Key Maize Drought-Responsive Genes and Pathways Revealed by Comparative Transcriptome and Physiological Analyses of Contrasting Inbred Lines

 ,
,
Abstract

1. Introduction
2. Results
2.1. Physiological Analysis of Two Contrasting Maize Inbred Line Seedlings Responses to Drought Stress
2.2. RNA Sequencing (RNA-seq) Analysis
2.3. Transcriptomic Responses
2.4. Differential Gene Expression Analysis
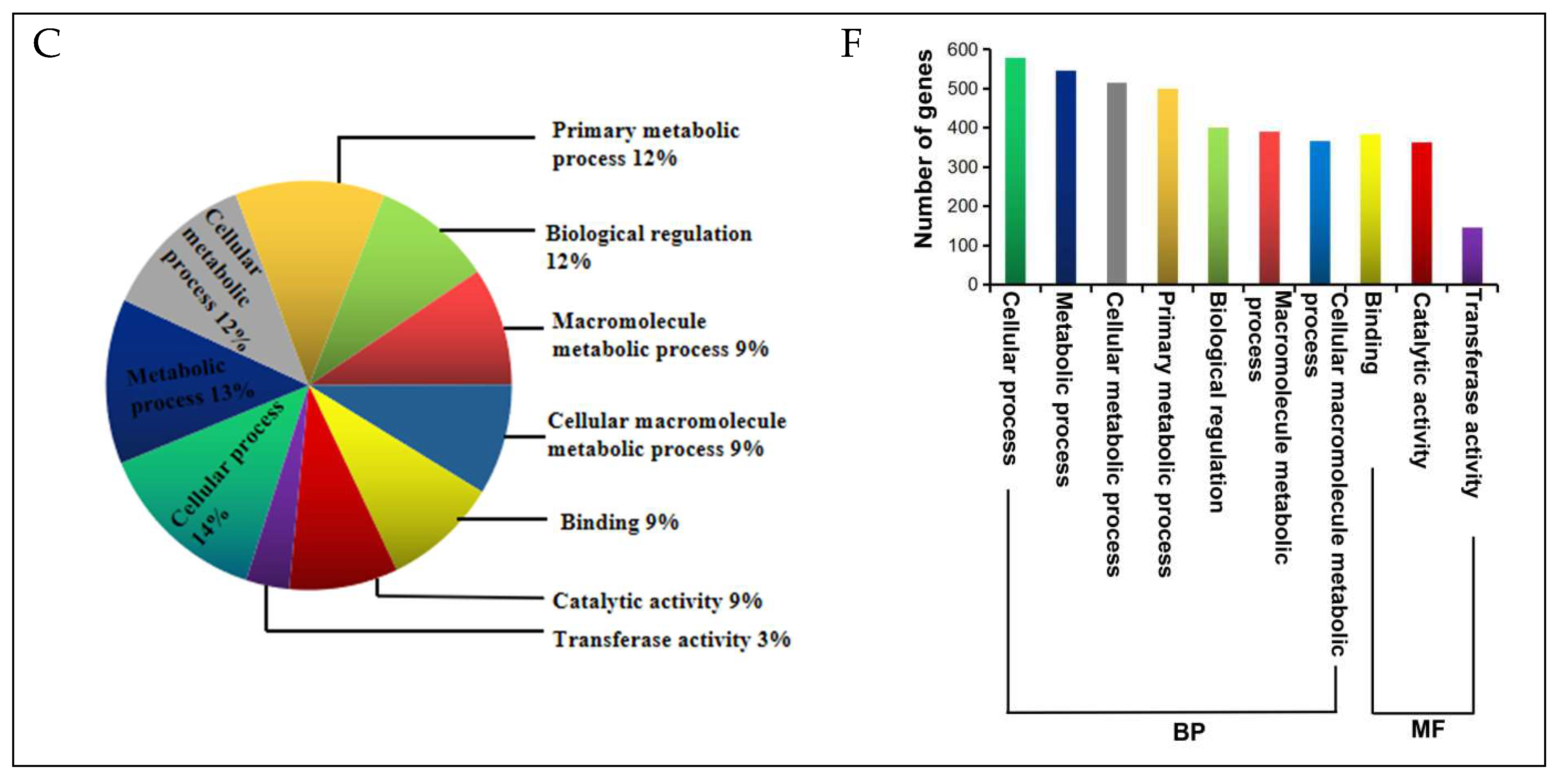
2.5. DEGs Annotation and Functional Categorization
2.6. Differentially Expressed Genes Encoding Transcription Factors
2.7. Metabolic Pathways Enrichment Analysis of the DEGs
2.8. DEGs Related to “Response to Stress” and “Response to Stimuli”
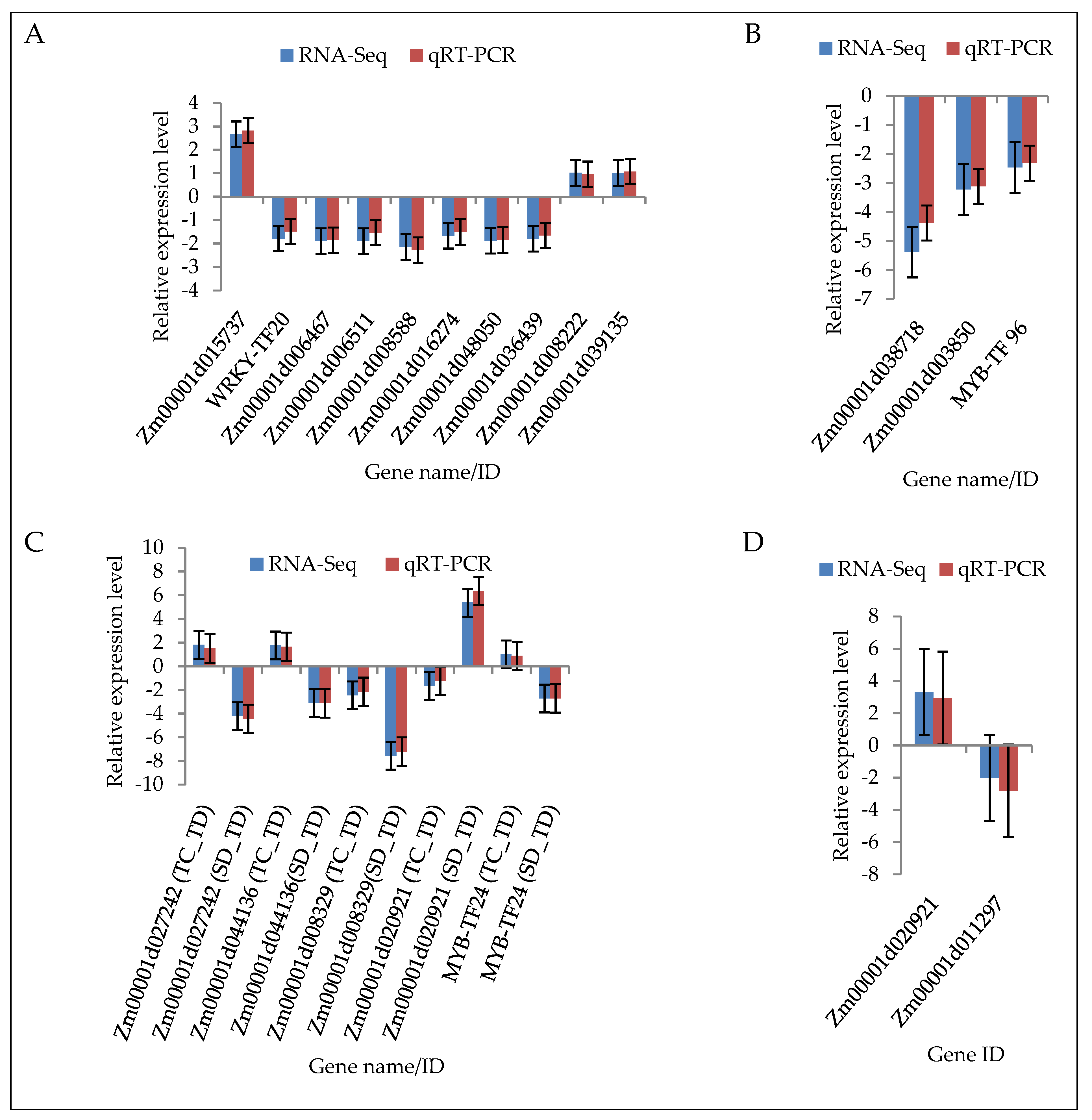
2.9. Validation of DEGs by Quantitative Real-Time PCR (qRT-PCR)
3. Discussion
3.1. Clear Divergence Exist Between Inbred Lines YE8112 and MO17 in Their Drought Stress Responses
3.2. Stress Signal Transduction and Protein Kinases under Drought Stress Conditions
3.3. Transcription Factor (TF) Related Genes Are a Critical Component of Drought Response Machinery
3.4. Enhanced Cellular Redox Homeostasis Is Essential for Plants to Tolerate Drought Stress
3.5. Carbohydrate Metabolism and Cell Growth Promotion Are Vital for Seedlings Survival under Drought
3.6. Protein Ubiquitination Plays a Significant Role in Drought Stress Response Regulation
3.7. Overlapping Drought Responsive DEGs between Inbred Lines under Drought Stress
3.8. Metabolic Pathways Significantly Enriched under Drought Stress Conditions
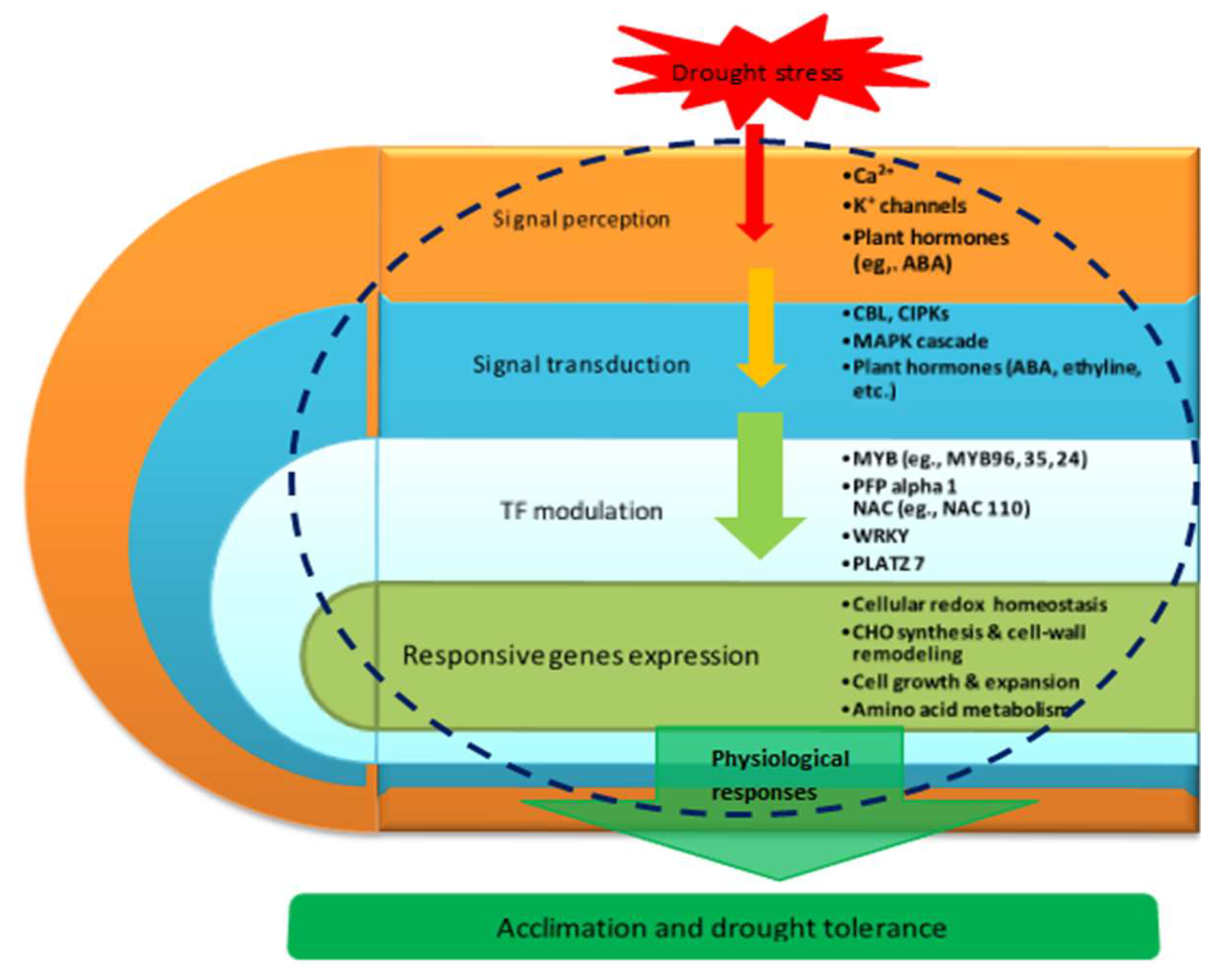
3.9. Proposed Molecular Model of Maize Seedling Drought Stress Tolerance
4. Materials and Methods
4.1. Plant Materials and Drought Stress Treatment
4.2. Physiological and Phenotypic Characterizations
4.3. Total RNA Extraction, cDNA Library Construction, and Transcriptome Sequencing
4.4. Sequencing Reads Processing, Mapping, and Gene Expression Quantification
4.5. Functional Annotation of Gene Transcripts
4.6. Differentially Expressed Genes (DEGs) Library Construction and Differential Analysis
4.7. Gene Ontology (GO) Enrichment and KEGG Pathway Enrichment Analyses
4.8. Quantitative Real Time-PCR (qRT-PCR) Analysis
4.9. Statistical Analysis of Physiological Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABA | Abscisic acid |
| ABF/AREB | ABA-responsive element binding factor/ABA-responsive element binding |
| Ca2+ | Calcium signals receptors/messengers |
| CBL | Calcineurin B-like |
| CDPKs | Calcium dependent protein kinases |
| CIPKs | CBL-interacting protein kinases |
| DEGs | Differentially expressed genes |
| DREB/CBF | Dehydration responsive element binding/C-repeat binding factor |
| ENTH/ANTH/VHS | Epsin N-terminal homology/AP180 N-terminal homology/Vps27, Hrs and STAM |
| GO | Gene ontology |
| HSPs | Heat shock proteins |
| K+ | Potassium channels signal receptors |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LEA | Late embryogenesis abundant proteins |
| LHY | Late hypocotyl elongation protein |
| LSH5 | Light dependent short hypocotyls 5 |
| MAPK | Mitogen-activated protein kinases |
| MDA | Malondialdehyde |
| MYB | Myeloblastosis oncogene |
| NAC | NAM, ATAF1/2, and CUC2 domain proteins |
| PFP alpha 1 | Pyrophosphate fructose-6-phosphate 1-phosphotransferase subunit alpha 1 |
| PLATZ | Plant AT-rich sequence and zinc binding protein |
| POD | Peroxidases |
| qRT-PCR | Quantitative real-time polymerase chain reaction |
| RNA-seq | RNA sequencing |
| ROS | Reactive oxygen species |
| SnRK2 | SNF1-related kinase 2 |
| SOD | Superoxide dismutase |
| TBL20 | Trichome birefringence-like 20 |
| TF | Transcription factor |
| TR | Thioredoxin reductase |
| WRKY | TF family denoted by protein domain composed of a conserved WRKYGQK motif and a zinc-finger domain |
References
- Ghatak, A.; Chaturvedi, P.; Weckwerth, W. Cereal crop proteomics: Systemic analysis of crop drought stress responses towards marker-assisted selection breeding. Front. Plant Sci. 2017, 8, 757. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Han, Z.; Zhang, T.; Chen, S.; Ma, C. A systems approach to spatio-temporal understanding of the drought stress response in maize. Sci. Rep. 2017, 7, 6590. [Google Scholar] [CrossRef] [PubMed]
- Min, H.; Chen, C.; Wei, S.; Shang, X.; Sun, M.; Xia, R.; Liu, X.; Hao, D.; Chen, H.; Xie, Q. Identification of drought tolerant mechanisms in maize seedlings based on transcriptome analysis of recombination inbred lines. Front. Plant Sci. 2016, 7, 1080. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Ning, F.; Zhang, Q.; Wu, X.; Wang, W. Enhancing omics research of crop responses to drought under field conditions. Front. Plant Sci. 2017, 8, 174. [Google Scholar] [CrossRef] [PubMed]
- Feller, U.; Vaseva, I.I. Extreme climatic events: Impacts of drought and high temperature on physiological processes in agronomically important plants. Front. Environ. Sci. 2014, 2, 39. [Google Scholar] [CrossRef]
- IPCC. Climate Change 2014: Synthesis Report; Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Pachauri, R.K., Meyer, L.A., Eds.; IPCC: Geneva, Switzerland, 2014; 151p. [Google Scholar]
- Andjelkovic, V. Introductory Chapter: Climate Changes and Abiotic Stress in Plants; IntechOpen Publishers: London, UK, 2018; p. 4. [Google Scholar]
- Thirunavukkarasu, N.; Sharma, R.; Singh, N.; Shiriga, K.; Mohan, S.; Mittal, S.; Mittal, S.; Mallikarjuna, M.G.; Rao, A.R.; Dash, P.K.; et al. Genomewide expression and functional interactions of genes under drought stress in maize. Int. J. Genet. 2017, 2017, 2568706. [Google Scholar] [CrossRef]
- Zheng, J.; Fu, J.; Gou, M.; Huai, J.; Liu, Y.; Jian, M.; Huang, Q.; Guo, X.; Dong, Z.; Wang, H.; et al. Genome-wide transcriptome analysis of two maize inbred lines under drought stress. Plant Mol. Biol. 2010, 72, 407–421. [Google Scholar] [CrossRef]
- Yin, X.G.; Jørgen, E.O.; Wang, M.; Kersebaum, K.-C.; Chen, H.; Baby, S.; Öztürk, I.; Chen, F. Adapting maize production to drought in the northeast farming region of China. Eur. J. Agron. 2016, 77, 47–58. [Google Scholar] [CrossRef]
- Maiti, R.K.; Maiti, L.E.; Sonia, M.; Maiti, A.M.; Maiti, M.; Maiti, H. Genotypic variability in maize (Zea mays L.) for resistance to drought and salinity at the seedling stage. J. Plant Physiol. 1996, 148, 741–744. [Google Scholar] [CrossRef]
- Li, Z.; Xu, W.; Xue, B.; Cao, P. Discuss on evaluating method to drought-resistance of maize in seedling stage. J. Maize Sci. 2004, 12, 73–75. [Google Scholar]
- Shan, X.; Li, Y.; Jiang, Y.; Jiang, Z.; Hao, W.; Yuan, Y. Transcriptome profile analysis of maize seedlings in response to high-salinity, drought and cold stresses by deep sequencing. Plant Mol. Biol. Rep. 2013, 31, 1485–1491. [Google Scholar] [CrossRef]
- Harb, A. Identification of candidate genes for drought stress tolerance. In Drought Stress Tolerance in Plants; Hossain, M.A., Wani, S.H., Bhattacharjee, S., Burritt, D.J., Tran, L.-S.P., Eds.; Springer International Publishing: Cham, Switzerland, 2016; Volume 2. [Google Scholar]
- Farooq, M.; Wahid, A.; Kobayashi, N.; Fujita, D.; Basra, S.M.A. Plant drought stress: Effects, mechanisms and management. In Sustainable Agriculture; Springer: Dordrecht, The Netherlands, 2009; Volume 29, pp. 185–212. [Google Scholar]
- Hanin, M.; Brini, F.; Ebel, C.; Toda, Y.; Takeda, S.; Masmoudi, K. Plant dehydrins and stress tolerance versatile proteins for complex mechanisms. Plant Signal. Behav. 2011, 6, 1503–1509. [Google Scholar] [CrossRef] [PubMed]
- Kosova, K.; Vitamvas, P.; Prasil, I.T. Wheat and barley dehydrins under cold, drought, and salinity—What can LEA-II proteins tell us about plant stress response? Front. Plant Sci. 2014, 5, 343. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Vinocur, B.; Shoseyov, O.; Altman, A. Role of plant heat-shock proteins and molecular chaperons in the abiotic stress response. Trends Plant Sci. 2004, 9, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Hasanuzzaman, M.; Nahar, K.; Mahabub Alam, M.; Roychowdhury, R.; Fujita, M. Physiological, biochemical, and molecular mechanisms of heat stress tolerance in plants. Int. J. Mol. Sci. 2013, 14, 9643–9684. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lei, L.; Lai, J.; Zhao, H.; Song, W. Effects of drought stress and water recovery on physiological responses and gene expression in maize seedlings. BMC Plant Biol. 2018, 18, 68. [Google Scholar] [CrossRef]
- Singh, D.; Laxmi, A. Transcriptional regulation of drought response: A tortuous network of transcriptional factors. Front. Plant Sci. 2015, 6, 895. [Google Scholar] [CrossRef] [PubMed]
- Mun, B.G.; Lee, S.U.; Park, E.J.; Kim, H.H.; Hussain, A.; Imran, Q.M.; Lee, I.J.; Yun, B.W. Analysis of transcription factors among differentially expressed genes induced by drought stress in Populus davidiana. 3 Biotech 2017, 7, 209. [Google Scholar] [CrossRef]
- Wang, C.T.; Ru, J.N.; Liu, Y.W.; Yang, J.F.; Li, M.; Xu, Z.S.; Fu, J.D. The maize WRKY transcription factor ZmWRKY40 confers drought resistance in transgenic Arabidopsis. Int. J. Mol. Sci. 2018, 19, 2580. [Google Scholar] [CrossRef]
- Liu, X.; Li, X.; Li, W.; Li, M.; Liu, X. Analysis on difference for drought responses of maize inbred lines at seedling stage. J. Maize Sci. 2004, 12, 63–65. [Google Scholar]
- Bin, W.; Xinhai, L.; Muji, X. Genetic variation in fifty-three maize inbred lines in relation to drought tolerance at seedling stage. Sci. Agric. Sin. 2007, 40, 665–676. [Google Scholar]
- Wang, G.; Zhu, Q.G.; Meng, Q.W.; Wu, C.A. Transcript profiling during salt stress of young cotton (Gossypium hirsutum) seedlings via Solexa sequencing. Acta Physiol. Plant. 2012, 34, 107–115. [Google Scholar] [CrossRef]
- Luz, H.P.; Juan, D.R. RNA-seq in kinetoplastids: A powerful tool for the understanding of the biology and host-pathogen interactions. Infect. Genet. Evol. 2017, 49, 273–282. [Google Scholar] [CrossRef]
- Kakumanu, A.; Ambavaram, M.M.R.; Klumas, C.; Krishnan, A.; Batlang, U.; Myers, E.; Grene, R.; Pereira, A. Effects of drought on gene expression in maize reproductive and leaf meristem tissue revealed by RNA-seq. Plant Physiol. 2012, 160, 846–867. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Z.H.; Feng, L.; Zheng, Y.; Li, D.D.; Li, X.B. Genome-wide functional analysis of cotton (Gossypium hirsutum) in response to drought. PLoS ONE 2013, 8, e80879. [Google Scholar] [CrossRef]
- Nakashima, K.; Ito, Y.; Yamaguchi-Shinozaki, K. Transcriptional regulatory networks in response to abiotic stresses in Arabidopsis and grasses. Plant Physiol. 2009, 149, 88–95. [Google Scholar] [CrossRef]
- Zhang, T.; Huang, L.; Wang, Y.; Wang, W.; Zhao, X.; Zhang, S.; Zhang, J.; Fengyi, H.; Fu, B.; Li, Z. Differential transcriptome profiling of chilling stress response between shoots and rhizomes of Oryza longistaminata using RNA sequencing. PLoS ONE 2017, 12, e0188625. [Google Scholar] [CrossRef]
- Zenda, T.; Liu, S.; Wang, X.; Jin, H.; Liu, G.; Duan, H. Comparative proteomic and physiological analyses of two divergent maize inbred lines provide more insights into drought-stress tolerance mechanisms. Int. J. Mol. Sci. 2018, 19, 3225. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat. Meth. 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Wang, P.; Wang, F.F.; Yang, J. De novo assembly and analysis of the Pugionium cornutum (L.) Gaertn. transcriptome and identification of genes involved in the drought response. Gene 2017, 626, 290–297. [Google Scholar] [CrossRef]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.; Su, Z. agriGO: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38, W64–W70. [Google Scholar] [CrossRef]
- Wang, H.; Wang, H.; Shao, H.; Tang, X. Recent advances in utilizing transcription factors to improve plant abiotic stress tolerance by transgenic technology. Front. Plant Sci. 2016, 7, 67. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L.P. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Edmeades, G.O. Progress in Achieving and Delivering Drought Tolerance in Maize—An Update; ISAA: Ithaca, NY, USA, 2013; pp. 1–39. [Google Scholar]
- Bhanu, B.D.; Ulaganathan, K.; Shanker, A.R.; Desai, S. RNA-seq analysis of irrigated vs. water stressed transcriptomes of Zea mays cultivar Z59. Front. Plant Sci. 2016, 7, 239. [Google Scholar] [CrossRef]
- Moussa, H.R.; Abdel-Aziz, S.M. Comparative response of drought tolerant and drought sensitive maize genotypes to water stress. Aust. J. Crop Sci. 2008, 1, 31–36. [Google Scholar]
- Ahmadi, A.; Emam, Y.; Pessarakli, M. Biochemical changes in maize seedlings exposed to drought stress conditions at different nitrogen levels. J. Plant Nutr. 2010, 33, 541–556. [Google Scholar] [CrossRef]
- Marenduzzo, D.; Finan, K.; Cook, P.R. The depletion attraction: An underappreciated force driving cellular organization. J. Cell Biol. 2006, 175, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Oliver, S.N.; Dennis, E.S.; Dolferus, R. ABA regulates apoplastic sugar transport and is a potential signal for cold-induced pollen sterility in rice. Plant Cell Physiol. 2007, 48, 1319–1330. [Google Scholar] [CrossRef]
- Barkla, B.; Pantoja, O. Plasma membrane and abiotic stress. Plant Cell Monogr. 2010, 19, 457–470. [Google Scholar] [CrossRef]
- Hyacinthe, L.G.; Philippe, F.; Domon, J.M.; Gillet, F.; Pelloux, J.; Rayon, C. Cell wall metabolism in response to abiotic stress. Plants 2015, 4, 112–166. [Google Scholar] [CrossRef]
- Sharma, P.; Jha, A.B.; Dubey, R.S.; Pessarakli, M. Reactive Oxygen Species, Oxidative Damage, and Antioxidative Defense Mechanism in Plants under Stressful Conditions. J. Bot. 2012, 10, 26. [Google Scholar] [CrossRef]
- Andrej, F.; Tatiana, B.; Gagan, P.; Robert, B.; Gerd, U.B.; Claudia, B.; Ludger, A.W. Early responses of mature arabidopsis thaliana plants to reduced water potential in the agar-based polyethylene glycol infusion drought model. J. Plant Physiol. 2017, 208, 70–83. [Google Scholar] [CrossRef]
- Mahajan, S.; Tuteja, N. Cold, salinity and drought stresses: An overview. Arch. Biochem. Biophys. 2005, 444, 139–158. [Google Scholar] [CrossRef]
- Chaves, M.M.; Flexas, J.; Pinheiro, C. Photosynthesis under drought and salt stress: Regulation mechanisms from whole plant to cell. Ann. Bot. 2009, 103, 551–560. [Google Scholar] [CrossRef]
- Jonak, C.; Kiergerl, S.; Ligterink, W.; Barker, P.J.; Huskisson, N.S.; Hirt, H. Stress signaling in plants: A mitogen-activated protein kinase pathway is activated by cold and drought. Plant Biology. Proc. Natl. Acad. Sci. USA 1996, 93, 11274–11279. [Google Scholar] [CrossRef]
- Dudhate, A.; Shinde, H.; Tsugama, D.; Liu, S.; Takano, T. Transcriptomic analysis reveals the differentially expressed genes and pathways involved in drought tolerance in pearl millet [Pennisetum glaucum (L.) R. Br]. PLoS ONE 2018, 13, e0195908. [Google Scholar] [CrossRef]
- Piao, H.L.; Xuan, Y.H.; Park, S.H.; Je, B.; Park, S.J.; Park, S.H.; Kim, C.M.; Huang, J.; Wang, G.K.; Kim, M.J.; et al. OsCIPK31, a CBL-interacting protein kinase is involved in germination and seedling growth under abiotic stress conditions in rice plants. Mol. Cells 2010, 30, 19–27. [Google Scholar] [CrossRef]
- Tai, F.; Yuan, Z.; Li, S.; Wang, Q.; Liu, F. ZmCIPK8, a CBL-interacting protein kinase, regulates maize response to drought stress. Plant Cell Tissue Organ Cult. 2016, 124, 59–469. [Google Scholar] [CrossRef]
- Kubilay, Y.; Zeki, K. Gene regulation network behind drought escape, avoidance and tolerance strategies in black poplar (Populus nigra L.). Plant Physiol. Biochem. 2017, 115, 183–199. [Google Scholar] [CrossRef]
- Shi, W.; Cheng, J.; Wen, X. Transcriptomic studies reveal a key metabolic pathway contributing to a well-maintained photosynthetic system under drought stress in foxtail millet (Setaria italica L.). PeerJ 2018, 6, e4752. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, V.J.; Rubio, M.; Trainotti, L.; Verde, I.; Bonghi, C.; Martínez-Gómez, P. Prunus transcription factors: Breeding perspectives. Front. Plant Sci. 2015, 6, 443. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Cruz de Carvalho, M.H.; Torres-Jerez, I.; Kang, Y.; Allen, S.N.; Huhman, D.V.; Tang, Y.; Murray, J.; Sumner, L.W.; Udvardi, M.K. Global reprogramming of transcription and metabolism in Medicago truncatula during progressive drought and after rewatering. Plant Cell Environ. 2014, 37, 2553–2576. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Jia, H.; Chen, X.; Hao, L.; An, H.; Guo, X. The cotton WRKY transcription factor GhWRKY17 functions in drought and salt stress in transgenic Nicotiana benthamiana through ABA signalling and the modulation of reactive oxygen species production. Plant Cell Physiol. 2014, 55, 2060–2076. [Google Scholar] [CrossRef]
- Meyer, Y.; Siala, W.; Bashandy, T.; Riondet, C.; Vignols, F.; Reichheld, J.P. Glutaredoxins and thioredoxins in plants. Biochem. Biophys. Acta. 2008, 1783, 589–600. [Google Scholar] [CrossRef]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Ann. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef]
- Gill, S.S.; Tuteja, N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol. Biochem. 2010, 48, 909–930. [Google Scholar] [CrossRef]
- Campbell, S.A.; Close, T.J. Dehydrins: Genes, proteins, and associations with phenotypic traits. New Phytol. 1997, 137, 61–74. [Google Scholar] [CrossRef]
- Reidy, B.; McQueen-Mason, S.; Nösberger, J.; Fleming, A. Differential expression of alpha- and beta-expansin genes in the elongating leaf of Festuca pratensis. Plant Mol. Biol. 2001, 46, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Marowa, P.; Ding, A.; Kong, Y. Expansins: Roles in plant growth and potential applications in crop improvement. Plant Cell Rep. 2016, 35, 949–965. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, V.; Selbig, J.; Scheible, W.R. Involvement of TBL/DUF231 proteins into cell wall biology. Plant Signal. Behav. 2010, 5, 1057–1059. [Google Scholar] [CrossRef]
- Opassiri, R.; Pomthong, B.; Onkoksoong, T.; Akiyama, T.; Esen, A.; Cairns, J.R.K. Analysis of rice glycosyl hydrolase family 1 and expression of Os4bglu l2 ß-glucosidase. BMC Plant Biol. 2006, 6, 33. [Google Scholar] [CrossRef]
- Zhang, C.; Shi, S. Physiological and proteomic responses of contrasting alfalfa (Medicago sativa L.) varieties to PEG-Induced osmotic stress. Front. Plant Sci. 2018, 9, 242. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.S.; Knowles, S.M.; Andronis, C.; Ong, M.S.; Tobin, E.M. CIRCADIAN CLOCK ASSOCIATED 1 and LATE ELONGATED HYPOCOTYL function synergistically in the circadian clock of Arabidopsis. Plant Physiol. 2009, 50, 834–843. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, J.; Liu, H.; Chong, K.; Xu, Y. Roles of ubiquitination-mediated protein degradation in plant responses to abiotic stresses. Environ. Exp. Bot. 2015, 114, 92–103. [Google Scholar] [CrossRef]
- Shinde, H.; Tanaka, K.; Dudhate, A.; Tsugama, D.; Mine, Y.; Kamiya, T.; Gupta, S.K.; Liu, S.; Takano, T. Comparative de novo transcriptomic profiling of the salinity stress responsiveness in contrasting pearl millet lines. Environ. Exp. Bot. 2018, 155, 619–627. [Google Scholar] [CrossRef]
- Serrano, I.; Campos, L.; Rivas, S. Roles of E3 ubiquitin-ligases in nuclear protein homeostasis during plant stress responses. Front. Plant Sci. 2018, 9, 139. [Google Scholar] [CrossRef]
- Lyzenga, W.J.; Stone, S.L. Abiotic stress tolerance mediated by protein ubiquitination. J. Exp. Bot. 2012, 63, 599–616. [Google Scholar] [CrossRef]
- Hu, X.; Wu, L.; Zhao, F.; Zhang, D.; Li, N.; Zhu, G.; Li, C.; Wang, W. Phosphoproteomic analysis of the response of maize leaves to drought, heat and their combination stress. Front. Plant Sci. 2015, 6, 298. [Google Scholar] [CrossRef]
- Sharma, M.; Pandey, A.; Pandey, G.K. Role of plant U-BOX (PUB) protein in stress and development. In Plant Stress, 7th ed.; Pandey, G.K., Ed.; Global Science Books: New Delhi, India, 2013; pp. 1–9. [Google Scholar]
- Lindemose, S.; O’Shea, C.; Jensen, M.K.; Skriver, K. Structure, function and networks of transcription factors involved in abiotic stress responses. Int. J. Mol. Sci. 2013, 14, 5842–5878. [Google Scholar] [CrossRef]
- Ambawat, S.; Sharma, P.; Yadav, N.R.; Yadav, R.C. MYB transcription factor genes as regulators for plant responses: An overview. Physiol. Mol. Biol. Plants 2013, 19, 307–321. [Google Scholar] [CrossRef]
- Seo, P.J.; Xiang, F.; Qiao, M.; Park, J.Y.; Lee, Y.N.; Kim, S.G.; Lee, Y.H.; Park, W.J.; Park, C.M. The MYB96 transcription factor mediates abscisic acid signaling during drought stress response in Arabidopsis. Plant Physiol. 2009, 151, 275–289. [Google Scholar] [CrossRef]
- Nielsen, T.H.; Rung, J.H.; Villadsen, D. Fructose-2,6-bisphosphate: A traffic signal in plant metabolism. Trends Plant Sci. 2004, 9, 556–563. [Google Scholar] [CrossRef]
- Scott, P.; Lange, A.J.; Pilkis, S.J.; Kruger, N.J. Carbon metabolism in leaves of transgenic tobacco (Nicotiana tabacum L.) containing elevated fructose 2,6-bisphosphate levels. Plant J. 1995, 7, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Kovács, G.; Sorvari, S.; Scott, P.; Toldi, O. Pyrophosphate:fructose 6-phosphate 1-phosphotransferase operates in net gluconeogenic direction in taproots of cold and drought stressed carrot plants. Acta Biol. Szeged. 2006, 50, 25–30. [Google Scholar]
- Yadav, B.S.; Sing, A.K.; Kushwaha, S.K. Systems-based approach to the analyses of plant functions: Conceptual understanding, implementation and analysis. In Plant Bionformatics; Hakeem, K., Malik, A., Varda-Sukan, F., Ozturk, M., Eds.; Springer: Cham, Switzerland, 2017; pp. 107–133. [Google Scholar]
- Luo, M.; Zhao, Y.; Wang, Y.; Shi, Z.; Zhang, P.; Zhang, Y.; Song, W.; Zhao, J. Comparative proteomics of contrasting maize genotypes provides insights into salt-stress tolerance mechanisms. J. Proteome Res. 2018, 17, 141–153. [Google Scholar] [CrossRef]
- Verberne, M.C.; Budi Muljono, R.A.; Verpoorte, R. Salicylic acid biosynthesis. New Compr. Biochem. 1999, 33, 295–312. [Google Scholar] [CrossRef]
- Wahid, A.; Gelani, S.; Ashraf, M.; Foolad, M.R. Heat tolerance in plants: An overview. Environ. Exp. Bot. 2007, 61, 199–223. [Google Scholar] [CrossRef]
- Cui, D.; Wu, D.; Liu, J.; Li, D.; Xu, C.; Li, S.; Li, P.; Zhang, H.; Liu, X.; Jiang, C.; et al. Proteomic analysis of seedling roots of two maize inbred lines that differ significantly in the salt stress response. PLoS ONE 2015, 10, e0116697. [Google Scholar] [CrossRef]
- Galmés, J.; Flexas, J.; Savé, R.; Medrano, H. Water relations and stomatal characteristics of Mediterranean plants with different growth forms and leaf habits: Responses to water stress and recovery. Plant Soil 2007, 290, 139–155. [Google Scholar] [CrossRef]
- Han, L.B.; Song, G.L.; Zhang, X. Preliminary observation of physiological responses of three turfgrass species to traffic stress. HortTechnology 2008, 18, 139–143. [Google Scholar] [CrossRef]
- Dhindsa, R.S.; Plumb-Dhindsa, P.; Thorpe, T.A. Leaf senescence: Correlated with increased leaves of membrane permeability and lipid peroxidation, and decreased levels of superoxide dismutase and catalase. J. Exp. Bot. 1981, 32, 93–101. [Google Scholar] [CrossRef]
- Bates, T.S.; Waldren, R.P.; Teare, I.D. Rapid determination of free proline for water-stress studies. Plant Soil 1973, 39, 205–207. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Alstchul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 389–3402. [Google Scholar]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 106, 10–11. [Google Scholar] [CrossRef]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D4884. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS2.0:a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−Δ∆CT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physiological Characteristics | Treatment Exposure Period (days) 1 | Sensitive Inbred Line MO17 | Tolerant Inbred Line YE8112 | ||
|---|---|---|---|---|---|
| Control | Stress | Control | Stress | ||
| Leaf relative water content (%) | 1 | 92.23 ± 0.1763 b | 90.33 ± 0.8819 c | 94.63 ± 0.2603 a | 92.53 ± 0.1202 b |
| 3 | 93.03 ± 0.0882 a | 88.53 ± 0.1202 b | 93.70 ± 0.4163 a | 89.90 ± 0.3056 b | |
| 5 | 92.83 ± 0.4910 a | 70.43 ± 0.1202 c | 94.60 ± 0.4042 a | 86.53 ± 0.2848 b | |
| 7 | 92.73 ± 0.2403 a | 65.40 ± 0.2082 c | 93.80 ± 0.3215 a | 81.80 ± 1.0214 b | |
| Proline content (µg·g−1 FW) | 1 | 88.62 ± 0.3358 a | 80.45 ± 0.6265 b | 73.99 ± 0.7420 c | 89.95 ± 0.3259 a |
| 3 | 88.78 ± 0.1981 c | 93.68 ± 0.5860 b | 74.29 ± 1.1873 d | 97.19 ± 1.0451 a | |
| 5 | 88.69 ± 0.5200 c | 99.78 ± 1.3309 b | 75.25 ± 0.6094 d | 108.00 ± 0.9824 a | |
| 7 | 87.67 ± 1.2440 c | 109.21 ± 0.7280 b | 78.07 ± 0.9696 d | 121.96 ± 0.7967 a | |
| Peroxidase activity (unit·mg−1 protein FW min−1) | 1 | 0.2653 ± 0.0028 b | 0.2747 ± 0.0023 b | 0.2590 ± 0.0012 c | 0.3083 ± 0.0035 a |
| 3 | 0.2630 ± 0.0059 c | 0.3147 ± 0.0015 b | 0.2557 ± 0.0008 c | 0.3553 ± 0.0047 a | |
| 5 | 0.2530 ± 0.0064 d | 0.3677 ± 0.0020 b | 0.2643 ± 0.0052 c | 0.3847 ± 0.1186 a | |
| 7 | 0.2563 ± 0.0043 c | 0.4033 ± 0.0039 b | 0.2653 ± 0.0073 c | 0.4350 ± 0.0012 a | |
| MDA content (µmol·g−1 FW) | 1 | 0.0119 ± 0.00003 b | 0.0128 ± 0.00024 a | 0.0118 ± 0.00067 b | 0.0119 ± 0.00006 b |
| 3 | 0.0117 ± 0.00031 c | 0.0152 ± 0.00015 a | 0.0117 ± 0.00018 c | 0.0123 ± 0.00028 b | |
| 5 | 0.0115 ± 0.00012 a | 0.0207 ±0.0002 8 c | 0.0119 ± 0.00023 a | 0.0153 ± 0.00057 b | |
| 7 | 0.0120 ± 0.00012 a | 0.0220 ± 0.00005 c | 0.0118 ± 0.00018 a | 0.0204 ± 0.00036 b | |
| Drought stress injury symptoms | At 7 days | Not visible | Leaves distinctly shriveled up | Not visible | Leaves remain green and relatively intact |
| Sample 1 | Rep 2 | Total Reads 3 | Clean Reads 4 | GC Content (%) 5 | % ≥ Q30 6 | Mapped Reads (%) 7 | Uniq. Map Reads (%) 8 | Multiple Map Reads (%) 9 |
|---|---|---|---|---|---|---|---|---|
| YE8112CK | 1 | 65,863,880 | 32,931,940 | 57.05 | 89.78 | 50,491,140 (76.66%) | 48,782,990 (74.07%) | 1,708,053 (2.59%) |
| YE8112CK | 2 | 64,181,688 | 32,090,844 | 56.15 | 89.07 | 49,434,118 (77.02%) | 47,831,879 (74.53%) | 1,708,053 (2.59%) |
| YE8112CK | 3 | 59,227,506 | 29,613,753 | 56.82 | 89.96 | 45,756,005 (77.25%) | 44,156,338 (74.55%) | 1,599,667 (2.70%) |
| YE8112D | 1 | 57,696,300 | 28,848,150 | 56.47 | 90.17 | 45,284,984 (78.49%) | 41,401,607 (77.82%) | 1,466,413 (2.54%) |
| YE8112D | 2 | 59,181,570 | 29,590,785 | 56.13 | 89.54 | 45,357,186 (76.67%) | 43,736,983 (73.90%) | 1,638,203 (2.77%) |
| YE8112D | 3 | 53,205,054 | 26,602,527 | 56.53 | 90.16 | 41,401,607 (77.82%) | 40,072,870 (75.32%) | 1,152,150 (2.47%) |
| MO17CK | 1 | 46,584,386 | 23,292,193 | 56.04 | 89.17 | 30,323,524 (65.09%) | 29,171,374 (62.62%) | 1,590,356 (3.34%) |
| MO17CK | 2 | 47,641,294 | 23,820,647 | 55.87 | 88.99 | 31,266,355 (65.63%) | 29,675,999 (62.29%) | 1,590,356 (3.34%) |
| MO17CK | 3 | 47,180,622 | 23,590,311 | 56.54 | 88.70 | 31,791,159 (67.38%) | 27,907,153 (59.15%) | 3,884,006 (8.23%) |
| MO17D | 1 | 44,255,824 | 22,127,912 | 56.10 | 86.79 | 26,090,436 (58.95%) | 24,995,448 (56.48%) | 1,094,998 (2.47%) |
| MO17D | 2 | 68,595,584 | 34,297,774 | 56.22 | 87.01 | 41,355,964 (60.29%) | 38,616,715 (56.30%) | 2,739,249 (3.99%) |
| MO17D | 3 | 47,342,680 | 23,671,340 | 56.38 | 85.02 | 26,906,481 (56.83%) | 24,564,920 (51.82%) | 2,371,561 (5.01%) |
| Comparison/Group 1 | DEG Number 2 | Up-Regulated 3 | Down-Regulated 4 |
|---|---|---|---|
| SC__TC | 4331 | 1964 | 2367 |
| SC__SD | 754 | 329 | 425 |
| TC__TD | 129 | 49 | 80 |
| SD__TD | 5398 | 2485 | 2913 |
| Gene ID 1 | Gene Name/Description 2 | log2 FC 3 | Expr. 4 | FDR 5 | p-Value 6 | KEGG Pathway 7 |
|---|---|---|---|---|---|---|
| Zm00001d027242 | Granule-bound starch synthase 1 | 4.3867007 | Up | 6.57 × 10−6 | 0.0001190 | -- |
| Zm00001d044136 | Glycerol-3-phosphate acyltransferase 1 | 3.814405 | Up | 1.09 × 10−5 | 0.0020520 | Glycerophospholipid metabolism |
| Zm00001d029906 | BETA_EXPANSIN7 | 2.8909994 | Up | 0.002383 | 0.0015080 | -- |
| Zm00001d036676 | Putative B-box type zinc finger family protein | 2.666452 | Up | 0.0034115 | 3.60 × 10−9 | -- |
| Zm00001d011473 | POTASSIUM_CHANNEL5 | 2.5990283 | Up | 0.0020524 | 0.0000007 | -- |
| Zm00001d013261 | Cysteine protease 1 | 1.9554585 | Up | 2.60 × 10−8 | 1.72 × 10−9 | -- |
| Zm00001d038199 | Mildew resistance locus O (MLO)-like protein 1 | 1.9445226 | Up | 3.07 × 10−6 | 0.0135900 | -- |
| Zm00001d044765 | Benzoate carboxyl methyltransferase | 1.7383231 | Up | 0.0004904 | 0.0148800 | -- |
| Zm00001d012220 | Putative ENTH/ANTH/VHS superfamily protein | 1.7341537 | Up | 0.0008504 | 0.0000584 | -- |
| 2626 | N/A | 1.7277288 | Up | 0.0039107 | 0.0061660 | -- |
| Zm00001d044285 | CALCINEURIN_B-LIKE10 | 1.6791443 | Up | 7.19 × 10−6 | 0.0042720 | -- |
| 1650 | N/A | 1.6197127 | Up | 0.0016858 | 0.0000231 | -- |
| Zm00001d033985 | N/A | 1.6129169 | Up | 0.0040434 | 0.0000005 | -- |
| Zm00001d046998 | LIGHT-DEPENDENT SHORT HYPOCOTYLS 5 | 1.5404762 | Up | 0.0025583 | 0.0011140 | -- |
| 2197 | N/A | 1.5333337 | Up | 7.19 × 10−6 | 0.0022920 | -- |
| Zm00001d028399 | Thaumatin-like protein 1 | 1.532225 | Up | 4.36 × 10−9 | 2.63 × 10−8 | -- |
| Zm00001d024268 | NAC-TRANSCRIPTION_FACTOR_110 | 1.5062543 | Up | 1.30 × 10−6 | 0.0001437 | -- |
| Zm00001d011297 | MYB-RELATED-TRANSCRIPTION_FACTOR_35 | 1.4840701 | Up | 0.0037662 | 4.04 × 10−9 | -- |
| Zm00001d024546 | LATE_HYPOCOTYL_ELONGATION_PROTEIN_ORTHOLOG1 | 1.4762326 | Up | 1.03 × 10−5 | 0.0001785 | Circadian rhythm—plant |
| Zm00001d031349 | Serine--glyoxylate aminotransferase | 1.415994 | Up | 8.99 × 10−7 | 0.0003943 | Alanine; glutamate; and carbohydrate metabolisms |
| Zm00001d048622 | N/A | 1.4131758 | Up | 1.02 × 10−6 | 0.0000001 | -- |
| 3628 | N/A | 1.3978699 | Up | 0.0025583 | 0.0081300 | -- |
| Zm00001d038049 | Putative O-glycosyl hydrolase | 1.2684934 | Up | 0.00603 | 0.0459900 | -- |
| Zm00001d017374 | Protein kinase superfamily protein | 1.2481588 | Up | 0.0062081 | 0.0042520 | -- |
| Zm00001d008222 | N/A | 1.1376143 | Up | 0.0026542 | 0.0001437 | -- |
| 1682 | N/A | 1.1285633 | Up | 0.0051255 | 1.04 × 10−10 | -- |
| Zm00001d008808 | MYB-RELATED-TRANSCRIPTION_FACTOR_24 | 1.1148561 | Up | 0.0069804 | 0.0000014 | -- |
| Zm00001d040639 | RING-H2 finger protein ATL3F | 1.0980329 | Up | 0.0039389 | 0.0089120 | -- |
| Zm00001d017918 | TRICHOME BIREFRINGENCE-LIKE 20 | 1.0976007 | Up | 0.0006213 | 1.40 × 10−8 | -- |
| Zm00001d029154 | Inactive beta-amylase 9 | 1.0845327 | Up | 0.0034368 | 0.0020340 | -- |
| Zm00001d003850 | Probable botrytis susceptible 1 interactor (BOI) -related E3 ubiquitin-protein ligase 2 | 1.0379841 | Up | 0.0008516 | 0.0001594 | -- |
| Zm00001d046318 | Putative flavin adenine dinucleotide (FAD)-binding berberine family protein | −1.0302981 | Down | 0.0034368 | 0.0001458 | -- |
| Zm00001d005118 | Sec14p-like phosphatidylinositol transfer family protein | −1.0555703 | Down | 0.0004904 | 0.0000696 | -- |
| Zm00001d011428 | Urophorphyrin methylase 1 | −1.0643172 | Down | 0.0051255 | 0.0002838 | -- |
| Zm00001d044228 | THIAMINE_BIOSYNTHESIS2 | −1.065855 | Down | 0.0097628 | 0.0002834 | Thiamine metabolism |
| Zm00001d015700 | Putative chloride channel-like protein CLC-g | −1.0848056 | Down | 0.0003217 | 0.0000007 | -- |
| Zm00001d015504 | Protein phosphatase 2C isoform gamma | −1.0960379 | Down | 0.0051889 | 0.0018030 | -- |
| Zm00001d034345 | FERREDOXIN_NADP_REDUCTASE1 | −1.11374 | Down | 9.83 × 10−5 | 0.0134500 | Photosynthesis |
| Zm00001d028164 | Sulfate transporter 2.2 | −1.1852218 | Down | 0.0008516 | 0.0002960 | -- |
| Zm00001d038186 | Protein NRT1/PTR FAMILY 3.1 | −1.2124124 | Down | 0.0001231 | 0.0216600 | -- |
| Zm00001d019312 | BETA_GLUCOSIDASE_AGGREGATING_FACTOR1 | −1.2524453 | Down | 0.0014524 | 0.0004082 | -- |
| Zm00001d013202 | G2-LIKE-TRANSCRIPTION_FACTOR_8 | −1.2938652 | Down | 0.0036596 | 0.0020330 | -- |
| Zm00001d011648 | Nuclear pore complex protein NUP50A | −1.2960047 | Down | 0.0022073 | 0.0222900 | -- |
| Zm00001d002126 | loricrin-related | −1.3017332 | Down | 0.0041743 | 0.0013570 | -- |
| Zm00001d015025 | Adenosine monophosphate (AMP) binding protein | −1.3030892 | Down | 4.79 × 10−6 | 0.0002818 | -- |
| Zm00001d049831 | Nodulin-like protein | −1.3069293 | Down | 1.24 × 10−5 | 7.30 × 10−9 | -- |
| Zm00001d003116 | Major facilitator superfamily protein | −1.3071872 | Down | 0.0023919 | 0.0071570 | -- |
| Zm00001d002069 | DNA topoisomerase 2 | −1.3224108 | Down | 0.0014831 | 0.0024900 | -- |
| Zm00001d023592 | Amino acid permease 2 | −1.4219628 | Down | 0.0014831 | 0.0240500 | -- |
| Zm00001d003457 | Plant AT-rich and zinc (PLATZ) domain-containing protein 3 | −1.4228022 | Down | 0.0029883 | 0.0151800 | -- |
| Zm00001d018738 | PATHOGENESIS_RELATED_PROTEIN4 | −1.5048829 | Down | 0.0003217 | 0.0010230 | Plant hormone signal transduction; plant–pathogen interaction |
| Zm00001d007517 | Subtilisin-like serine endopeptidase family protein | −1.5493362 | Down | 0.0011401 | 0.0005202 | -- |
| Zm00001d021653 | Glucose-6-phosphate/phosphate translocator 2 | −1.5573945 | Down | 0.0040043 | 4.00 × 10−7 | -- |
| Zm00001d017279 | PHENYLALANINE_AMMONIA_LYASE7 | −1.7207427 | Down | 0.0070058 | 2.595 × 10−7 | Phenylalanine metabolism; phenylpropanoid biosynthesis |
| Gene ID | Gene Name/Description | Expression in TC_TD | Expression in SD_TD | KEGG Pathway | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Expr. | Log2FC | p-Value | FDR | Expr. | Log2FC | p-Value | FDR | |||
| Zm00001d051511 | PLATZ-TF7 | Up | 1.03869 | 1.287 × 10−9 | 0.00747 | Down | −1.73755 | 1.03 × 10−4 | 8.48 × 10−12 | _ |
| Zm00001d052139 | Nitrate reductase [NAD(P)H] | Down | −1.64472 | 5.367 × 10−8 | 0.000867 | Down | −7.52945 | 1.95 × 10−32 | 1.92 × 10−9 | Nitrogen metabolism |
| Zm00001d052164 | Ferredoxin—nitrite reductase | Down | −1.0642 | 0.000162 | 0.003449 | Up | 4.192623 | 0.000165 | 0.007743 | Nitrogen metabolism |
| Zm00001d052247 | Shikimate kinase 1 chloroplastic | Down | −1.18798 | 2.60 × 10−6 | 0.009651 | Down | −7.09198 | 2.37 × 10−11 | 2.00 × 10−23 | Phenylalanine, biosynthesis; biosynthesis of amino acids |
| Zm00001d053568 | N/A | Up | 1.754627 | 1.83 × 10−7 | 2.6 × 10−6 | Down | −1.75125 | 1.74 × 10−3 | 7.69 × 10−7 | _ |
| Gene ID | Gene Name/Description | Expression in TC_TD | Expression in SC_SD | KEGG Pathway | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| log2 FC | Expres. | FDR | p-Value | log2 FC | Expres. | FDR | p-Value | |||
| Zm00001d007012 | Ribonucleoprotein A | −1.10345 | Down | 0.003932 | 3.979 × 10−7 | −1.95005 | Down | 2.79 × 10−6 | 0.000617 | -- |
| Zm00001d014863 | MYB- RELATED TF 96 | 1.121959 | Up | 8.85 × 10−5 | 0.002432 | −1.19141 | Down | 0.001988 | 3.68 × 10−6 | -- |
| Zm00001d026501 | GLUTAMINE_SYNTHETASE1 | −1.92817 | Down | 3.62 × 10−6 | 0.00001242 | −2.62577 | Down | 9.96 × 10−19 | 0.000174 | -- |
| Zm00001d045919 | Pyrophosphate—fructose-6-phosphate 1-phosphotransferase subunit alpha 2 | 1.510247 | Up | 0.004543 | 0.00001574 | −2.95099 | Down | 0.00041 | 1.118 × 10−9 | Fructose and mannose metabolism |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zenda, T.; Liu, S.; Wang, X.; Liu, G.; Jin, H.; Dong, A.; Yang, Y.; Duan, H. Key Maize Drought-Responsive Genes and Pathways Revealed by Comparative Transcriptome and Physiological Analyses of Contrasting Inbred Lines. Int. J. Mol. Sci. 2019, 20, 1268. https://doi.org/10.3390/ijms20061268
Zenda T, Liu S, Wang X, Liu G, Jin H, Dong A, Yang Y, Duan H. Key Maize Drought-Responsive Genes and Pathways Revealed by Comparative Transcriptome and Physiological Analyses of Contrasting Inbred Lines. International Journal of Molecular Sciences. 2019; 20(6):1268. https://doi.org/10.3390/ijms20061268
Chicago/Turabian StyleZenda, Tinashe, Songtao Liu, Xuan Wang, Guo Liu, Hongyu Jin, Anyi Dong, Yatong Yang, and Huijun Duan. 2019. "Key Maize Drought-Responsive Genes and Pathways Revealed by Comparative Transcriptome and Physiological Analyses of Contrasting Inbred Lines" International Journal of Molecular Sciences 20, no. 6: 1268. https://doi.org/10.3390/ijms20061268
APA StyleZenda, T., Liu, S., Wang, X., Liu, G., Jin, H., Dong, A., Yang, Y., & Duan, H. (2019). Key Maize Drought-Responsive Genes and Pathways Revealed by Comparative Transcriptome and Physiological Analyses of Contrasting Inbred Lines. International Journal of Molecular Sciences, 20(6), 1268. https://doi.org/10.3390/ijms20061268