Phylogeography of the Assassin Bug Sphedanolestes impressicollis in East Asia Inferred From Mitochondrial and Nuclear Gene Sequences

 ,
, 

Abstract
:
1. Introduction
2. Results
2.1. Genetic Diversity
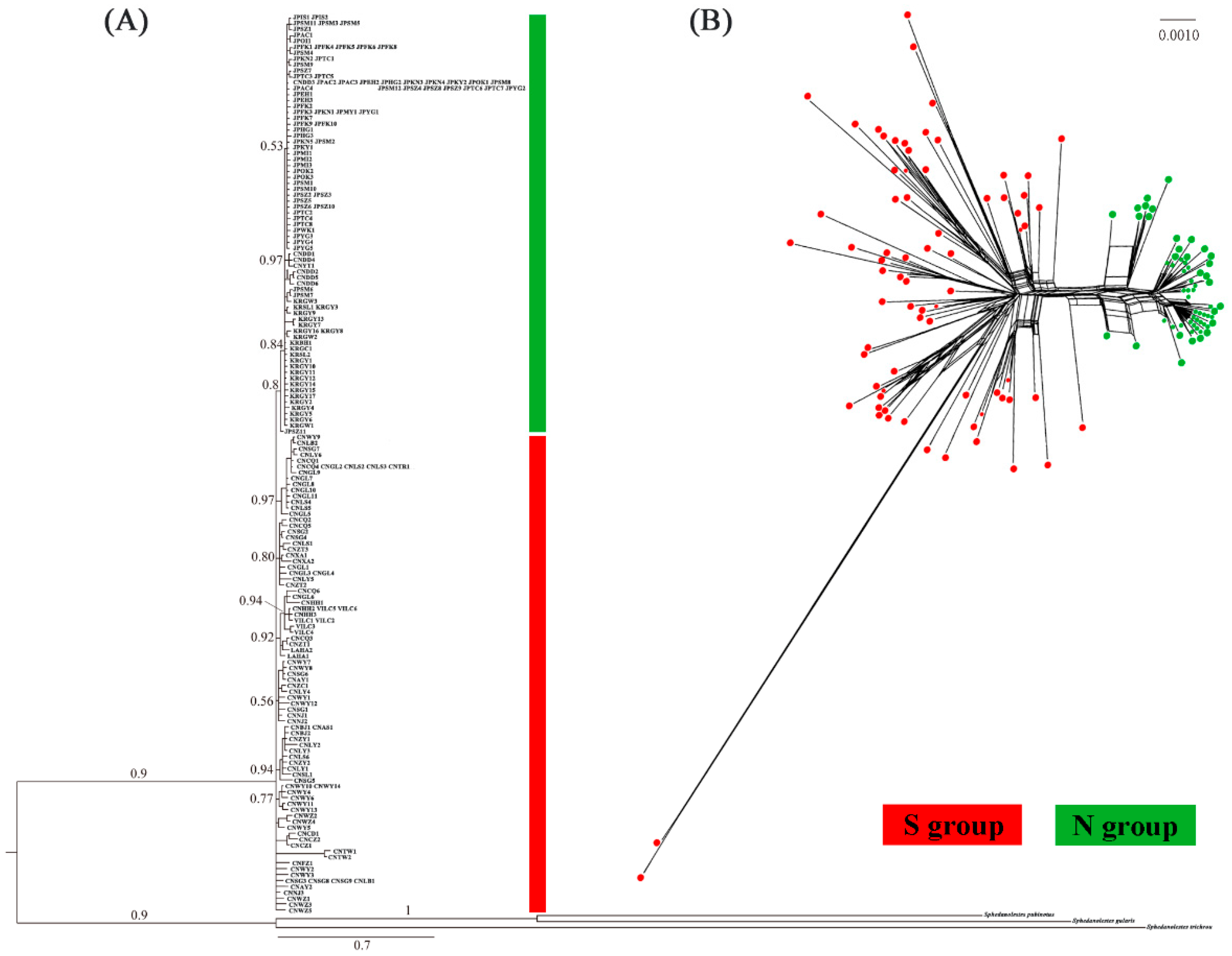
2.2. Population Genetic Structure
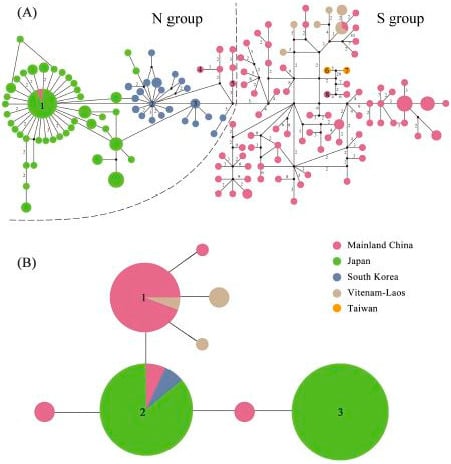
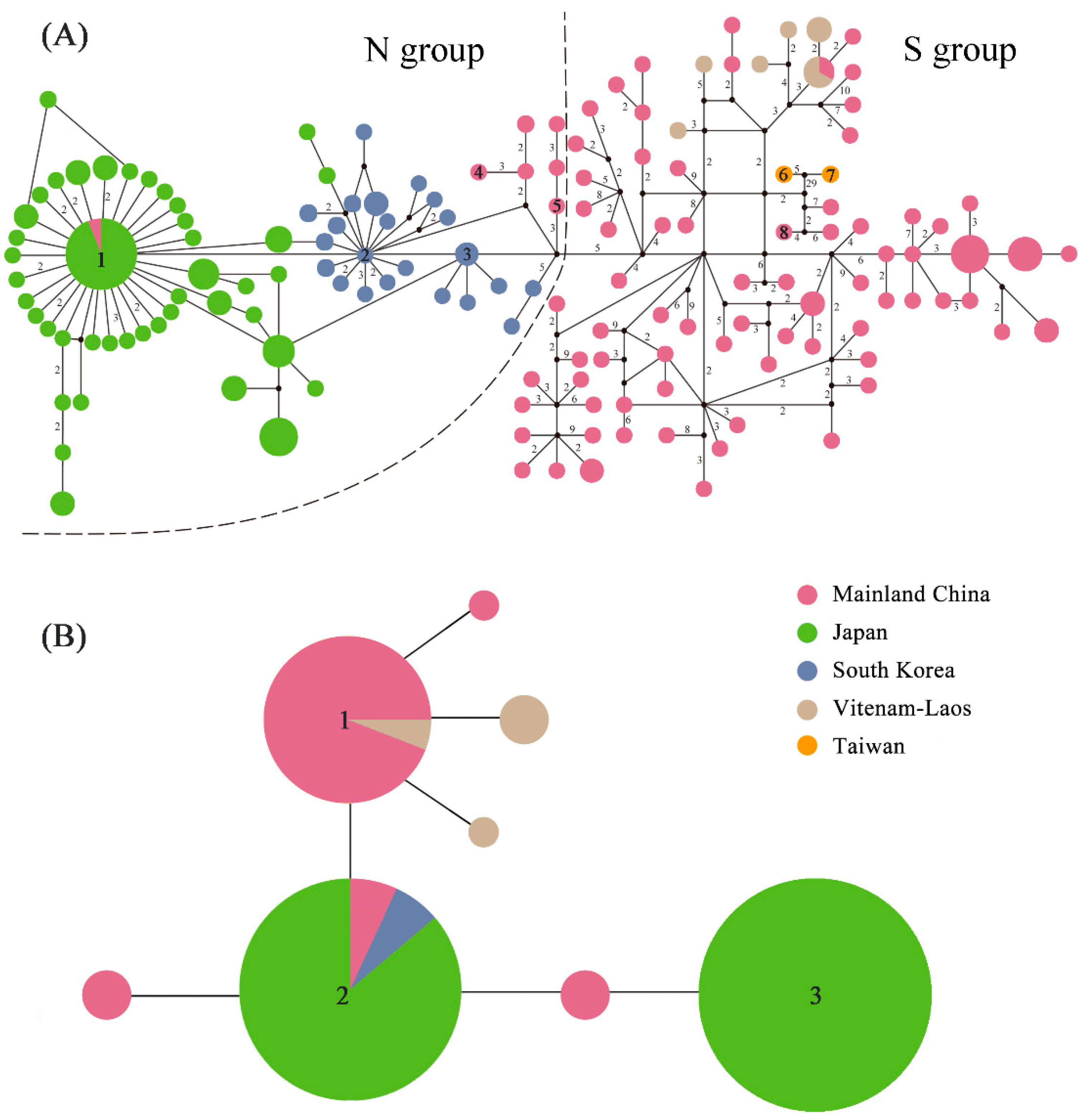
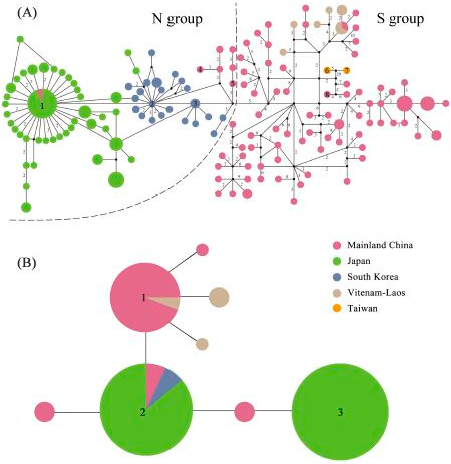
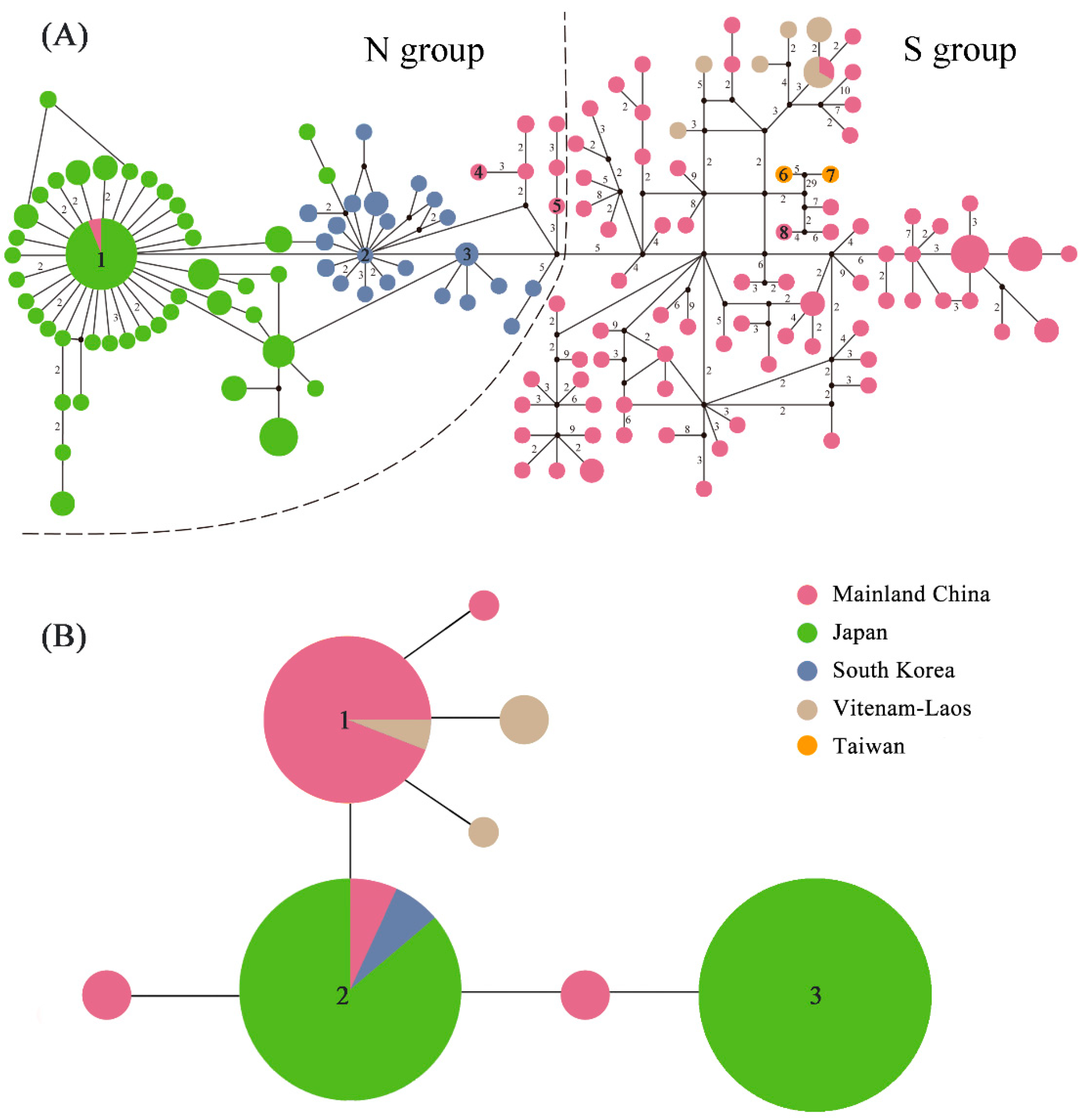
2.3. Haplotype Distribution
2.4. Estimated Divergence Time
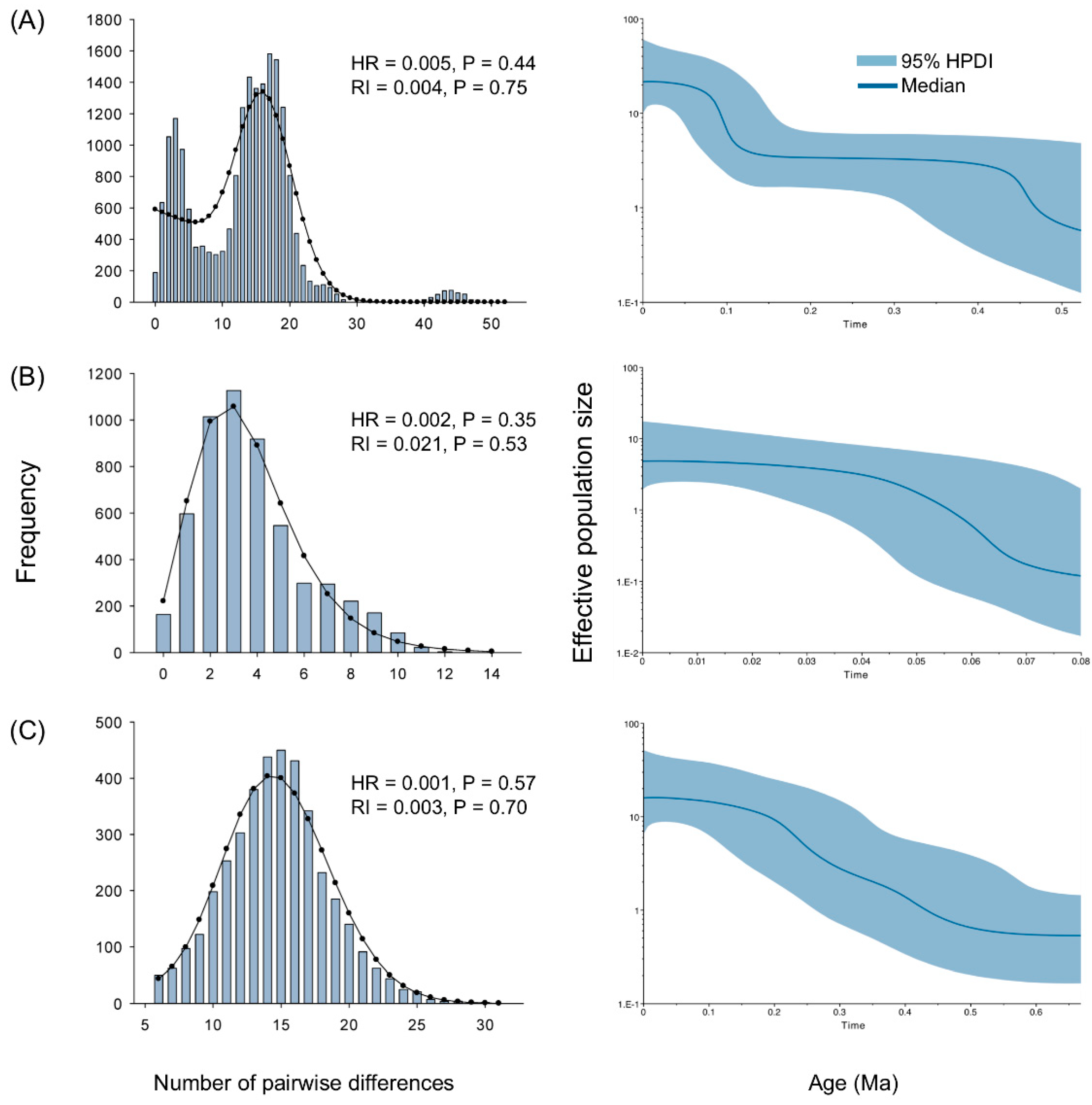
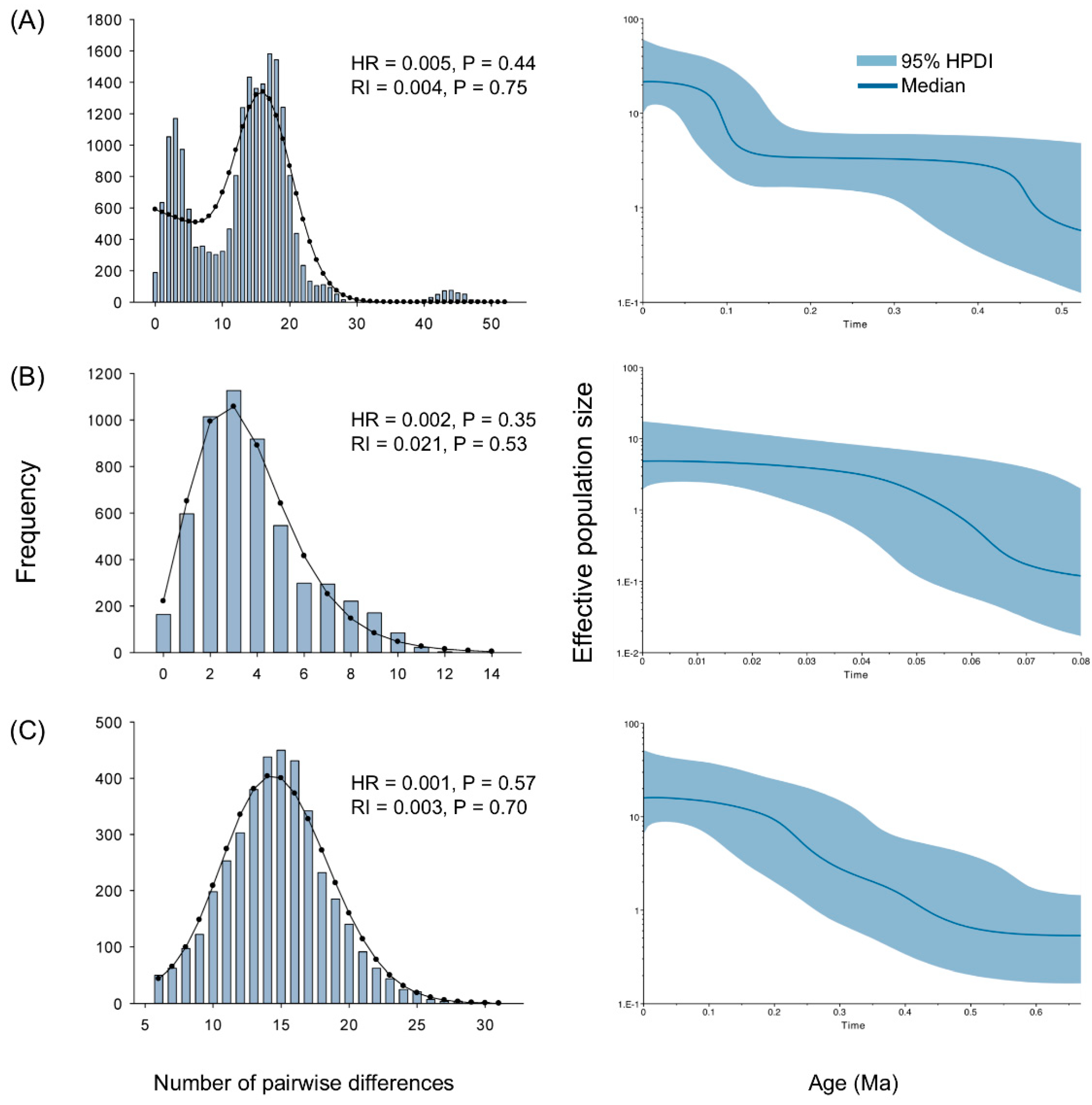
2.5. Demographic History
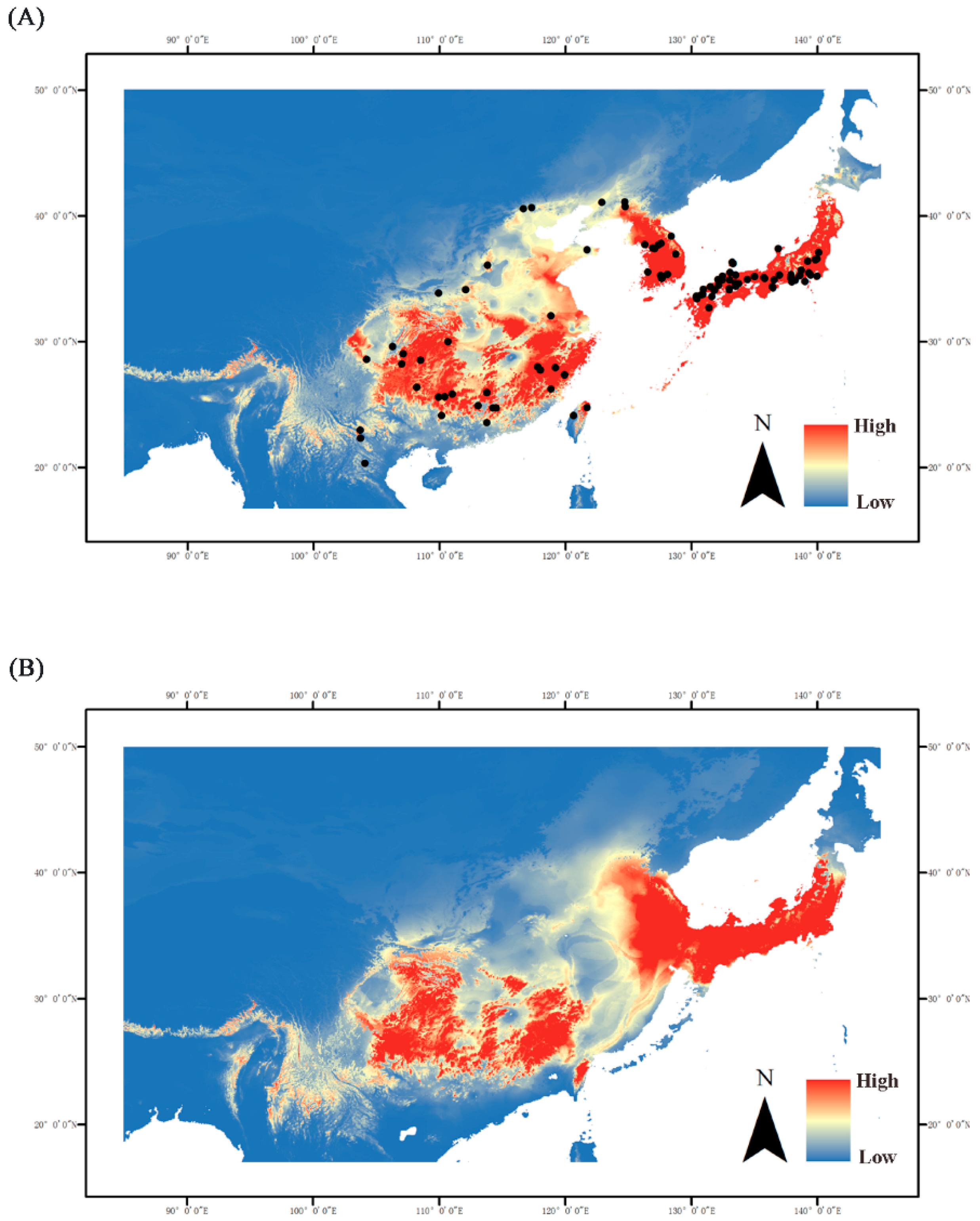
2.6. Ecological Niche Modeling
3. Discussion
3.1. Sphedanolestes Impressicollis Distribution Pattern in East Asia
3.2. Historical Demography in the Sino-Japanese Region
3.3. Phylogeography of Native Chinese Populations
4. Material and Methods
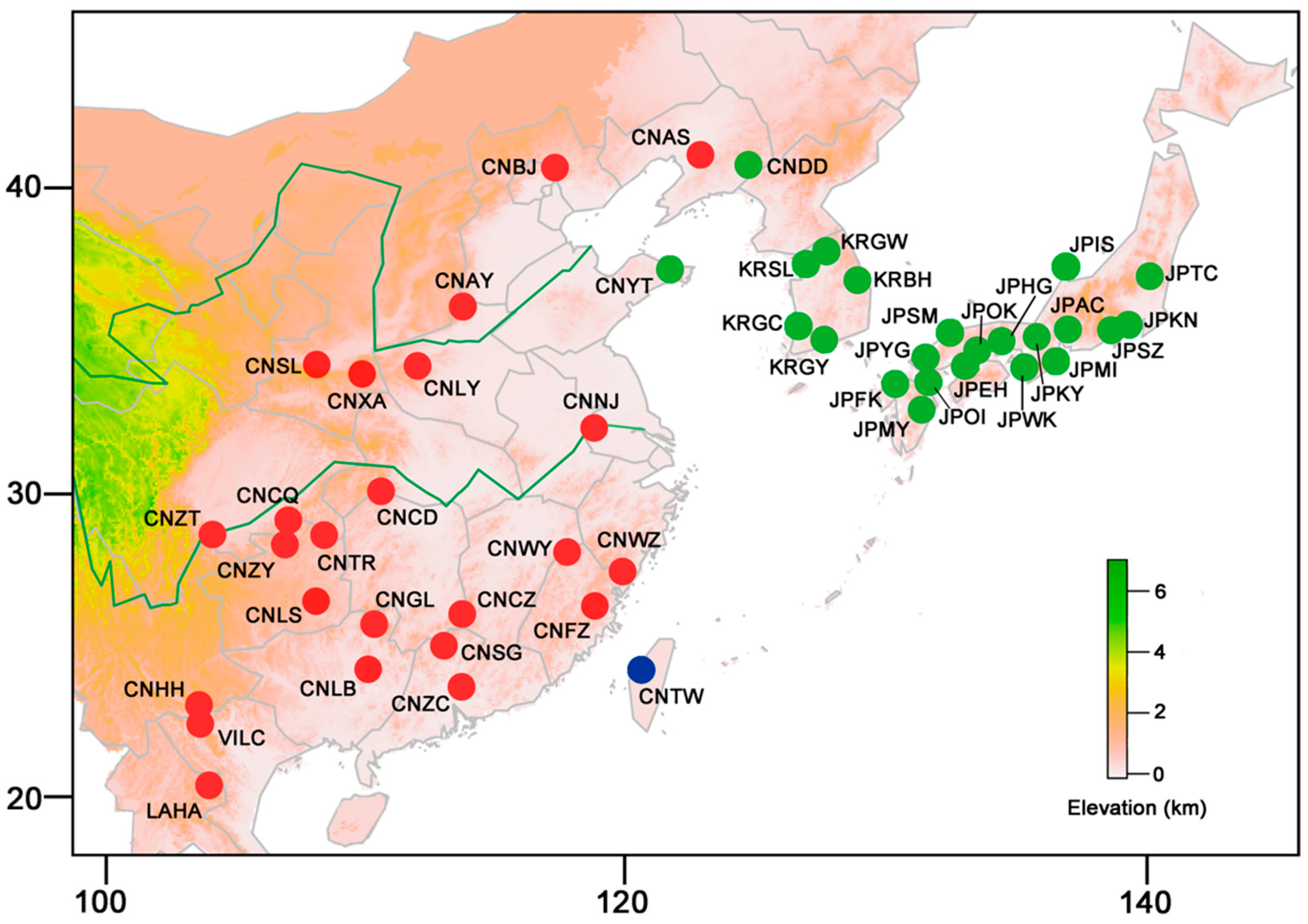
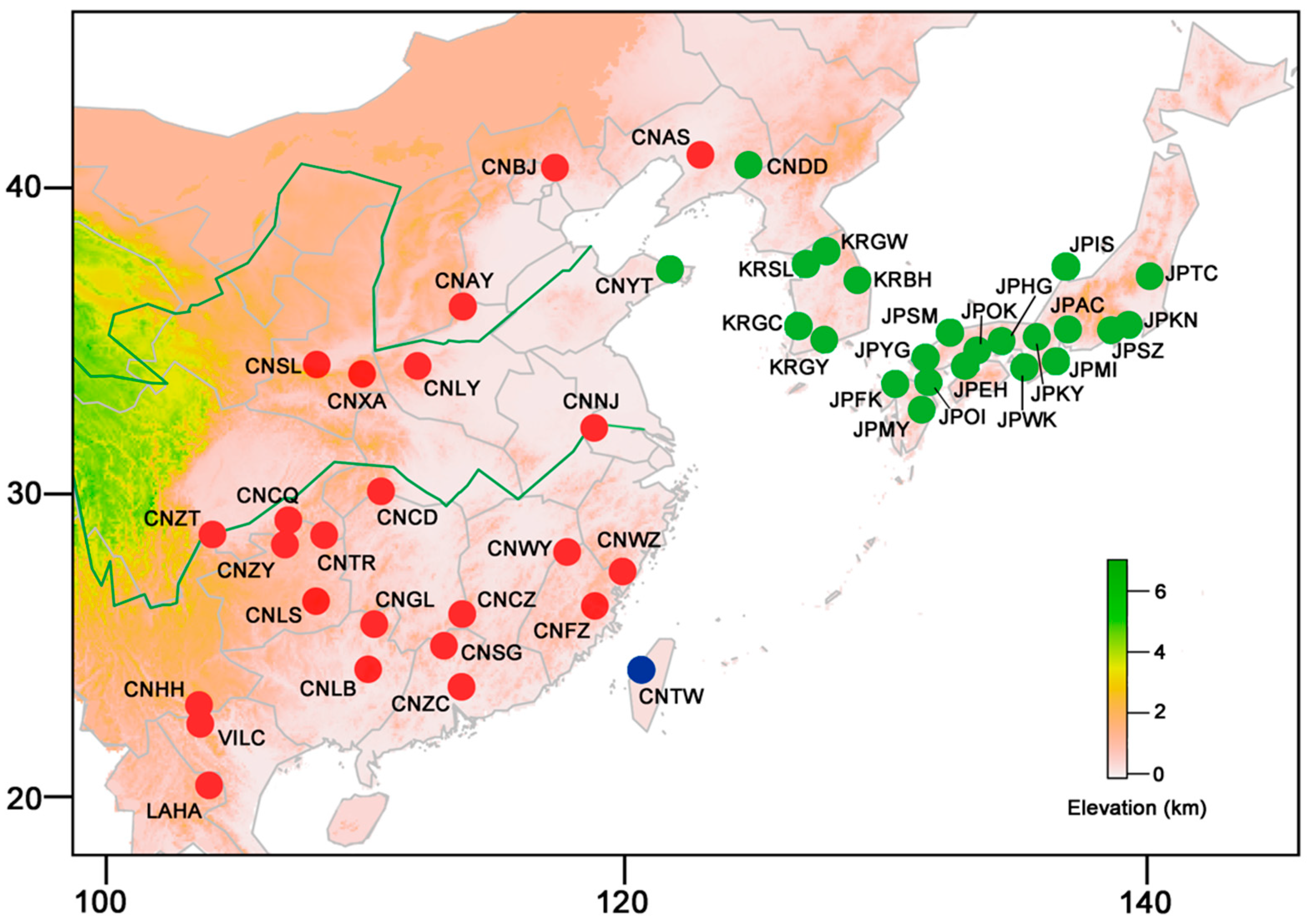
4.1. Sampling, DNA Extraction, Amplification, and Sequencing
4.2. Phylogenetic Analysis
4.3. Genetic Diversity and Population Differentiation
4.4. Divergence Time Estimation
4.5. Demographic History
4.6. Ecological Niche Modeling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cox, C.B.; Moore, P.D.; Ladle, R. Biogeography: An Ecological and Evolutionary Approach, 9th ed.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2016. [Google Scholar]
- Voris, H.K. Maps of Pleistocene sea levels in Southeast Asia: Shorelines, river systems and time durations. J. Biogeogr. 2000, 27, 1153–1167. [Google Scholar] [CrossRef]
- Hewitt, G.M. The genetic legacy of the Quaternary ice ages. Nature 2000, 405, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, G.M. Genetic consequences of climatic oscillations in the Quaternary. Philos. Trans. R. Soc. B 2004, 359, 183–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taberlet, P. Biodiversity at the intraspecific level: The comparative phylogeographic approach. J. Biotechnol. 1998, 64, 91–100. [Google Scholar] [CrossRef]
- Hewitt, G.M. Speciation, hybrid zones and phylogeography—Or seeing genes in space and time. Mol. Ecol. 2001, 10, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Avise, J. Phylogeography: The History and Formation of Species; Harvard University Press: Cambridge, MA, USA; London, UK, 2000. [Google Scholar]
- An, Z.; Liu, T.; Lu, Y.; Porter, S.; Kukla, G.; Xihao, W.; Yingming, H. The long-term paleomonsoon variation recorded by the loess-paleosol sequence in central China. Quat. Int. 1990, 7, 91–95. [Google Scholar]
- Liu, D.; Li, Z. Geography of Asia; Commercial Press: Beijing, China, 1996. [Google Scholar]
- Zhang, R. Geological events and mammalian distribution in China. Acta Zool. Sin. 2002, 48, 141–153. [Google Scholar]
- Ding, L.; Gan, X.N.; He, S.-P.; Zhao, E.-M. A phylogeographic, demographic and historical analysis of the short-tailed pit viper (Gloydius brevicaudus): Evidence for early divergence and late expansion during the Pleistocene. Mol. Ecol. 2011, 20, 1905–1922. [Google Scholar] [CrossRef]
- Axelrod, D. Floristic Characteristics Diversity of East Asia Plants; ScienceOpen, Inc.: Burlington, MA, USA, 1996; pp. 43–55. [Google Scholar]
- Millien-Parra, V.; Jaeger, J.J. Island biogeography of the Japanese terrestrial mammal assemblages: An example of a relict fauna. J. Biogeogr. 1999, 26, 959–972. [Google Scholar] [CrossRef]
- Harrison, S.; Yu, G.; Takahara, H.; Prentice, I. Palaeovegetation (Communications arising): Diversity of temperate plants in east Asia. Nature 2001, 413, 129–130. [Google Scholar] [CrossRef]
- Qi, X.; Yuan, N.; Comes, H.P.; Sakaguchi, S.; Qiu, Y. A strong ‘filter’ effect of the East China Sea land bridge for East Asia’s temperate plant species: Inferences from molecular phylogeography and ecological niche modelling of Platycrater arguta (Hydrangeaceae). BMC Evol. Biol. 2014, 14, 41. [Google Scholar] [CrossRef]
- Zhang, D.; Ye, Z.; Yamada, K.; Zhen, Y.; Zheng, C.; Bu, W. Pleistocene sea level fluctuation and host plant habitat requirement influenced the historical phylogeography of the invasive species Amphiareus obscuriceps (Hemiptera: Anthocoridae) in its native range. BMC Evol. Biol. 2016, 16, 174. [Google Scholar] [CrossRef]
- Park, Y.C.; Maekawa, K.; Matsumoto, T.; Santoni, R.; Choe, J.C. Molecular phylogeny and biogeography of the Korean woodroaches Cryptocercus spp. Mol. Phylogenet. Evol. 2004, 30, 450–464. [Google Scholar] [CrossRef]
- Wei, S.; Cao, L.; Gong, Y.; Shi, B.; Wang, S.; Zhang, F.; Guo, X.; Wang, Y.; Chen, X. Population genetic structure and approximate Bayesian computation analyses reveal the southern origin and northward dispersal of the oriental fruit moth Grapholita molesta (Lepidoptera: Tortricidae) in its native range. Mol. Ecol. 2015, 24, 4094–4111. [Google Scholar] [CrossRef]
- Xun, H.; Li, H.; Li, S.; Wei, S.; Zhang, L.; Song, F.; Jiang, P.; Yang, H.; Han, F.; Cai, W. Population genetic structure and post-LGM expansion of the plant bug Nesidiocoris tenuis (Hemiptera: Miridae) in China. Sci. Rep. 2016, 6, 26755. [Google Scholar] [CrossRef]
- Xue, J.; Zhang, H.; Ning, X.; Bu, W.; Yu, X. Evolutionary history of a beautiful damselfly, Matrona basilaris, revealed by phylogeographic analyses: The first study of an odonate species in mainland China. Heredity 2018. [Google Scholar] [CrossRef]
- Cai, W.; Cai, X.; Wang, Y. Notes on the genus Sphedanolestes Stål (Heteroptera: Reduviidae: Harpactorinae) from China, with the description of three new species. Raffles Bull. Zool. 2004, 52, 379–388. [Google Scholar]
- Ambrose, D.P. Assassin Bugs; Oxford & IBH Publ. Co. Pvt. Ltd.: New Delhi, India; Science Publishers, Inc.: Enfield, NH, USA, 1999; pp. 220–250. [Google Scholar]
- Du, Z.; Liu, H.; Li, H.; Ishikawa, T.; Su, Z.-H.; Cai, W.; Kamitani, S.; Tadauchi, O. Invasion of the assassin bug Agriosphodrus dohrni (Hemiptera: Reduviidae) to Japan: Source estimation inferred from mitochondrial and nuclear gene sequences. Int. J. Biol. Macromol. 2018, 118, 1565–1573. [Google Scholar] [CrossRef]
- Peretolchina, T.; Pavan, M.G.; Correa-Antonio, J.; Gurgel-Goncalves, R.; Lima, M.M.; Monteiro, F.A. Phylogeography and demographic history of the Chagas disease vector Rhodnius nasutus (Hemiptera: Reduviidae) in the Brazilian Caatinga biome. PLoS Negl. Trop. Dis. 2018, 12, e0006731. [Google Scholar] [CrossRef]
- Liu, Y.; Qiu, Y.U.E.; Wang, X.U.; Yang, H.; Hayashi, M.; Wei, C. Morphological variation, genetic differentiation and phylogeography of the East Asia cicada Hyalessa maculaticollis (Hemiptera: Cicadidae). Syst. Entomol. 2018, 43, 308–329. [Google Scholar] [CrossRef]
- Varvio, S.-L.; Chakraborty, R.; Nei, M. Genetic variation in subdivided populations and conservation genetics. Heredity 1986, 57, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Chiang, Y.-C.; Schaal, B.A.; Chou, C.-H.; Chiang, T.Y. Organelle DNA phylogeography of Cycas taitungensis, a relict species in Taiwan. Mol. Ecol. 2001, 10, 2669–2681. [Google Scholar] [CrossRef]
- Provan, J.; Murphy, S.; Maggs, C.A. Tracking the invasive history of the green alga Codium fragile ssp. tomentosoides. Mol. Ecol. 2005, 14, 189–194. [Google Scholar] [CrossRef]
- Arnaud-Haond, S.; Teixeira, S.; Massa, S.I.; Billot, C.; Saenger, P.; Coupland, G.; Duarte, C.M.; Serrao, E.A. Genetic structure at range edge: Low diversity and high inbreeding in Southeast Asian mangrove (Avicennia marina) populations. Mol. Ecol. 2006, 15, 3515–3525. [Google Scholar] [CrossRef]
- Bittkau, C.; Comes, H.P. Evolutionary processes in a continental island system: Molecular phylogeography of the Aegean Nigella arvensis alliance (Ranunculaceae) inferred from chloroplast DNA. Mol. Ecol. 2005, 14, 4065–4083. [Google Scholar] [CrossRef]
- Kimura, M. Land connections between Eurasian continent and Japanese Islands related to human migration. Migr. Diffus. 2003, 4, 14–33. [Google Scholar]
- Kimura, M. Quaternary paleogeography of the Ryukyu Arc. J. Geogr. 1996, 105, 259–285. [Google Scholar] [CrossRef]
- Simpson, G.G. Mammals and land bridges. J. Washington Acad. Sci. 1940, 30, 137–163. [Google Scholar]
- Lomolino, M.V.; Riddle, B.R.; Brown, J.H. Biogeography, 3rd ed.; Sinauer Associates Inc.: Sunderland, MA, USA, 2006. [Google Scholar]
- Ikeda, H.; Cho, Y.-B.; Sota, T. Colonization history of the carrion beetle Necrophila jakowlewi (Coleoptera: Silphidae) in Japan inferred from phylogeographic analysis. Zool. Sci. 2013, 30, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Saul, H. The Nepalese Himalayas in prehistoric context: A review of their role in Eurasian exchange networks and domestication. Hist. Compass 2016, 14, 191–205. [Google Scholar] [CrossRef]
- Petit, R.J.; Aguinagalde, I.; de Beaulieu, J.-L.; Bittkau, C.; Brewer, S.; Cheddadi, R.; Ennos, R.; Fineschi, S.; Grivet, D.; Lascoux, M. Glacial refugia: Hotspots but not melting pots of genetic diversity. Science 2003, 300, 1563–1565. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shu, Q.; Zhou, S.; Zhao, Z.; Zhang, J. Review and prospects of Quaternary glaciation research in China. J. Glaciol. Geocryol. 2004, 26, 235–243. [Google Scholar]
- Song, W.; Cao, L.; Li, B.; Gong, Y.; Hoffmann, A.A.; Wei, S. Multiple refugia from penultimate glaciations in East Asia demonstrated by phylogeography and ecological modelling of an insect pest. BMC Evol. Biol. 2018, 18, 152. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Xu, Q.; Shen, Y. The relationship between climate change and Quaternary glacial cycles on the Qinghai–Tibetan Plateau: Review and speculation. Quat. Int. 2002, 97, 93–101. [Google Scholar] [CrossRef]
- Yi, C.-L.; Cui, Z.-J.; Xiong, H.-G. Numerical periods of Quaternary glaciations in China. Quat. Sci. 2005, 25, 609–619. [Google Scholar]
- Zhang, H.; Yan, J.; Zhang, G.; Zhou, K. Phylogeography and demographic history of Chinese black-spotted frog populations (Pelophylax nigromaculata): Evidence for independent refugia expansion and secondary contact. BMC Evol. Biol. 2008, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-H.; Yeung, C.K.L.; Feinstein, J.; Han, L.; Le, M.H.; Wang, C.X.; Ding, P. Sailing through the Late Pleistocene: Unusual historical demography of an East Asian endemic, the Chinese Hwamei (Leucodioptron canorum canorum), during the last glacial period. Mol. Ecol. 2009, 18, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Frati, F.; Beckenbach, A.; Crespi, B.; Liu, H.; Flook, P. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 1994, 87, 651–701. [Google Scholar] [CrossRef]
- Monteiro, F.A.; Barrett, T.V.; Fitzpatrick, S.; Cordon-Rosales, C.; Feliciangeli, D.; Beard, C.B. Molecular phylogeography of the Amazonian Chagas disease vectors Rhodnius prolixus and R. robustus. Mol. Ecol. 2003, 12, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Kawakita, A.; Sota, T.; Ascher, J.S.; Ito, M.; Tanaka, H.; Kato, M. Evolution and phylogenetic utility of alignment gaps within intron sequences of three nuclear genes in bumble bees (Bombus). Mol. Biol. Evol. 2003, 20, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Connor, T.R.; Siren, J.; Aanensen, D.M.; Corander, J. Hierarchical and spatially explicit clustering of DNA sequences with BAPS software. Mol. Biol. Evol. 2013, 30, 1224–1228. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. MrBayes: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Posada, D. Selection of Models of DNA Evolution with jModelTest. In Bioinformatics for DNA Sequence Analysis; Posada, D., Ed.; Humana Press: Totowa, NJ, USA, 2009; pp. 93–112. [Google Scholar]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2005, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Bandelt, H.J.; Macaulay, V.; Richards, M. Median networks: Speedy construction and greedy reduction, one simulation, and two case studies from human mtDNA. Mol. Phylogenet. Evol. 2000, 16, 8–28. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Bouckaert, R.; Heled, J.; Kuhnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef]
- Brower, A. Rapid morphological radiation and convergence among races of the butterfly Heliconius erato inferred from patterns of mitochondrial DNA evolution. Proc. Natl. Acad. Sci. USA 1994, 91, 6491–6495. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [PubMed]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [PubMed]
- Rogers, A.R.; Harpending, H. Population growth makes waves in the distribution of pairwise genetic differences. Mol. Biol. Evol. 1992, 9, 552–569. [Google Scholar] [PubMed]
- Clark, P.U.; Dyke, A.S.; Shakun, J.D.; Carlson, A.E.; Clark, J.; Wohlfarth, B.; Mitrovica, J.X.; Hostetler, S.W.; McCabe, A.M. The Last Glacial Maximum. Science 2009, 325, 710–714. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.J.; Anderson, R.P.; Schapire, R.E. Maximum entropy modeling of species geographic distributions. Ecol. Model. 2006, 190, 231–259. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | N | Nh | S | π | Tajima’s D | Fu’s Fs | τ |
|---|---|---|---|---|---|---|---|
| All samples | 199 | 152 | 225 | 0.00744 | −2.087 * | −23.945 * | 16.90000 |
| N group | 105 | 71 | 83 | 0.00215 | −2.476 * | −25.989 * | 2.38867 |
| S group | 94 | 81 | 189 | 0.00865 | −1.972 * | −24.186 * | 14.97266 |
| Dataset | Source of variation | d.f. | SSD | Percentage | Fixation Indices |
|---|---|---|---|---|---|
| Mitochondrial dataset | Among groups | 1 | 392.480 | 44.70 | ΦCT = 0.44698 * |
| Among populations within groups | 46 | 436.791 | 18.77 | ΦSC = 0.33949 * | |
| Within populations | 151 | 469.413 | 36.53 | ΦST = 0.63473 * | |
| Nuclear gene | Among groups | 1 | 21.423 | 51.14 | ΦCT = 0.56121 * |
| Among populations within groups | 22 | 27.434 | 27.42 | ΦSC = 0.51144 * | |
| Within populations | 63 | 14.361 | 21.44 | ΦST = 0.78562 * |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, Z.; Ishikawa, T.; Liu, H.; Kamitani, S.; Tadauchi, O.; Cai, W.; Li, H. Phylogeography of the Assassin Bug Sphedanolestes impressicollis in East Asia Inferred From Mitochondrial and Nuclear Gene Sequences. Int. J. Mol. Sci. 2019, 20, 1234. https://doi.org/10.3390/ijms20051234
Du Z, Ishikawa T, Liu H, Kamitani S, Tadauchi O, Cai W, Li H. Phylogeography of the Assassin Bug Sphedanolestes impressicollis in East Asia Inferred From Mitochondrial and Nuclear Gene Sequences. International Journal of Molecular Sciences. 2019; 20(5):1234. https://doi.org/10.3390/ijms20051234
Chicago/Turabian StyleDu, Zhenyong, Tadashi Ishikawa, Hui Liu, Satoshi Kamitani, Osamu Tadauchi, Wanzhi Cai, and Hu Li. 2019. "Phylogeography of the Assassin Bug Sphedanolestes impressicollis in East Asia Inferred From Mitochondrial and Nuclear Gene Sequences" International Journal of Molecular Sciences 20, no. 5: 1234. https://doi.org/10.3390/ijms20051234
APA StyleDu, Z., Ishikawa, T., Liu, H., Kamitani, S., Tadauchi, O., Cai, W., & Li, H. (2019). Phylogeography of the Assassin Bug Sphedanolestes impressicollis in East Asia Inferred From Mitochondrial and Nuclear Gene Sequences. International Journal of Molecular Sciences, 20(5), 1234. https://doi.org/10.3390/ijms20051234