a common1 modifications, Oxidation (+15.994915 @ M) was set as common2 modification, and Acetylation (+42.010565 @ Protein N-term) and Methylation (+14.01565 @ K, R) were set as rare1 modifications. Byonic was set to allow a maximum of two common modifications and one rare modification and to allow a maximum of two missed cleavages. MS/MS spectra were matched with a tolerance of 12 ppm on precursor mass and 0.4 Da on fragment mass.
Common contaminants were filtered automatically by Byonic and include TRYP_PIG, ALBU_BOVIN, ALBU_HUMAN, CASB_BOVIN, CASK_BOVIN, CAS1_BOVIN, CO3_HUMAN, HBA_HUMAN, HBB_HUMAN, K1M1_SHEEP, K2C1_HUMAN, K22E_HUMAN, K1C10_HUMAN, K1C15_SHEEP, K1C9_HUMAN, KRHB1_HUMAN, KRHB3_HUMAN, KRHB5_HUMAN, KRHB6_HUMAN, TRFE_HUMAN.
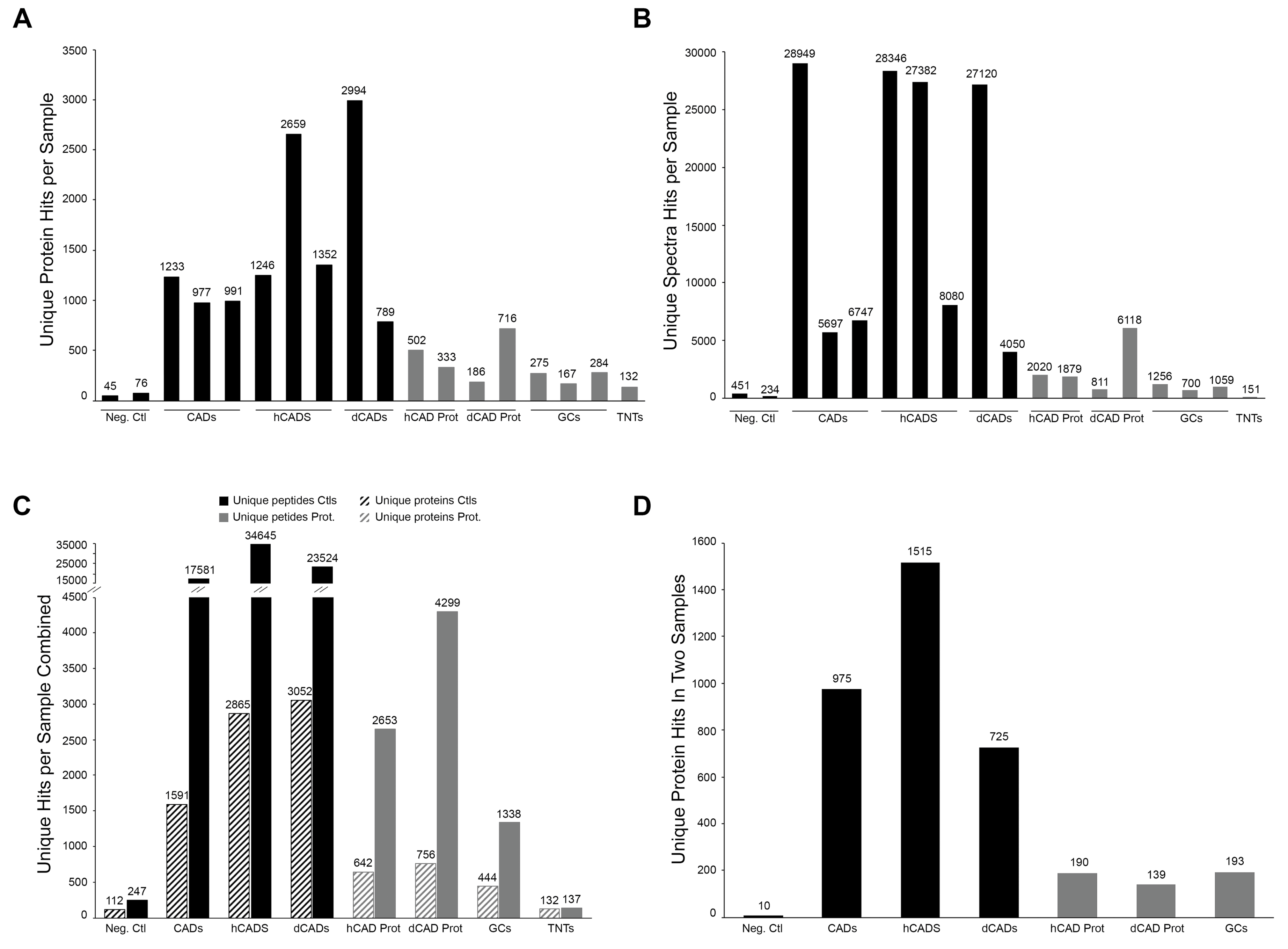
The growth cone samples and their respective controls were repeated in triplicate. Since this study sought to validate our LCM collection/enrichment method against existing methods, triplicate samples (with an “in two” exclusion criteria) were chosen to match the studies of Nozumi et al. and Estrada et al. Due to the nature of microproteomic studies, technical replicates are not possible since near full injection is necessary to increase the sensitivity and coverage of microproteomic samples.
By using Byonic, the proteome was searched with a reverse-decoy strategy and all data filtered and presented at a 1% false discovery rate. Byonic calculates a Byonic score that is an indicator of the correctness of our peptide-spectrum matches (PSM). Byonic scores reflect the absolute quality of the PSM, and have proved more useful than p-values [
59]. Byonic scores range from 0 to 1000, with 300 being a good score, 400 a very good score, and scores over 500 reflecting near perfect matches. Thus, all filtered protein identification hits have an FDR rate ≤ 1%, and a Byonic score >250 or a log probability >3.
The Raw MS/MS files and Byonic search results files have been deposited in the UCSD MassIVE repository with the dataset identifier MSV000082576 and can be accessed at
ftp://massive.ucsd.edu/MSV000082576.
To analyze the MS data and the reproducibility of our replicates, we used a spectral counting method known as normalized spectral abundance factor (NSAF) [
29]. We have previously used this label-free quantitation method to determine the relative abundance of proteins in 1000 whole cell samples [
26]. By taking into account the fact that longer proteins have more spectra available to be identified, raw MS spectral data can be normalized. Thus, using NSAF quantification we were able to analyze the abundance of proteins caught in our various samples and plotted correlation graphs. In cases where multiple isoforms of an individual protein were identified those isoforms were treated as separate proteins for NSAF quantification and correlation analysis. Negative controls (10 proteins were found in at least two samples) served as a baseline NSAF value for all other datasets.
In order to compare the different sample sets, DESeq [
30] was used to estimate the variance mean in count data, allowing the use of a negative binomial model to test differential expression. NSAF/DESeq normalization resulted in non-normal data. In order to approximate normality and to use a parametric test such as Pearson, we used Johnson transformations.
All plots and data analysis were obtained using R and the statistical analyses were done using StatPlusPro, JMP, or SuperExactTest R software package [
31].
4.8. Quantitative Overlap
The Quantitative overlap analyses were performed as previously described using the SuperExactTest R software package [
31]. The SuperExactTest was specifically developed to visualize multi-set intersections. We used this software to compare and visualize the lists of proteins identified for each MS sample and published protein lists [
12,
13,
22]. Circular plots show the intersections and the corresponding statistics of the different protein sets. For each lot, the middle tracks represent the protein sets analyzed and the green blocks highlight the proteins that were “present”, compared to the white or “absent” proteins in each intersection. The height of the bars in the outer layer is proportional to the intersection sizes, and the number of proteins is indicated on the top of the bars. The color intensity of the bars is a representation of the P value significance of the intersections.
4.9. Immunofluorescence (IF) Validation Using the Human Protein Atlas (HPA) Subcell Database
We used the IF images from the 12073 proteins in the HPA Subcell database [
39,
40] to analyze our MS data for its localization to protrusions. IF images for all proteins were catalogued as being observed or not in protrusions. Positive hits were counted only when they were clearly identifiable in protrusions (
Figure S3). These data are compiled in a publicly accessible database, PROTPR (
https://goussetlab.shinyapps.io/PROTPR/).
A 2 × 2 contingency table with odds ratio was used to calculate fold enrichment over expected. Significance was tested using two separate chi square tests (Pearson’s chi-square test and Yates’s correction for continuity).
4.10. Immunofluorescence (IF)
Among all the proteins obtained by MS in the protrusion samples, we identified proteins that we would “expect” or “not expect” to be found in protrusions based on their known function and/or localization. These proteins were then visualized by IF in order to determine their localization within cells and whether or not they were present within protrusions. For these IF experiments, 60,000 CAD cells were plated the day before on coverslips. The next day, cells were fixed and permeabilized with methanol (7 min at −20 °C) or Triton (0.1% for 10 min at RT), depending on primary antibody used. The next steps were: blocking with 2% BSA for 1 h, incubation with primary antibody (in blocking solution) for 1 h at RT, washes with PBS, wash with blocking solution, incubation with secondary antibody (1:400 in blocking solution) for 1 h at RT, washes with PBS, mounting on slides and sealed with Aqua-poly mount (PolySciences, Warrington, PA, USA). The primary antibodies used were: rabbit anti-Grk5 (sc-11396), rabbit anti-Cd47 (sc-25773), mouse anti-Anxa2 (sc-28385), goat anti-Hist1h3b (sc-8654), and goat anti-Tenm2 (sc-165674) from Santa Cruz Biotechnology (Dallas, TX, USA) and rabbit anti-Cobl (NBP1-89615), rabbit anti-Arg1 (NBP1-32731), and mouse anti-Hspa1b (H00003304-M02) from Novus Biologicals (Centennial, CO, USA). The secondary antibodies used were Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 546; Donkey anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, FITC or Rabbit anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 (ThermoFisher Scientific). Negative controls: cells were permeabilized with either MeOH or Triton X-100 and incubated only with secondary antibodies to ensure that under our experimental conditions; we did not have nonspecific fluorescent signals from the secondary antibodies used. For each antibody, high resolution images were acquired using a widefield inverted Leica microscope (DMI3000) controlled by Metamorph acquisition software (Molecular Devices, San Jose, CA, USA), a 63×/1.25 oil objective and a Leica DFC300 FX camera. For each condition, Z-stacks were acquired and 3D cell volume rendering was obtained. Representative pictures are shown. The analyses of the Z-stacks were obtained using the Image J software (
http://rsb.info.nih.gov/ij/).
4.11. Myosin-X Transfection
150,000 CAD cells, plated on a 35 mm dish the day before transfection, were transiently transfected with EGFPC1-hMyosin-X plasmid [
60] using Lipofectamine 2000 (ThermoFisher Scientific) in accordance with the manufacturer’s instructions. 48 h after transfection, cells were split, counted and seeded (70,000 cells) on a MMI Live cell Chamber dish and fixed 3 h later as described above. The GFP-Myo10 transfected cells were identified using the FITC filter of the LCM and subsequently, membrane structures, such as tip complex, were delimited with the FITC channel and cut with the laser.
4.12. Localization Enrichment Analysis
The localization of the proteins identified in our samples was analyzed using a free, in-house developed R software tool called COMPLEAT (COMPutational Localization Enrichment Analysis Tool). This software identifies the enriched/depleted localizations of lists of proteins using the COMPARTMENTS and HPA Subcellular Atlas localization databases.
A 2 × 2 contingency table with odds ratio was used to determine enrichment/depletion of localizations. The significance of the difference in background frequency versus the sample frequency was analyzed with two separate chi square tests (Pearson’s chi-square test and Yates’s correction for continuity). The chi-square value was then converted using R to a p-value using the command pchisq (Chi-square Value, Degrees of Freedom).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







