Hypoxia Induces Astrocyte-Derived Lipocalin-2 in Ischemic Stroke


 ,
,  , and
, and
Abstract
1. Introduction
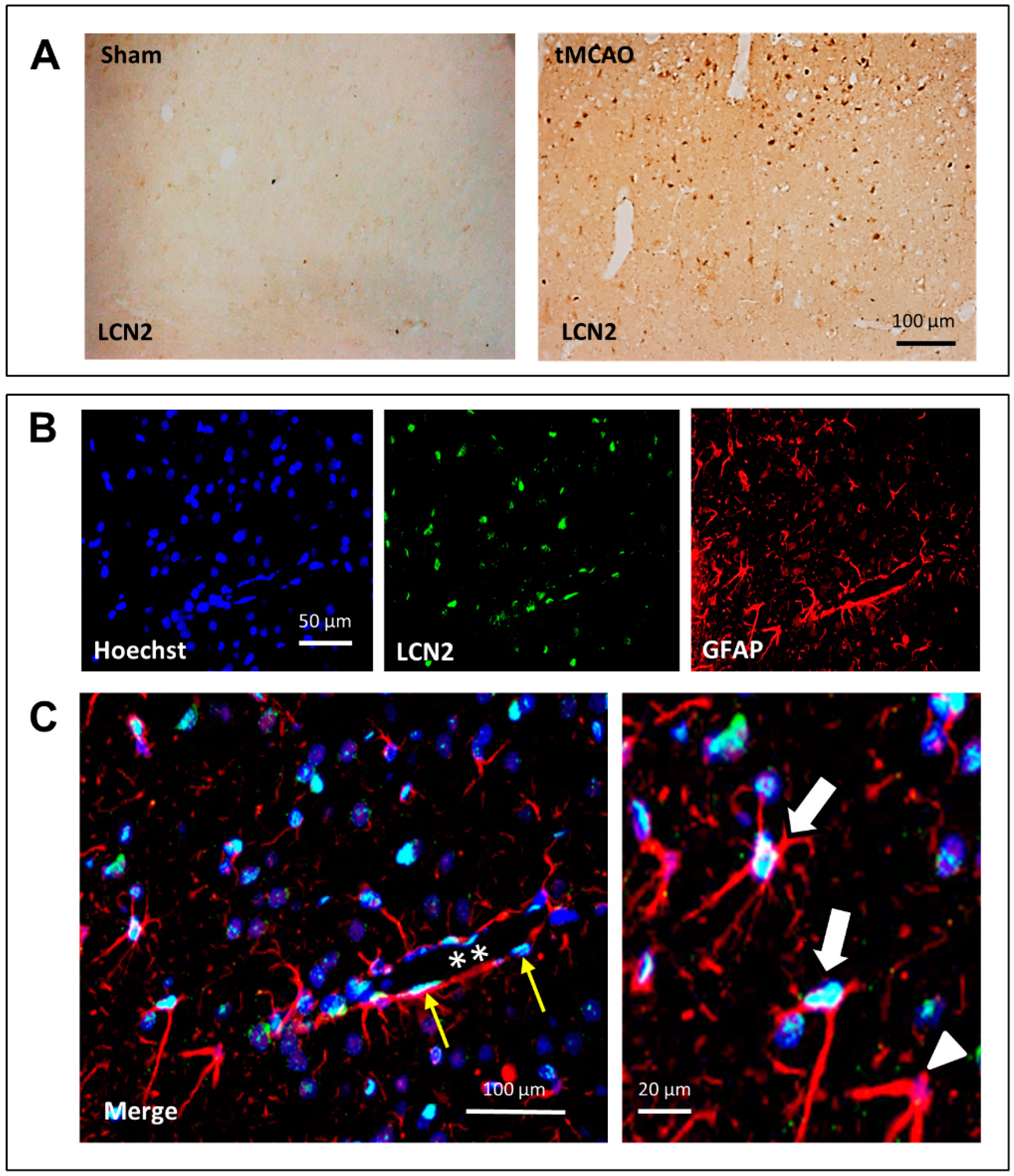
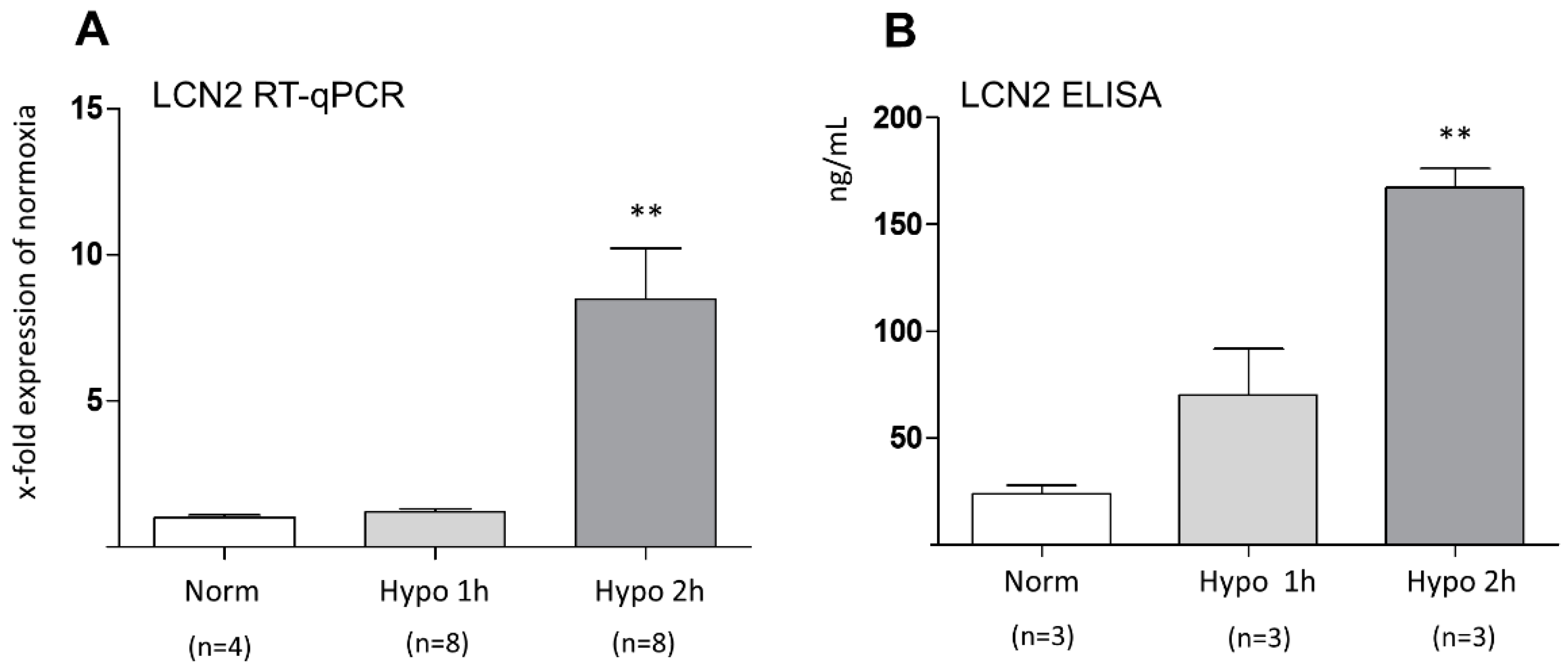
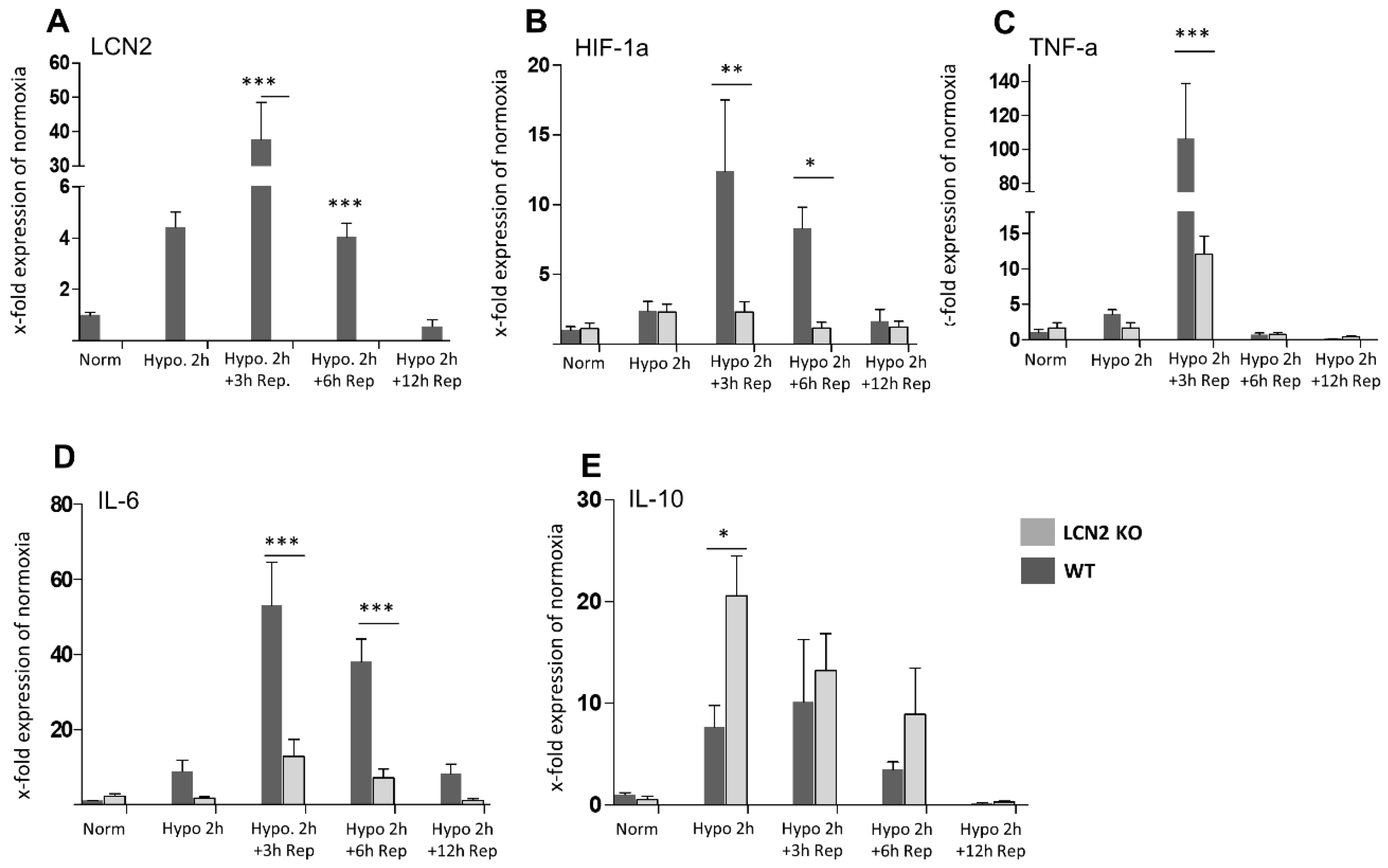
2. Results
3. Discussion
4. Material and Methods
4.1. Animals and Surgery
4.2. Blood Serum Analysis and Enzyme-Linked Immune Adsorbent Assay (ELISA)
4.3. TTC Staining, RNA Extraction, and Real-Time Quantitative PCR
4.4. Immunohistochemistry and Immunofluorescence Double-Labeling
4.5. SDS-PAGE and Western Blot Analysis
4.6. Primary Astrocyte Cultures from LCN2-Deficient and Wild Type Mice
4.7. In Vitro Hypoxia
4.8. Data Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sarkar, S.; Chakraborty, D.; Bhowmik, A.; Ghosh, M.K. Cerebral ischemic stroke: Cellular fate and therapeutic opportunities. Front. Biosci. (Landmark Ed.) 2019, 24, 435–450. [Google Scholar] [PubMed]
- Zhang, Q.Y.; Wang, Z.J.; Sun, D.M.; Wang, Y. Novel therapeutic effects of leonurine on ischemic stroke: New mechanisms of bbb integrity. Oxidative Med. Cell. Longev. 2017, 2017, 7150376. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 2015, 16, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Gabryel, B.; Kasprowska, D.; Kost, A.; Labuzek, K.; Urbanek, T. Astrocytes in ischemic stroke—A potential target for neuroprotective strategies. Postepy Hig. Med. Dosw. (Online) 2015, 69, 384–397. [Google Scholar] [CrossRef] [PubMed]
- Chistyakov, D.V.; Azbukina, N.V.; Lopachev, A.V.; Kulichenkova, K.N.; Astakhova, A.A.; Sergeeva, M.G. Rosiglitazone as a modulator of tlr4 and tlr3 signaling pathways in rat primary neurons and astrocytes. Int. J. Mol. Sci. 2018, 19, 113. [Google Scholar] [CrossRef] [PubMed]
- Mander, P.K.; Jekabsone, A.; Brown, G.C. Microglia proliferation is regulated by hydrogen peroxide from nadph oxidase. J. Immunol. (Balt. Md.: 1950) 2006, 176, 1046–1052. [Google Scholar] [CrossRef]
- Suzuki, A.; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 2011, 144, 810–823. [Google Scholar] [CrossRef]
- Hirayama, Y.; Koizumi, S. Astrocytes and ischemic tolerance. Neurosci. Res. 2018, 126, 53–59. [Google Scholar] [CrossRef]
- Hochmeister, S.; Engel, O.; Adzemovic, M.Z.; Pekar, T.; Kendlbacher, P.; Zeitelhofer, M.; Haindl, M.; Meisel, A.; Fazekas, F.; Seifert-Held, T. Lipocalin-2 as an infection-related biomarker to predict clinical outcome in ischemic stroke. PLoS ONE 2016, 11, e0154797. [Google Scholar] [CrossRef]
- Wang, G.; Weng, Y.C.; Han, X.; Whaley, J.D.; McCrae, K.R.; Chou, W.H. Lipocalin-2 released in response to cerebral ischaemia mediates reperfusion injury in mice. J. Cell. Mol. Med. 2015, 19, 1637–1645. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Jha, M.K.; Suk, K. Lipocalin-2 in the inflammatory activation of brain astrocytes. Crit. Rev. Immunol. 2015, 35, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Xing, C.; Wang, X.; Cheng, C.; Montaner, J.; Mandeville, E.; Leung, W.; van Leyen, K.; Lok, J.; Wang, X.; Lo, E.H. Neuronal production of lipocalin-2 as a help-me signal for glial activation. Stroke 2014, 45, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Zendedel, A.; Habib, P.; Dang, J.; Lammerding, L.; Hoffmann, S.; Beyer, C.; Slowik, A. Omega-3 polyunsaturated fatty acids ameliorate neuroinflammation and mitigate ischemic stroke damage through interactions with astrocytes and microglia. J. Neuroimmunol. 2015, 278, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Habib, P.; Slowik, A.; Zendedel, A.; Johann, S.; Dang, J.; Beyer, C. Regulation of hypoxia-induced inflammatory responses and m1-m2 phenotype switch of primary rat microglia by sex steroids. J. Mol. Neurosci. 2014, 52, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Tajalli-Nezhad, S.; Karimian, M.; Beyer, C.; Atlasi, M.A.; Azami Tameh, A. The regulatory role of toll-like receptors after ischemic stroke: Neurosteroids as tlr modulators with the focus on tlr2/4. Cell. Mol. Life Sci. 2018, 76, 523–537. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef]
- Macrez, R.; Ali, C.; Toutirais, O.; Le Mauff, B.; Defer, G.; Dirnagl, U.; Vivien, D. Stroke and the immune system: From pathophysiology to new therapeutic strategies. Lancet Neurol. 2011, 10, 471–480. [Google Scholar] [CrossRef]
- Slowik, A.; Lammerding, L.; Zendedel, A.; Habib, P.; Beyer, C. Impact of steroid hormones e2 and p on the nlrp3/asc/casp1 axis in primary mouse astroglia and bv-2 cells after in vitro hypoxia. J. Steroid Biochem. Mol. Biol. 2018, 183, 18–26. [Google Scholar] [CrossRef]
- Scheld, M.; Ruther, B.J.; Grosse-Veldmann, R.; Ohl, K.; Tenbrock, K.; Dreymuller, D.; Fallier-Becker, P.; Zendedel, A.; Beyer, C.; Clarner, T.; et al. Neurodegeneration triggers peripheral immune cell recruitment into the forebrain. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 1410–1415. [Google Scholar] [CrossRef]
- Mortezaee, K.; Khanlarkhani, N.; Beyer, C.; Zendedel, A. Inflammasome: Its role in traumatic brain and spinal cord injury. J. Cell. Physiol. 2018, 233, 5160–5169. [Google Scholar] [CrossRef] [PubMed]
- Puig, B.; Brenna, S.; Magnus, T. Molecular communication of a dying neuron in stroke. Int. J. Mol. Sci. 2018, 19, 2834. [Google Scholar] [CrossRef] [PubMed]
- Zagrean, A.M.; Hermann, D.M.; Opris, I.; Zagrean, L.; Popa-Wagner, A. Multicellular crosstalk between exosomes and the neurovascular unit after cerebral ischemia. Therapeutic implications. Front. Neurosci. 2018, 12, 811. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, J.; Yang, B.; Zheng, Y.; Yao, M.; Sun, M.; Xu, L.; Lin, C.; Chang, D.; Tian, F. Ginkgo biloba extract inhibits astrocytic lipocalin-2 expression and alleviates neuroinflammatory injury via the jak2/stat3 pathway after ischemic brain stroke. Front. Pharmacol. 2018, 9, 518. [Google Scholar] [CrossRef] [PubMed]
- Asimakopoulou, A.; Weiskirchen, S.; Weiskirchen, R. Lipocalin 2 (lcn2) expression in hepatic malfunction and therapy. Front. Physiol. 2016, 7, 430. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Yeoh, B.S.; Vijay-Kumar, M. Lipocalin 2: An emerging player in iron homeostasis and inflammation. Annu. Rev. Nutr. 2017, 37, 103–130. [Google Scholar] [CrossRef] [PubMed]
- Lattke, M.; Reichel, S.N.; Magnutzki, A.; Abaei, A.; Rasche, V.; Walther, P.; Calado, D.P.; Ferger, B.; Wirth, T.; Baumann, B. Transient ikk2 activation in astrocytes initiates selective non-cell-autonomous neurodegeneration. Mol. Neurodegen. 2017, 12, 16. [Google Scholar] [CrossRef]
- Moschen, A.R.; Adolph, T.E.; Gerner, R.R.; Wieser, V.; Tilg, H. Lipocalin-2: A master mediator of intestinal and metabolic inflammation. Trends Endocrinol. Metab. 2017, 28, 388–397. [Google Scholar] [CrossRef]
- Kang, S.S.; Ren, Y.; Liu, C.C.; Kurti, A.; Baker, K.E.; Bu, G.; Asmann, Y.; Fryer, J.D. Lipocalin-2 protects the brain during inflammatory conditions. Mol. Psychiatry 2018, 23, 344–350. [Google Scholar] [CrossRef]
- Suk, K. Lipocalin-2 as a therapeutic target for brain injury: An astrocentric perspective. Progress Neurobiol. 2016, 144, 158–172. [Google Scholar] [CrossRef]
- Song, J.; Kim, O.Y. Perspectives in lipocalin-2: Emerging biomarker for medical diagnosis and prognosis for Alzheimer’s disease. Clin. Nutr. Res. 2018, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dang, J.; Mitkari, B.; Kipp, M.; Beyer, C. Gonadal steroids prevent cell damage and stimulate behavioral recovery after transient middle cerebral artery occlusion in male and female rats. Brain Behav. Immunity 2011, 25, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Habib, P.; Dang, J.; Slowik, A.; Victor, M.; Beyer, C. Hypoxia-induced gene expression of aquaporin-4, cyclooxygenase-2 and hypoxia-inducible factor 1alpha in rat cortical astroglia is inhibited by 17beta-estradiol and progesterone. Neuroendocrinology 2014, 99, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Zheng, M.; Xi, G.; Keep, R.F.; Hua, Y. Role of lipocalin-2 in brain injury after intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 2015, 35, 1454–1461. [Google Scholar] [CrossRef]
- Jin, M.; Jang, E.; Suk, K. Lipocalin-2 acts as a neuroinflammatogen in lipopolysaccharide-injected mice. Exp. Neurobiol. 2014, 23, 155–162. [Google Scholar] [CrossRef]
- Rathore, K.I.; Berard, J.L.; Redensek, A.; Chierzi, S.; Lopez-Vales, R.; Santos, M.; Akira, S.; David, S. Lipocalin 2 plays an immunomodulatory role and has detrimental effects after spinal cord injury. J. Neurosci. 2011, 31, 13412–13419. [Google Scholar] [CrossRef] [PubMed]
- Nam, Y.; Kim, J.H.; Seo, M.; Kim, J.H.; Jin, M.; Jeon, S.; Seo, J.W.; Lee, W.H.; Bing, S.J.; Jee, Y.; et al. Lipocalin-2 protein deficiency ameliorates experimental autoimmune encephalomyelitis: The pathogenic role of lipocalin-2 in the central nervous system and peripheral lymphoid tissues. J. Biol. Chem. 2014, 289, 16773–16789. [Google Scholar] [CrossRef] [PubMed]
- Eller, K.; Schroll, A.; Banas, M.; Kirsch, A.H.; Huber, J.M.; Nairz, M.; Skvortsov, S.; Weiss, G.; Rosenkranz, A.R.; Theurl, I. Lipocalin-2 expressed in innate immune cells is an endogenous inhibitor of inflammation in murine nephrotoxic serum nephritis. PLoS ONE 2013, 8, e67693. [Google Scholar] [CrossRef]
- Offner, H.; Subramanian, S.; Parker, S.M.; Afentoulis, M.E.; Vandenbark, A.A.; Hurn, P.D. Experimental stroke induces massive, rapid activation of the peripheral immune system. J. Cereb. Blood Flow Metab. 2006, 26, 654–665. [Google Scholar] [CrossRef]
- Finsterer, J.; Wahbi, K. Cns-disease affecting the heart: Brain-heart disorders. J. Neurol. Sci. 2014, 345, 8–14. [Google Scholar] [CrossRef]
- DeMars, K.M.; Yang, C.; Hawkins, K.E.; McCrea, A.O.; Siwarski, D.M.; Candelario-Jalil, E. Spatiotemporal changes in p-glycoprotein levels in brain and peripheral tissues following ischemic stroke in rats. J. Exp. Neurosci. 2017, 11, 1179069517701741. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Zheng, W.; Dong, Y.; Wang, J.; Garstka, M.A.; Li, R.; An, J.; Ma, H. Serum levels of adipokines and cytokines in psoriasis patients: A systematic review and meta-analysis. Oncotarget 2018, 9, 1266–1278. [Google Scholar] [CrossRef] [PubMed]
- Hutanu, A.; Iancu, M.; Balasa, R.; Maier, S.; Dobreanu, M. Predicting functional outcome of ischemic stroke patients in romania based on plasma crp, stnfr-1, d-dimers, ngal and nse measured using a biochip array. Acta Pharmacol. Sin. 2018, 39, 1228–1236. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Zhang, J.; Li, X.; Xia, C.; Han, Y. Protein microarray analysis identifies key cytokines associated with malignant middle cerebral artery infarction. Brain Behav. 2017, 7, e00746. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Ko, P.W.; Lee, H.W.; Jeong, J.Y.; Lee, M.G.; Kim, J.H.; Lee, W.H.; Yu, R.; Oh, W.J.; Suk, K. Astrocyte-derived lipocalin-2 mediates hippocampal damage and cognitive deficits in experimental models of vascular dementia. Glia 2017, 65, 1471–1490. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D. Impact of inflammation on the blood-neural barrier and blood-nerve interface: From review to therapeutic preview. Int. Rev. Neurobiol. 2017, 137, 29–45. [Google Scholar] [PubMed]
- Shindo, A.; Maki, T.; Mandeville, E.T.; Liang, A.C.; Egawa, N.; Itoh, K.; Itoh, N.; Borlongan, M.; Holder, J.C.; Chuang, T.T.; et al. Astrocyte-derived pentraxin 3 supports blood-brain barrier integrity under acute phase of stroke. Stroke 2016, 47, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.; Bix, G.J. Roles of barrier integrins and extracellular matrix in stroke. Am. J. Physiol. Cell Physiol. 2019, 316, 252–263. [Google Scholar] [CrossRef]
- Strecker, J.K.; Schmidt, A.; Schabitz, W.R.; Minnerup, J. Neutrophil granulocytes in cerebral ischemia—Evolution from killers to key players. Neurochem. Int. 2017, 107, 117–126. [Google Scholar] [CrossRef]
- Lee, S.; Kim, J.H.; Kim, J.H.; Seo, J.W.; Han, H.S.; Lee, W.H.; Mori, K.; Nakao, K.; Barasch, J.; Suk, K. Lipocalin-2 is a chemokine inducer in the central nervous system: Role of chemokine ligand 10 (cxcl10) in lipocalin-2-induced cell migration. J. Boil. Chem. 2011, 286, 43855–43870. [Google Scholar] [CrossRef]
- Egashira, Y.; Hua, Y.; Keep, R.F.; Iwama, T.; Xi, G. Lipocalin 2 and blood-brain barrier disruption in white matter after experimental subarachnoid hemorrhage. Acta Neurochir. Suppl. 2016, 121, 131–134. [Google Scholar]
- Jin, M.; Kim, J.H.; Jang, E.; Lee, Y.M.; Soo Han, H.; Woo, D.K.; Park, D.H.; Kook, H.; Suk, K. Lipocalin-2 deficiency attenuates neuroinflammation and brain injury after transient middle cerebral artery occlusion in mice. J. Cereb. Blood Flow Metab. 2014, 34, 1306–1314. [Google Scholar] [CrossRef] [PubMed]
- Jha, M.K.; Jo, M.; Kim, J.H.; Suk, K. Microglia-astrocyte crosstalk: An intimate molecular conversation. Neuroscientist 2018, 1073858418783959. [Google Scholar] [CrossRef]
- Du, Y.; Deng, W.; Wang, Z.; Ning, M.; Zhang, W.; Zhou, Y.; Lo, E.H.; Xing, C. Differential subnetwork of chemokines/cytokines in human, mouse, and rat brain cells after oxygen-glucose deprivation. J. Cereb. Blood Flow Metab. 2017, 37, 1425–1434. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.; Vakilian, A.; Mahmoodi, M.H.; Hassanshahi, G.; Falahati-Pour, S.K.; Dolatabadi, M.R.; Nadimi, A.E. Circulatory levels of c-x-c motif chemokine ligands 1, 9, and 10 are elevated in patients with ischemic stroke. Eurasian J. Med. 2017, 49, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, I.; Hama, S.; Itakura, S.; Takasaki, I.; Nishi, T.; Tabuchi, Y.; Kogure, K. Lipocalin2 as a plasma marker for tumors with hypoxic regions. Sci. Rep. 2014, 4, 7235. [Google Scholar] [CrossRef] [PubMed]
- Leung, L.; Radulovich, N.; Zhu, C.Q.; Organ, S.; Bandarchi, B.; Pintilie, M.; To, C.; Panchal, D.; Tsao, M.S. Lipocalin2 promotes invasion, tumorigenicity and gemcitabine resistance in pancreatic ductal adenocarcinoma. PLoS ONE 2012, 7, e46677. [Google Scholar] [CrossRef] [PubMed]
- Lammerding, L.; Slowik, A.; Johann, S.; Beyer, C.; Zendedel, A. Poststroke inflammasome expression and regulation in the peri-infarct area by gonadal steroids after transient focal ischemia in the rat brain. Neuroendocrinology 2016, 103, 460–475. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.; Dang, J.; Baertling, F.; Denecke, B.; Clarner, T.; Kirsch, C.; Beyer, C.; Kipp, M. Ttc staining of damaged brain areas after mca occlusion in the rat does not constrict quantitative gene and protein analyses. J. Neurosci. Methods 2010, 187, 84–89. [Google Scholar] [CrossRef]
- Zendedel, A.; Monnink, F.; Hassanzadeh, G.; Zaminy, A.; Ansar, M.M.; Habib, P.; Slowik, A.; Kipp, M.; Beyer, C. Estrogen attenuates local inflammasome expression and activation after spinal cord injury. Mol. Neurobiol. 2018, 55, 1364–1375. [Google Scholar] [CrossRef]
- Berger, T.; Togawa, A.; Duncan, G.S.; Elia, A.J.; You-Ten, A.; Wakeham, A.; Fong, H.E.; Cheung, C.C.; Mak, T.W. Lipocalin 2-deficient mice exhibit increased sensitivity to escherichia coli infection but not to ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2006, 103, 1834–1839. [Google Scholar] [CrossRef] [PubMed]
- Clarner, T.; Janssen, K.; Nellessen, L.; Stangel, M.; Skripuletz, T.; Krauspe, B.; Hess, F.M.; Denecke, B.; Beutner, C.; Linnartz-Gerlach, B.; et al. Cxcl10 triggers early microglial activation in the cuprizone model. J. Immunol. (Balt. Md.: 1950) 2015, 194, 3400–3413. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence | Product Size (bp) | AT °C |
|---|---|---|---|
| LCN2 (mouse) | 5′ GCAGGTGGTACGTTGTGGG 3′ 3′ CTCTTGTAGCTCATAGATGGTGC 5′ | 95 | 65 |
| LCN2 (rat) | 5′ CAGAACTTGATCCCTGCCCC 3′ 3′ GCTGTACATGGTAAAGCGGC 5′ | 147 | 59 |
| HIF-1α | 5′ TCAAGTCAGCAACGTGGAAG 3′ 3′ TATCGAGGCTGTGTCGACTG 5′ | 198 | 65 |
| TNF-α | 5′ ACCCCTTTACTCTGACCCC 3′ 3′ GAGTCCTTGATGGTGGTGC 5′ | 189 | 62 |
| IL-6 | 5′ GATACCACTCCCAACAGACCTG 3′ 3′ GGTACTCCAGAAGACCAGAGGA 5′ | 223 | 64 |
| IL-10 | 5′ GCTCTTGCACTACCAAAGCC 3′ 3′ CTGCTGATCCTCATGCCAGT 5′ | 112 | 65 |
| CycloA (rat) | 5′ GGCAAATGCTGGACCAAACAC 3′ 3′ TTAGAGTTGTCCACAGTCGGAGAT 5′G | 196 | 65 |
| CycloA (mouse) | 5′ TTGGGTCCAGGAATGGCAAGA 3′ 3′ ACATTGCGGGAGCAGATGGGGT 5′ | 148 | 64 |
| Antibody | Company | WB | IHC | IF |
|---|---|---|---|---|
| LCN2 | Santa Cruz Biotechnology, Santa Cruz, CA, USA | 1:1000 | 1:500 | 1:300 |
| GFAP | Bioss, Woburn, MA, USA | --------- | ---------- | 1:300 |
| GAPDH | Sigma-AldrichTM, Merck, Darmstadt, Germany | 1:4000 | ---------- | ------ |
| Goat-anti-rabbit (488) | Invitrogen TM, Thermo Fisher Scientific, Langerwehe, Germany | ---------- | ---------- | 1:500 |
| Goat-anti-chicken (594) | Invitrogen TM, Thermo Fisher Scientific, Langerwehe, Germany | ---------- | ---------- | 1:500 |
| Goat-anti-rabbit | Vector Laboratories, Burlingame, CA, USA | ---------- | 1:50 | ------ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranjbar Taklimie, F.; Gasterich, N.; Scheld, M.; Weiskirchen, R.; Beyer, C.; Clarner, T.; Zendedel, A. Hypoxia Induces Astrocyte-Derived Lipocalin-2 in Ischemic Stroke. Int. J. Mol. Sci. 2019, 20, 1271. https://doi.org/10.3390/ijms20061271
Ranjbar Taklimie F, Gasterich N, Scheld M, Weiskirchen R, Beyer C, Clarner T, Zendedel A. Hypoxia Induces Astrocyte-Derived Lipocalin-2 in Ischemic Stroke. International Journal of Molecular Sciences. 2019; 20(6):1271. https://doi.org/10.3390/ijms20061271
Chicago/Turabian StyleRanjbar Taklimie, Fatemeh, Natalie Gasterich, Miriam Scheld, Ralf Weiskirchen, Cordian Beyer, Tim Clarner, and Adib Zendedel. 2019. "Hypoxia Induces Astrocyte-Derived Lipocalin-2 in Ischemic Stroke" International Journal of Molecular Sciences 20, no. 6: 1271. https://doi.org/10.3390/ijms20061271
APA StyleRanjbar Taklimie, F., Gasterich, N., Scheld, M., Weiskirchen, R., Beyer, C., Clarner, T., & Zendedel, A. (2019). Hypoxia Induces Astrocyte-Derived Lipocalin-2 in Ischemic Stroke. International Journal of Molecular Sciences, 20(6), 1271. https://doi.org/10.3390/ijms20061271