Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells

 , and
, and
Abstract
1. Introduction
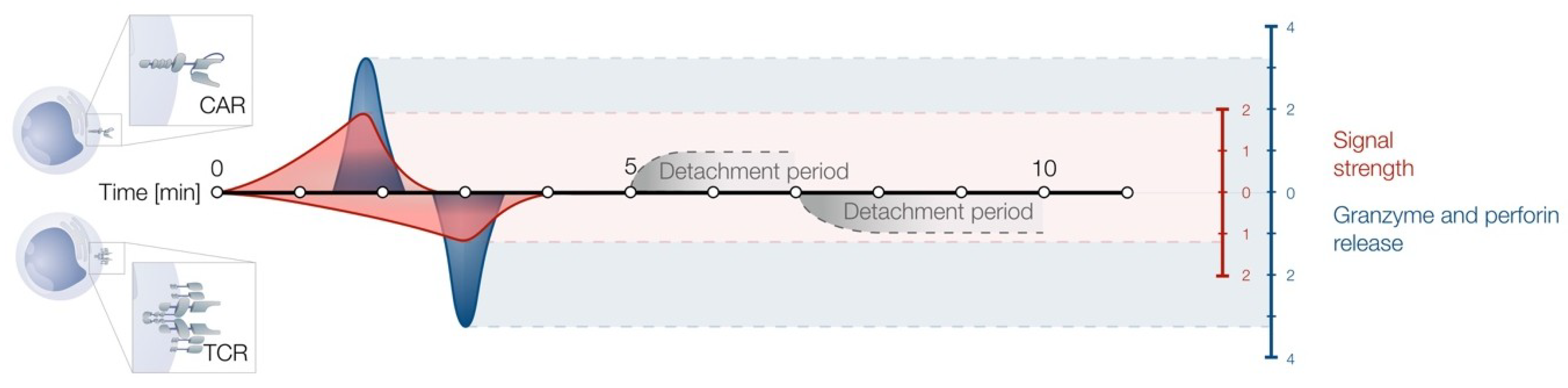
2. Non-Classical Immune Synapse Formation
3. Perforin and Granzyme
4. Fas and Fas Ligand (FasL) Axis
5. Cytokine Production
6. CAR T Cells as Serial Killers
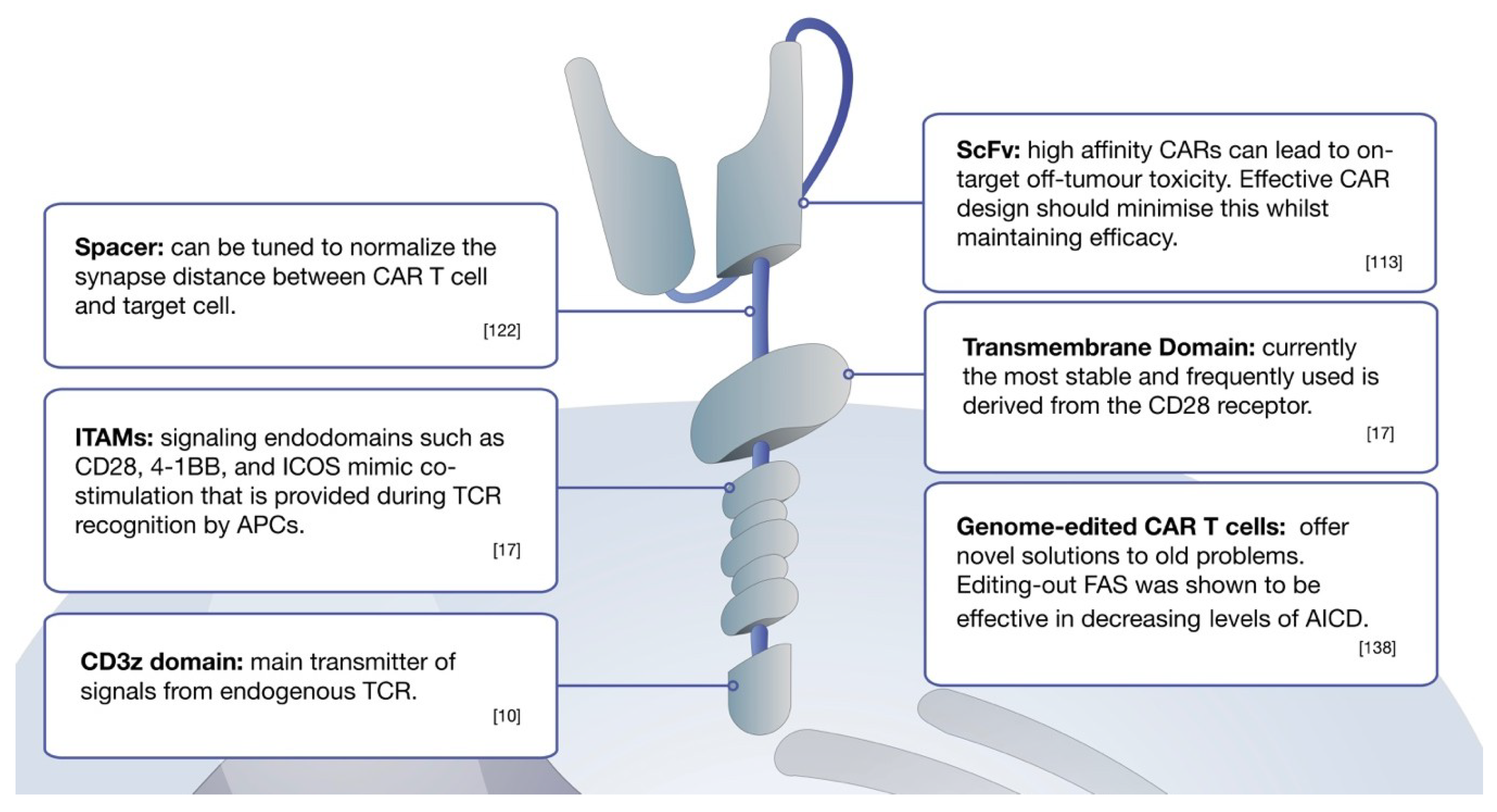
7. Affinity Variations of CAR Design Can Maximize Killing Efficiency
8. Optimizing CAR T Cell Functionality and Killing Potential
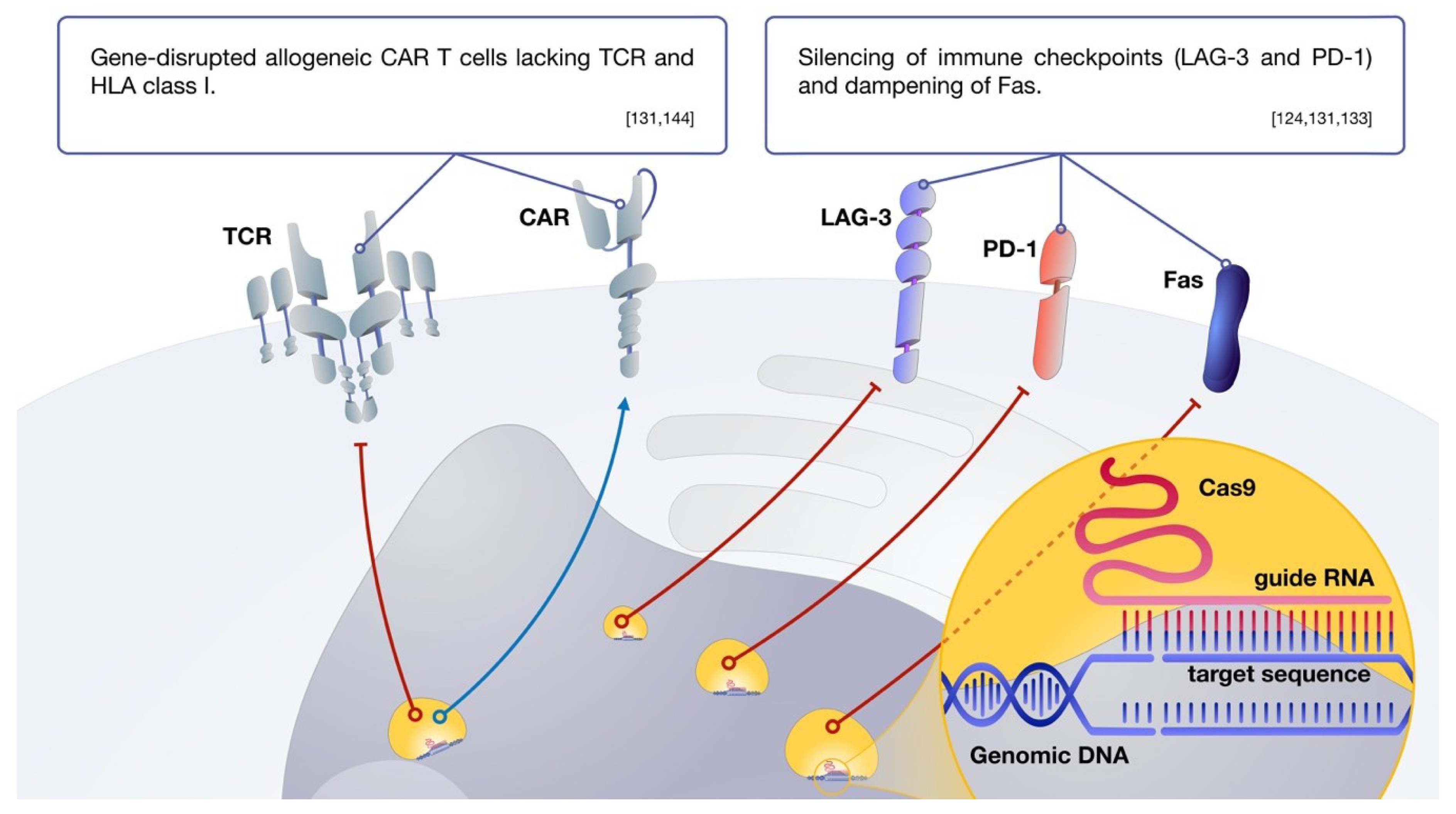
9. CAR Delivery and Genetic Modifications of CAR T Cells
10. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Melief, C.J.M. Tumor Eradication By Adoptive Transfer of Cytotoxic T Lymphocytes. Adv. Cancer Res. 1992, 58, 143–175. [Google Scholar] [PubMed]
- Rammensee, H.-G.; Bevan, M.J. Evidence from in Vitro Studies That Tolerance to Self Antigens Is MHC-Restricted. Nature 1984, 308, 741–744. [Google Scholar] [CrossRef] [PubMed]
- Kobold, S.; Steffen, J.; Chaloupka, M.; Grassmann, S.; Henkel, J.; Castoldi, R.; Zeng, Y.; Chmielewski, M.; Schmollinger, J.C.; Schnurr, M.; et al. Selective Bispecific T Cell Recruiting Antibody and Antitumor Activity of Adoptive T Cell Transfer. J. Natl. Cancer Inst. 2015, 107, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rapp, M.; Grassmann, S.; Chaloupka, M.; Layritz, P.; Kruger, S.; Ormanns, S.; Rataj, F.; Janssen, K.P.; Endres, S.; Anz, D.; et al. C-C Chemokine Receptor Type-4 Transduction of T Cells Enhances Interaction with Dendritic Cells, Tumor Infiltration and Therapeutic Efficacy of Adoptive T Cell Transfer. Oncoimmunology 2016, 5, 1–12. [Google Scholar] [CrossRef] [PubMed]
- NIH. Tumor Infiltrating Lymphocytes Adjuvant Therapy of Melanoma (TIL)—NCT00200577. Available online: https://clinicaltrials.gov/ct2/show/NCT00200577?cond=til+melanoma&rank=2 (accessed on 10 March 2019).
- Harris, D.T.; Kranz, D.M. Adoptive T Cell Therapies: A Comparison of T Cell Receptors and Chimeric Antigen Receptors. Trends Pharmacol. Sci. 2016, 37, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.H.J.; Willemsen, R.; van Elzakker, P.; van Steenbergen-Langeveld, S.; Broertjes, M.; Oosterwijk-wakka, J.; Oosterwijk, E.; Sleijfer, S.; Debets, R.; Gratama, J.W. Immune Responses to Transgene and Retroviral Vector in Patients Treated with Ex Vivo—Engineered T Cells. Blood 2011, 117, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Rogers-freezer, L.; Chen, C.C.; Yang, J.C.; et al. A Phase I Study on Adoptive Immunotherapy Using Gene- Modified T Cells for Ovarian Cancer. Clin. Cancer Res. 2007, 12, 6106–6115. [Google Scholar] [CrossRef]
- Ajina, A.; Maher, J. Strategies to Address Chimeric Antigen Receptor Tonic Signaling. Mol. Cancer Ther. 2018, 17, 1795–1815. [Google Scholar] [CrossRef] [PubMed]
- Eshhar, Z.; Waks, T.; Grosst, G.; Schindler, D.G. Specific Activation and Targeting of Cytotoxic Lymphocytes through Chimeric Single Chains Consisting of Antibody-Binding Domains and the y or C Subunits of the Immunoglobulin and T-Cell Receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef]
- Jensen, M.C.; Riddell, S.R. Designing Chimeric Antigen Receptors to Effectively and Safely Target Tumors. Curr. Opin. Immunol. 2015, 33, 9–15. [Google Scholar] [CrossRef]
- Wange, R.L.; Samelson, L.E. Complex Complexes: Signaling at the TCR. Immunity 1996, 5, 197–205. [Google Scholar] [CrossRef]
- Geiger, T.L.; Leitenberg, D.; Flavell, R.A. The TCR ζ -Chain Immunoreceptor Tyrosine-Based Activation Motifs Are Sufficient for the Activation and Differentiation of Primary T Lymphocytes. J. Immunol. 1999, 162, 5931–5939. [Google Scholar]
- Haynes, N.M.; Snook, M.B.; Trapani, J.A.; Cerruti, L.; Jane, S.M.; Smyth, M.J.; Darcy, P.K. Redirecting Mouse CTL Against Colon Carcinoma: Superior Signaling Efficacy of Single-Chain Variable Domain Chimeras Containing TCR- vs Fc RI-. J. Immunol. 2001, 166, 182–187. [Google Scholar] [CrossRef]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef]
- Guedan, S.; Chen, X.; Madar, A.; Carpenito, C.; McGettigan, S.E.; Frigault, M.; Lee, J.; Posey, A.; Scholler, J.; Scholler, N.; et al. ICOS-Based Chimeric Antigen Receptors Program Bipolar TH17/TH1 Cells. Blood 2014, 124, 1070–1080. [Google Scholar] [CrossRef]
- Krause, A.; Guo, H.; Latouche, J.; Tan, C.; Cheung, N.V.; Sadelain, M. Antigen-Dependent CD28 Signaling Selectively Enhances Survival and Proliferation in Genetically Modified Activated Human Primary T Lymphocytes. J. Exp. Med. 1998, 188, 619–626. [Google Scholar] [CrossRef]
- Krogsgaard, M.; Davis, M.M. How T Cells “see” Antigen. Nat. Immunol. 2005, 6, 239–245. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Zhong, J.F.; Zhang, X. Engineering CAR-T Cells. Biomark Res. 2017, 5, 1–6. [Google Scholar] [CrossRef]
- Love, P.E.; Hayes, S.M. ITAM-Mediated Signaling by the T-Cell Antigen Receptor. Cold Spring Harb. Perspect. Biol. 2010, 1–11. [Google Scholar] [CrossRef]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-Lymphocyte Cytotoxicity and Proliferation Directed by a Single Chimeric TCR ζ / CD28 Receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef]
- Kerkar, S.P.; Muranski, P.; Kaiser, A.; Boni, A.; Sanchez-Perez, L.; Yu, Z.; Palmer, D.C.; Reger, R.N.; Borman, Z.A.; Zhang, L.; et al. Tumor-Specific CD8+T Cells Expressing Interleukin-12 Eradicate Established Cancers in Lymphodepleted Hosts. Cancer Res. 2010, 70, 6725–6734. [Google Scholar] [CrossRef]
- Chmielewski, M.; Hombach, H.A. Of CARs and TRUCKs: Chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol. Rev. 2013, 257, 83–90. [Google Scholar] [CrossRef]
- Srivastava, S.; Riddell, S.R. Engineering CAR-T Cells: Design Concepts. Trends Immunol. 2015, 36, 494–502. [Google Scholar] [CrossRef]
- Yang, Y.; Jacoby, E.; Fry, T.J. Challenges and Opportunities of Allogeneic Donor-Derived CAR T Cells. Curr. Opin. Hematol. 2015, 22, 509–515. [Google Scholar] [CrossRef]
- Brentjens, R.; Latouche, J.-B. Eradication of Systemic B-Cell Tumors by Genetically Targeted Human T Lymphocytes Co-Stimulated by CD80 and Interleukin-15. Nat. Med. 2003, 9, 279–286. [Google Scholar] [CrossRef]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef]
- Maude, S.L.; Teachey, D.T.; Porter, D.L.; Grupp, S.A. CD19-Targeted Chimeric Antigen Receptor T-Cell Therapy for Acute Lymphoblastic Leukemia. Blood 2016, 128, 1141. [Google Scholar] [CrossRef]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T Cells Expressing CD19 Chimeric Antigen Receptors for Acute Lymphoblastic Leukaemia in Children and Young Adults: A Phase 1 Dose-Escalation Trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Novartis. Novartis (2017) Prescribing Information (KymriahTM). Available online: https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/kymriah.pdf (accessed on 13 January 2019).
- FDA. YescartaTM (2017) Prescribing Information. Available online: https://www.fda.gov/downloads/ BiologicsBloodVaccines/CellularGeneTherapyProducts/ApprovedProducts/UCM581226.pdf (accessed on 13 January 2019).
- Zheng, P.P.; Kros, J.M.; Li, J. Approved CAR T Cell Therapies: Ice Bucket Challenges on Glaring Safety Risks and Long-Term Impacts. Drug Discov. Today 2018, 23, 1175–1182. [Google Scholar] [CrossRef]
- D’Aloia, M.M.; Zizzari, I.G.; Sacchetti, B.; Pierelli, L.; Alimandi, M. CAR-T Cells: The Long and Winding Road to Solid Tumors Review-Article. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Boyiadzis, M.M.; Dhodapkar, M.V.; Brentjens, R.J.; Kochenderfer, J.N.; Neelapu, S.S.; Maus, M.V.; Porter, D.J.; Maloney, D.G.; Grupp, S.A.; Mackall, C.L.; et al. Chimeric Antigen Receptor (CAR) T Therapies for the Treatment of Hematologic Malignancies: Clinical Perspective and Significance. J. Immunother. Cancer 2018, 5, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-Targeted CAR T Cells Induce Remission in B-ALL That Is Naive or Resistant to CD19-Targeted CAR Immunotherapy. Nat. Med. 2017, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Labanieh, L.; Majzner, R.G.; Mackall, C.L. Programming CAR-T Cells to Kill Cancer. Nat. Biomed. Eng. 2018, 2, 377–391. [Google Scholar] [CrossRef]
- Lamers, C.H.J.; Sleijfer, S.; Vulto, A.G.; Kruit, W.H.J.; Kliffen, M.; Debets, R.; Gratama, J.W.; Stoter, G.; Oosterwijk, E. Treatment of Metastatic Renal Cell Carcinoma With Autologous T-Lymphocytes Genetically Retargeted Against Carbonic Anhydrase IX: First Clinical Experience. J. Clin. Oncol. 2006, 24, e20–e22. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.A.N.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T Cells Targeting Carcinoembryonic Antigen Can Mediate Regression of Metastatic Colorectal Cancer but Induce Severe Transient Colitis. Mol. Ther. 2011, 19, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer Regression and Neurologic Toxicity Following Anti-MAGE- A3 TCR Gene Therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef] [PubMed]
- Linette, G.P.; Stadtmauer, E.A.; Maus, M.V.; Rapoport, A.P.; Levine, B.L.; Emery, L.; Litzky, L.; Bagg, A.; Carreno, B.M.; Cimino, P.J.; et al. Cardiovascular Toxicity and Titin Cross-Reactivity of Affinity-Enhanced T Cells in Myeloma and Melanoma. Blood 2013, 122, 863–871. [Google Scholar] [CrossRef]
- Muller, P.Y.; Milton, M.N. The Determination and Interpretation of the Therapeutic Index in Drug Development. Nat. Rev. Drug Discov. 2012, 11, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Paszkiewicz, P.J.; Fräßle, S.P.; Srivastava, S.; Sommermeyer, D.; Hudecek, M.; Drexler, I.; Sadelain, M.; Liu, L.; Jensen, M.C.; Riddell, S.R.; et al. Targeted Antibody-Mediated Depletion of Murine CD19 CAR T Cells Permanently Reverses B Cell Aplasia. J. Clin. Investig. 2016, 126, 4262–4272. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric Antigen Receptor T-Cell Therapy-Assessment and Management of Toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Norelli, M.; Camisa, B.; Barbiera, G.; Falcone, L.; Purevdorj, A.; Genua, M.; Sanvito, F.; Ponzoni, M.; Doglioni, C.; Cristofori, P.; et al. Monocyte-Derived IL-1 and IL-6 Are Differentially Required for Cytokine-Release Syndrome and Neurotoxicity Due to CAR T Cells. Nat. Med. 2018, 24, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Giavridis, T.; Van Der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T Cell-Induced Cytokine Release Syndrome Is Mediated by Macrophages and Abated by IL-1 Blockade Letter. Nat. Med. 2018, 24, 731–738. [Google Scholar] [CrossRef]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2014, 6, 224ra25. [Google Scholar] [CrossRef] [PubMed]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and Management in CAR T-Cell Therapy. Mol. Ther. Oncolytics 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Monks, C.R.F.; Freiberg, B.A.; Kupfer, H.; Sciaky, N.; Kupfer, A. Three-Dimensional Segregation of Supramolecular Activation Clusters in T Cells. Nature 1998, 395, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L. The Immunological Synapse. Cancer Immunol. Res. 2014, 11, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Davenport, A.J.; Cross, R.S.; Watson, K.A.; Liao, Y.; Shi, W.; Prince, H.M.; Beavis, P.A.; Trapani, J.A.; Kershaw, M.H.; Ritchie, D.S.; et al. Chimeric Antigen Receptor T Cells Form Nonclassical and Potent Immune Synapses Driving Rapid Cytotoxicity. Proc. Natl. Acad. Sci. USA 2018, 115, 201716266. [Google Scholar] [CrossRef]
- Xiong, W.; Chen, Y.; Kang, X.; Chen, Z.; Zheng, P.; Hsu, Y.H.; Jang, J.H.; Qin, L.; Liu, H.; Dotti, G.; et al. Immunological Synapse Predicts Effectiveness of Chimeric Antigen Receptor Cells. Mol. Ther. 2018, 26, 963–975. [Google Scholar] [CrossRef] [PubMed]
- Meiraz, A.; Garber, O.G.; Harari, S.; Hassin, D.; Berke, G. Switch from Perforin-Expressing to Perforin-Deficient CD8+ T Cells Accounts for Two Distinct Types of Effector Cytotoxic T Lymphocytes in Vivo. Immunology 2009, 128, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Stinchcombe, J.C.; Majorovits, E.; Bossi, G.; Fuller, S.; Griffiths, G.M. Centrosome Polarization Delivers Secretory Granules to the Immunological Synapse. Nature 2006, 443, 462–465. [Google Scholar] [CrossRef]
- Cullen, S.P.; Martin, S.J. Mechanisms of Granule-Dependent Killing. Cell Death Differ. 2008, 15, 251–262. [Google Scholar] [CrossRef] [PubMed]
- De Saint Basile, G.; Ménasché, G.; Fischer, A. Molecular Mechanisms of Biogenesis and Exocytosis of Cytotoxic Granules. Nat. Rev. Immunol. 2010, 10, 568–579. [Google Scholar] [CrossRef]
- Kägi, D.; Ledermann, B.; Bürki, K.; Seiler, P.; Odermatt, B.; Olsen, K.J.; Podack, E.R.; Zinkernagel, R.M.; Hengartner, H. Cytotoxicity Mediated by T Cells and Natural Killer Cells Is Greatly Impaired in Perforin-Deficient Mice. Nature 1994, 369, 31–37. [Google Scholar] [CrossRef]
- Stalder, T.; Hahn, S.; Erb, P.; Paya, C.V.; Celis, E. Fas Antigen Is the Major Target Molecule for CD4+ T Cell-Mediated Cytotoxicity. J. Immunol. 1994, 152, 1127–1133. [Google Scholar] [CrossRef]
- Yasukawa, M.; Ohminami, H.; Arai, J.; Kasahara, Y.; Ishida, Y.; Fujita, S. Granule Exocytosis, and Not the Fas/Fas Ligand System, Is the Main Pathway of Cytotoxicity Mediated by Alloantigen-Specific CD4(+) as well as CD8(+) Cytotoxic T Lymphocytes in Humans. Blood 2000, 95, 2352–2355. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.; Wieczarkowiecz, A.; Marquardt, T.; Heuser, C.; Usai, L.; Pohl, C.; Seliger, B.; Abken, H. Tumor-Specific T Cell Activation by Recombinant Immunoreceptors: CD3 Signaling and CD28 Costimulation Are Simultaneously Required for Efficient IL-2 Secretion and Can Be Integrated Into One Combined CD28/CD3 Signaling Receptor Molecule. J. Immunol. 2001, 167, 6123–6131. [Google Scholar] [CrossRef]
- Hombach, A.; Köhler, H.; Rappl, G.; Abken, H. Human CD4+ T Cells Lyse Target Cells via Granzyme/Perforin upon Circumvention of MHC Class II Restriction by an Antibody-like Immunoreceptor. J. Immunol. 2006, 177, 5668–5675. [Google Scholar] [CrossRef]
- Kumaresan, P.R.; Manuri, P.R.; Albert, N.D.; Maiti, S.; Singh, H.; Mi, T.; Roszik, J.; Rabinovich, B.; Olivares, S.; Krishnamurthy, J.; et al. Bioengineering T Cells to Target Carbohydrate to Treat Opportunistic Fungal Infection. Proc. Natl. Acad. Sci. USA 2014, 111, 10660–10665. [Google Scholar] [CrossRef]
- Davenport, A.J.; Jenkins, M.R.; Cross, R.S.; Yong, C.S.; Prince, H.M.; Ritchie, D.; Trapani, J.; Kershaw, M.; Darcy, P.; Neeson, P. CAR-T Cells Inflict Sequential Killing of Multiple Tumor Target Cells. Cancer Immunol. Res. 2015, 3, 483–494. [Google Scholar] [CrossRef]
- Koehler, H.; Kofler, D.; Hombach, A.; Abken, H. CD28 Costimulation Overcomes Transforming Growth Factor-β-Mediated Repression of Proliferation of Redirected Human CD4+and CD8+T Cells in an Antitumor Cell Attack. Cancer Res. 2007, 67, 2265–2273. [Google Scholar] [CrossRef]
- Mamonkin, M.; Rouce, R.H.; Tashiro, H.; Brenner, M.K. A T-Cell-Directed Chimeric Antigen Receptor for the Selective Treatment of T-Cell Malignancies. Blood 2015, 126, 983–992. [Google Scholar] [CrossRef]
- Liadi, I.; Singh, H.; Romain, G.; Rey-Villamizar, N.; Merouane, A.; Adolacion, J.R.T.; Kebriaei, P.; Huls, H.; Qiu, P.; Roysam, B.; et al. Individual Motile CD4+ T Cells Can Participate in Efficient Multikilling through Conjugation to Multiple Tumor Cells. Cancer Immunol. Res. 2015, 3, 473–482. [Google Scholar] [CrossRef]
- Jenkins, M.R.; Rudd-Schmidt, J.A.; Lopez, J.A.; Ramsbottom, K.M.; Mannering, S.I.; Andrews, D.M.; Voskoboinik, I.; Trapani, J.A. Failed CTL/NK Cell Killing and Cytokine Hypersecretion Are Directly Linked through Prolonged Synapse Time. J. Exp. Med. 2015, 212, 307–317. [Google Scholar] [CrossRef]
- Chen, Y.L.; Chen, S.-H.; Wang, J.-Y.; Yang, B.-C. Fas Ligand on Tumor Cells Mediates Inactivation of Neutrophils. J. Immunol. 2003, 171, 1183–1191. [Google Scholar] [CrossRef]
- O’Connell, J.; O’Sullivan, G.C.; Collins, J.K.; Shanahan, F. The Fas Counterattack: Fas-Mediated T Cell Killing By Colon Cancer Cells Expressing Fas Ligand. J. Exp. Med. 1996, 184, 1075–1082. [Google Scholar] [CrossRef]
- Hassin, D.; Garber, O.G.; Meiraz, A.; Yael, S.; Berke, G. Cytotoxic T Lymphocyte Perforin and Fas Ligand Working in Concert Even When Fas Ligand Lytic Action Is Still Not Detectable. Immunology 2011, 2, 190–196. [Google Scholar] [CrossRef]
- Morales-Kastresana, A.; Miguel, S.F.; Rodriguez, I.; Palazon, A.; Martinez-Forero, I.; Labiano, S.; Hervas-Stubbs, S.; Sangro, B.; Ochoa, C.; Rouzaut, A.; et al. Therapeutic Activity of a Combination of Immunostimulatory Monoclonal Antibodies (Anti-B7-H1, CD137 and OX40) on a c-Myc-Driven Spontaneous Transgenic Model of Hepatocellular Carcinoma. J. Immunother. Cancer 2013, 1, O7. [Google Scholar] [CrossRef]
- Peter, M.E.; Hadji, A.; Murmann, A.E.; Brockway, S.; Putzbach, W.; Pattanayak, A.; Ceppi, P. The Role of CD95 and CD95 Ligand in Cancer. Cell Death Differ. 2015, 22, 885–886. [Google Scholar] [CrossRef]
- Lyubchenko, T.A.; Wurth, G.A.; Zweifach, A. Role of Calcium Influx in Cytotoxic T Lymphocyte Lytic Granule Exocytosis during Target Cell Killing. Immunity 2001, 15, 847–859. [Google Scholar] [CrossRef]
- Kagi, D.; Vignaux, F.; Ledermann, B.; Borki, K.; Depraetere, V.; Nagata, S.; Hengartner, H. Fas and Perforin Pathways as Major Mechanisms of T cell-mediated cytotoxicity. Science 1994, 265, 528–530. [Google Scholar] [CrossRef]
- Lowin, B.; Hahne, M.; Mattmann, C.; Tschopp, J. Cytolytic T-Cell Cytotoxicity Is Mediated through Perforin and Fas Lytic Pathways. Nature 1994, 370, 650–652. [Google Scholar] [CrossRef]
- Martínez-Lostao, L.; Anel, A.; Pardo, J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Fu, T.-M.; Cruz, A.; Sengupta, P.; Thomas, S.; Wang, S. Structural Basis and Functional Role of Intramembrane Trimerization of the Fas/CD95 Death Receptor. Mol. Cell 2016, 61, 602–613. [Google Scholar] [CrossRef]
- Walczak, H. Death Receptor-Ligand Systems in Cancer, Cell Death, and Inflammation. Cold Spring Harb. Perspect. Biol. 2013, 5, 1–18. [Google Scholar] [CrossRef]
- Nagata, S.; Tanaka, M. Programmed Cell Death and the Immune System. Nat. Rev. Immunol. 2017, 17, 333–340. [Google Scholar] [CrossRef]
- Waring, P.; Mullbacher, A. Cell Death Induced by the Fas/Fas Ligand Pathway and Its Role in Pathology. Immunol. Cell Biol. 1999, 77, 312–317. [Google Scholar] [CrossRef]
- Hong, L.K.; Chen, Y.; Smith, C.C.; Montgomery, S.A.; Vincent, B.G.; Dotti, G.; Savoldo, B. CD30-Redirected Chimeric Antigen Receptor T Cells Target CD30+ and CD30− Embryonal Carcinoma via Antigen-Dependent and Fas/FasL Interactions. Cancer Immunol. Res. 2018, 6, 1274–1287. [Google Scholar] [CrossRef]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A Novel Chimeric Antigen Receptor Containing a JAK—STAT Signaling Domain Mediates Superior Antitumor Effects. Nat. Publ. Gr. 2018, 24, 352–359. [Google Scholar] [CrossRef]
- Textor, A.; Listopad, J.J.; Le Wührmann, L.; Perez, C.; Kruschinski, A.; Chmielewski, M.; Abken, H.; Blankenstein, T.; Charo, J. Efficacy of CAR T-Cell Therapy in Large Tumors Relies upon Stromal Targeting by IFNγ. Cancer Res. 2014, 74, 6796–6805. [Google Scholar] [CrossRef]
- Viaud, S.; Ma, J.S.Y.; Hardy, I.R.; Hampton, E.N.; Benish, B.; Sherwood, L.; Nunez, V.; Ackerman, C.J.; Khialeeva, E.; Weglarz, M.; et al. Switchable Control over in Vivo CAR T Expansion, B Cell Depletion, and Induction of Memory. Proc. Natl. Acad. Sci. USA 2018, 115, E10898–E10906. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Collins, J.J.; Wong, W.W. Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 2018, 173, 1426–1438.e11. [Google Scholar] [CrossRef] [PubMed]
- Van Herpen, C.M.L.; Van Der Laak, J.A.W.M.; De Vries, I.J.M.; Van Krieken, J.H.; De Wilde, P.C.; Balvers, M.G.J.; Adema, G.J.; De Mulder, P.H.M. Intratumoral Recombinant Human Interleukin-12 Administration in Head and Neck Squamous Cell Carcinoma Patients Modifies Locoregional Lymph Node Architecture and Induces Natural Killer Cell Infiltration in the Primary Tumor. Clin. Cancer Res. 2005, 11, 1899–1909. [Google Scholar] [CrossRef] [PubMed]
- Car, B.; Vicki, M.; Review, I.-A.; Lipman, J.M.; Anderson, T.D. The Toxicology of Interleukin-12: A Review. Toxicol. Pathol. 1999, 27, 58–63. [Google Scholar] [CrossRef]
- Hung, K.; Hayashi, R.; Lafond-Walker, A.; Lowenstein, C.; Pardoll, D.; Levitsky, H. The Central Role of CD4+ T Cells in the Antitumor Immune Response. J. Exp. Med. 1998, 188, 2357–2368. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, T.; Huang, J.; Gooding, W.E.; Gambotto, A.; Robbins, P.D.; Vujanovic, N.L.; Alber, S.M.; Watkins, S.C.; Okada, H.; Storkus, W.J. Intratumoral Delivery of Dendritic Cells Engineered to Secrete Both Interleukin (IL)-12 and IL-18 Effectively Treats Local and Distant Disease in Association with Broadly Reactive Tc1-Type Immunity. Cancer Res. 2003, 63, 6378–6386. [Google Scholar] [CrossRef]
- Pegram, H.J.; Lee, J.C.; Hayman, E.G.; Imperato, G.H.; Tedder, T.F.; Sadelain, M.; Brentjens, R.J. Tumor-Targeted T Cells Modified to Secrete IL-12 Eradicate Systemic Tumors without Need for Prior Conditioning. Blood 2012, 119, 4133–4141. [Google Scholar] [CrossRef]
- Kerkar, S.; Rosenberg, S.; Restifo, N. IL-12 Triggers a Programmatic Change in Dysfunctional Myeloid-Derived Cells within Mouse Tumors. J. Clin. Investig. 2011, 121, 4746–4757. [Google Scholar] [CrossRef]
- Curtsinger, J.M.; Lins, D.C.; Mescher, M.F. Signal 3 Determines Tolerance versus Full Activation of Naive CD8 T Cells. J. Exp. Med. 2003, 197, 1141–1151. [Google Scholar] [CrossRef]
- Simpson-Abelson, M.R.; Purohit, V.S.; Pang, W.M.; Iyer, V.; Odunsi, K.; Demmy, T.L.; Yokota, S.J.; Loyall, J.L.; Kelleher R.J., Jr.; Balu-Iyer, S.; et al. IL-12 Delivered Intratumorally by Multilamellar Liposomes Reactivates Memory T Cells in Human Tumor Microenvironments. Clin. Immunol. 2009, 132, 71–82. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Robbins, P.F.; Wunderlich, J.R.; Sherry, R.M.; Schwartzentruber, D.J.; Topalian, S.L.; Nicholas, P.; Filie, A.; Chang, R.; et al. Durable Complete Responses in Heavily Pretreated Patients with Metastatic Melanoma Using T Cell Transfer Immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef]
- Kearney, C.J.; Vervoort, S.J.; Hogg, S.J.; Ramsbottom, K.M.; Freeman, A.J.; Lalaoui, N.; Pijpers, L.; Michie, J.; Brown, K.K.; Knight, D.A.; et al. Tumor Immune Evasion Arises through Loss of TNF Sensitivity. Sci. Immunol. 2018, 3, eaar3451. [Google Scholar] [CrossRef]
- Zaidi, M.R.; Merlino, G. The Two Faces of Interferon-γ in Cancer. Clin. Cancer Res. 2011, 17, 6118–6124. [Google Scholar] [CrossRef]
- Majzner, R.; Mackall, C. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1218–1226. [Google Scholar] [CrossRef]
- June, C.H.; Riddell, S.R.; Schumacher, T.N. Adoptive Cellular Therapy: A Race to the Finish Line. Sci. Transl. Med. 2015, 7, 1–9. [Google Scholar] [CrossRef]
- Kearney, C.J.; Lalaoui, N.; Freeman, A.J.; Ramsbottom, K.M.; Silke, J.; Oliaro, J. PD-L1 and IAPs Co-Operate to Protect Tumors from Cytotoxic Lymphocyte-Derived TNF. Cell Death Differ. 2017, 24, 1705–1716. [Google Scholar] [CrossRef]
- Patel, S.J.; Sanjana, N.E.; Kishton, R.J.; Eidizadeh, A.; Vodnala, S.K.; Cam, M.; Gartner, J.J.; Jia, L.; Steinberg, S.M.; Yamamoto, T.N.; et al. Identification of Essential Genes for Cancer Immunotherapy. Nat. Publ. Gr. 2017, 548, 537–542. [Google Scholar] [CrossRef]
- Choi, P.J.; Mitchison, T.J. Imaging Burst Kinetics and Spatial Coordination during Serial Killing by Single Natural Killer Cells. Proc. Natl. Acad. Sci. USA 2013, 110, 6488–6493. [Google Scholar] [CrossRef]
- Janssen, E.M.; Lemmens, E.E.; Wolfe, T.; Christen, U.; Von Herrath, M.G.; Schoenberger, S.P. CD4+ T Cells Are Required for Secondary Expansion and Memory in CD8+ T Lymphocytes. Nature 2003, 421, 852–856. [Google Scholar] [CrossRef]
- Antony, P.A.; Piccirillo, C.A.; Akpinarli, A.; Finkelstein, S.E.; Speiss, P.J.; Surman, D.R.; Palmer, D.C.; Chan, C.C.; Klebanoff, C.A.; Overwijk, W.W.; et al. CD8+ T Cell Immunity Against a Tumor/Self-Antigen Is Augmented by CD4+ T Helper Cells and Hindered by Naturally Occurring T Regulatory Cells. J. Immunol. 2005, 174, 2591–2601. [Google Scholar] [CrossRef]
- Waterhouse, N.J.; Sutton, V.R.; Sedelies, K.A.; Ciccone, A.; Jenkins, M.; Turner, S.J.; Bird, P.I.; Trapani, J.A. Cytotoxic T Lymphocyte-Induced Killing in the Absence of Granzymes A and B Is Unique and Distinct from Both Apoptosis and Perforin-Dependent Lysis. J. Cell Biol. 2006, 173, 133–144. [Google Scholar] [CrossRef]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric Antigen Receptor-Modified T Cells Derived from Defined CD8+ and CD4+ Subsets Confer Superior Antitumor Reactivity in Vivo. Leukemia 2016, 30, 492–500. [Google Scholar] [CrossRef]
- Lerner, R.A. Combinatorial Antibody Libraries: New Advances, New Immunological Insights. Nat. Rev. Immunol. 2016, 16, 498–508. [Google Scholar] [CrossRef]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB Costimulation Ameliorates T Cell Exhaustion Induced by Tonic Signaling of Chimeric Antigen Receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef]
- Gomes-Silva, D.; Srinivasan, M.; Orange, J.S.; Brenner, M.K. Tonic 4-1BB Costimulation in Chimeric Antigen Receptors Impedes T Cell Survival and Is Vector-Dependent. Cell Rep. 2017, 21, 17–26. [Google Scholar] [CrossRef]
- Zhong, S.; Malecek, K.; Johnson, L.A.; Yu, Z.; Vega-Saenz de Miera, E.; Darvishian, F.; McGary-Shipper, K.; Huang, K.; Boyer, J.; Corse, E.; et al. T Cell Receptor Affinity and Avidity Defines Antitumor Response and Autoimmunity in T Cell Immunotherapy. Proc. Natl. Acad. Sci. USA 2013, 110, 6973–6978. [Google Scholar] [CrossRef]
- Schmid, D.A.; Irving, M.B.; Posevitz, V.; Hebeisen, M.; Posevitz-Fejfar, A.; Sarria, J.C.F.; Gomez-Eerland, R.; Thome, M.; Schumacher, T.N.M.; Romero, P.; et al. Evidence for a TCR Affinity Threshold Delimiting Maximal CD8 T Cell Function. J. Immunol. 2010, 184, 4936–4946. [Google Scholar] [CrossRef]
- Chmielewski, M.; Hombach, A.; Heuser, C.; Adams, G.P.; Abken, H. T Cell Activation by Antibody-Like Immunoreceptors: Increase in Affinity of the Single-Chain Fragment Domain above Threshold Does Not Increase T Cell Activation against Antigen-Positive Target Cells but Decreases Selectivity. J. Immunol. 2004, 173, 7647–7653. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case Report of a Serious Adverse Event Following the Administration of t Cells Transduced with a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Liu, X.; Shuguang, J.; Fang, C.; Yang, S.; Olalere, D.; Pequignot, E.; Cogdill, A.; Li, N.; Ramones, M.; Granda, B.; et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res. 2015, 75, 3596–3607. [Google Scholar] [CrossRef]
- Carter, P.; Presta, L.E.N.; Gormant, C.M.; Ridgwayt, J.B.B.; Hennert, D.; Wong, W.L.; Rowland, A.M.; Kotts, C.; Carvert, M.E.; Shepard, H.M. Humanization of an Anti-P185HER2 Antibody for Human Cancer Therapy. Proc. Natl. Acad. Sci. USA 1992, 89, 4285–4289. [Google Scholar] [CrossRef]
- Finn, O.J. Human Tumor Antigens Yesterday, Today, and Tomorrow. Cancer Immunol. Res. 2017, 5, 347–354. [Google Scholar] [CrossRef]
- Dotti, G.; Gottschalk, S.; Savoldo, B.; Brenner, M.K. Design and Development of Therapies Using Chimeric Antigen Receptor-Expressing T Cells. Immunol. Rev. 2014, 257, 151–155. [Google Scholar] [CrossRef]
- Harris, D.T.; Hager, M.V.; Smith, S.N.; Cai, Q.; Stone, J.D.; Kruger, P.; Lever, M.; Dushek, O.; Schmitt, T.M.; Greenberg, P.D.; et al. Comparison of T Cell Activities Mediated by Human TCRs and CARs That Use the Same Recognition Domains. J. Immunol. 2017, ji1700236. [Google Scholar] [CrossRef]
- He, J.; Zhang, Z.; Lv, S.; Liu, X.; Cui, L.; Jiang, D.; Zhang, Q.; Li, L.; Qin, W.; Jin, H.; et al. Engineered CAR T Cells Targeting Mesothelin by PiggyBac Transposon System for the Treatment of Pancreatic Cancer. Cell. Immunol. 2018, 329, 31–40. [Google Scholar] [CrossRef]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; Van Der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC Locus with CRISPR/Cas9 Enhances Tumour Rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef]
- James, J.R.; Vale, R.D. Biophysical Mechanism of T-Cell Receptor Triggering in a Reconstituted System. Nature 2012, 487, 64–69. [Google Scholar] [CrossRef]
- James, S.E.; Greenberg, P.D.; Jensen, M.C.; Lin, Y.; Wang, J.; Till, B.G.; Raubitschek, A.A.; Forman, S.J.; Press, O.W. Antigen Sensitivity of CD22-Specific Chimeric TCR Is Modulated by Target Epitope Distance from the Cell Membrane. J. Immunol. 2008, 180, 7028–7038. [Google Scholar] [CrossRef]
- Kunkele, A.; Johnson, A.J.; Rolczynski, L.S.; Chang, C.A.; Hoglund, V.; Kelly-Spratt, K.S.; Jensen, M.C. Functional Tuning of CARs Reveals Signaling Threshold above Which CD8+ CTL Antitumor Potency Is Attenuated Due to Cell Fas-FasL-Dependent AICD. Cancer Immunol. Res. 2015, 3, 368–379. [Google Scholar] [CrossRef]
- Kramer, J.A.; Sagartz, J.E.; Morris, D.L. The Application of Discovery Toxicology and Pathology towards the Design of Safer Pharmaceutical Lead Candidates. Nat. Rev. Drug Discov. 2007, 6, 636–649. [Google Scholar] [CrossRef]
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; McNally, K.A.; Park, J.S.; Lim, W.A. Precision Tumor Recognition by T Cells with Combinatorial Antigen-Sensing Circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted Delivery of a PD-1-Blocking ScFV by CAR-T Cells Enhances Anti-Tumor Efficacy in Vivo. Nat. Biotechnol. 2018, 36, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Kano, Y.; Nagai, T.; Okuyama, N.; Sakoda, Y.; Tamada, K. IL-7 and CCL19 Expression in CAR-T Cells Improves Immune Cell Infiltration and CAR-T Cell Survival in the Tumor. Nat. Biotechnol. 2018, 36, 346–351. [Google Scholar] [CrossRef]
- Salter, A.I.; Ivey, R.G.; Kennedy, J.J.; Voillet, V.; Rajan, A.; Alderman, E.J.; Voytovich, U.J.; Lin, C.; Sommermeyer, D.; Liu, L.; et al. Phosphoproteomic Analysis of Chimeric Antigen Receptor Signaling Reveals Kinetic and Quantitative Differences That Affect Cell Function. Sci. Signal. 2018, 11, eaat6753. [Google Scholar] [CrossRef]
- Guedan, S.; Posey, A.D.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T Cell Persistence through ICOS and 4-1BB Costimulation. JCI Insight 2018, 3, 1–17. [Google Scholar] [CrossRef]
- Fedorov, V.D.; Themeli, M.; Sadelain, M. PD-1- and CTLA-4-Based Inhibitory Chimeric Antigen Receptors (ICARs) Divert Off-Target Immunotherapy Responses. Sci. Transl. Med. 2013, 5, 1–13. [Google Scholar] [CrossRef]
- Grada, Z.; Hegde, M.; Byrd, T.; Shaffer, D.; Ghazi, A.; Brawley, A.; Koch, J.; Dotti, G.; Heslop, H.; Gottschalk, S.; et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol. Ther. Nucleic Acids 2013, 2, e105. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Zhang, X.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. A Versatile System for Rapid Multiplex Genome-Edited CAR T Cell Generation. Oncotarget 2017, 8, 17002–17011. [Google Scholar] [CrossRef]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-Mediated PD-1 Disruption Enhances Anti-Tumor Efficacy of Human Chimeric Antigen Receptor T Cells. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Zhang, Y.; Cheng, C.; Liu, X.; Mu, W.; Xia, C.; Wang, H.; Zhang, X.; Wei, X.; Liu, X.; Li, N. CRISPR-Cas9 Mediated LAG-3 Disruption in CAR-T Cells. Front. Med. 2017, 11, 554–562. [Google Scholar] [CrossRef]
- Valton, J.; Guyot, V.; Marechal, A.; Filhol, J.M.; Juillerat, A.; Duclert, A.; Duchateau, P.; Poirot, L. A Multidrug-Resistant Engineered CAR T Cell for Allogeneic Combination Immunotherapy. Mol. Ther. 2015, 23, 1507–1518. [Google Scholar] [CrossRef]
- Gargett, T.; Brown, M. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front. Pharmacol. 2014, 5, 235. [Google Scholar] [CrossRef]
- Bachmann, D.; Aliperta, R.; Bergmann, R.; Feldmann, A.; Koristka, S.; Arndt, C.; Loff, S.; Welzel, P.; Albert, S.; Kegler, A.; et al. Retargeting of UniCAR T Cells with an in Vivo Synthesized Target Module Directed against CD19 Positive Tumor Cells. Oncotarget 2018, 9, 7487–7500. [Google Scholar] [CrossRef]
- Wu, C.-Y.; Roybal, K.T.; Puchner, E.M.; Onuffer, J.; Lim, W.A. Remote Control of Therapeutic T Cells through a Small Molecule-Gated Chimeric Receptor. Science 2015, 350, 293. [Google Scholar] [CrossRef]
- Ellis, J. Silencing and Variegation of Gammaretrovirus and Lentivirus Vectors. Hum. Gene Ther. 2005, 16, 1241–1246. [Google Scholar] [CrossRef]
- Witting, S.R.; Vallanda, P.; Gamble, A.L. Characterization of a Third Generation Lentiviral Vector Pseudotyped with Nipah Virus Envelope Proteins for Endothelial Cell Transduction. Gene Ther. 2013, 20, 997–1005. [Google Scholar] [CrossRef]
- Plesa, G.; Lacey, S.F.; Marcucci, K.T.; June, C.H.; Melenhorst, J.J.; Hwang, W.-T.; Levine, B.L.; Suhoski-Davis, M.; Kulikovskaya, I.; Gupta, M.; et al. Retroviral and Lentiviral Safety Analysis of Gene-Modified T Cell Products and Infused HIV and Oncology Patients. Mol. Ther. 2017, 26, 269–279. [Google Scholar] [CrossRef]
- Hackett, P.B.; Largaespada, D.A.; Cooper, L.J.N. A Transposon and Transposase System for Human Application. Mol. Ther. 2010, 18, 674–683. [Google Scholar] [CrossRef]
- Chabot, S.; Orio, J.; Schmeer, M.; Schleef, M.; Golzio, M.; Teissié, J. Minicircle DNA Electrotransfer for Efficient Tissue-Targeted Gene Delivery. Gene Ther. 2013, 20, 62–68. [Google Scholar] [CrossRef]
- Kebriaei, P.; Singh, H.; Huls, M.H.; Figliola, M.J.; Bassett, R.; Olivares, S.; Jena, B.; Dawson, M.J.; Kumaresan, P.R.; Su, S.; et al. Phase I Trials Using Sleeping Beauty to Generate CD19-Specific CAR T Cells. J. Clin. Investig. 2016, 126, 3363–3376. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’connor, R.S.; Hwang, W.T.; et al. Determinants of Response and Resistance to CD19 Chimeric Antigen Receptor (CAR) T Cell Therapy of Chronic Lymphocytic Leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F. ZFN, TALEN, and CRISPR/Cas-Based Methods for Genome Engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef]
- Kim, S.; Lee, M.J.; Kim, H.; Kang, M.; Kim, J.S. Preassembled Zinc-Finger Arrays for Rapid Construction of ZFNs. Nat. Methods 2011, 8, 7. [Google Scholar] [CrossRef]
- Deng, D.; Yan, C.; Pan, X.; Mahfouz, M.; Wang, J.; Zhu, J.; Shi, Y.; Yan, N. Structural Basis for Sequence-Specific Recognition Of DNA by TAL effectors. Science 2012, 335, 720–723. [Google Scholar] [CrossRef]
- Christian, M.; Cermak, T.; Doyle, E.L.; Schmidt, C.; Zhang, F.; Hummel, A.; Bogdanove, A.J.; Voytas, D.F. Targeting DNA Double-Strand Breaks with TAL Effector Nucleases. Genetics 2010, 186, 757–761. [Google Scholar] [CrossRef]
- Torikai, H.; Reik, A.; Yuen, C.; Zhang, Z.; Rebar, E.J.; Holmes, M.C.; Zhou, Y.; Lee, D.A.; Crossland, D.L.; Jaenisch, R. Toward Eliminating HLA Class I Expression to Generate Universal Cells from Allogeneic Donors. Blood 2013, 122, 1341–1349. [Google Scholar] [CrossRef]
- Torikai, H.; Reik, A.; Liu, P.-Q.; Zhou, Y.; Zhang, L.; Maiti, S. A Foundation for Universal T-Cell Based Immunotherapy: T Cells Engineered to Express a CD19-Specific Chimeric-Antigen-Receptor and Eliminate Expression of Endogenous TCR. Blood 2012, 119, 5697–5705. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- Cadilha, B.; Dorman, K.; Rataj, F.; Endres, S.; Kobold, S. Enabling T Cell Recruitment to Tumours as a Strategy for Improving Adoptive T Cell Therapy. Eur. Oncol. Haematol. 2017, 13, 66–73. [Google Scholar] [CrossRef]
- Tokarew, N.; Ogonek, J.; Endres, S.; Von Bergwelt-baildon, M.; Kobold, S. Teaching an Old Dog New Tricks: Next-Generation CAR T Cells. Br. J. Cancer 2018, 120, 26–37. [Google Scholar] [CrossRef]
- Rataj, F.; Kraus, F.B.T.; Chaloupka, M.; Grassmann, S.; Heise, C.; Cadilha, B.L.; Duewell, P.; Endres, S.; Kobold, S. PD1-CD28 Fusion Protein Enables CD4+ T Cell Help for Adoptive T Cell Therapy in Models of Pancreatic Cancer and Non-Hodgkin Lymphoma. Front. Immunol. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Zhang, J.; Endres, S.; Kobold, S. Enhancing Tumor T Cell Infiltration to Enable Cancer Immunotherapy. Immunotherapy 2019, 11, 201–213. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, S.; Brahmer, J.R.; Gettinger, S.; Smith, D.; McDermott, D.; Powderly, J.; Carvajal, R. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Zhao, Z.; Shi, L.; Zhang, W.; Han, J.; Zhang, S.; Fu, Z.; Cai, J. CRISPR Knock out of Programmed Cell Death Protein 1 Enhances Anti-Tumor Activity of Cytotoxic T Lymphocytes. Oncotarget 2018, 9, 5208–5215. [Google Scholar] [CrossRef]
- Chow, M.; Luster, A. Chemokines in Cancer. Cancer Immunol. Res. 2014, 2, 1125–1131. [Google Scholar] [CrossRef]
- Di Stasi, A.; De Angelis, B.; Rooney, C.M.; Zhang, L.; Mahendravada, A.; Foster, A.E.; Heslop, H.E.; Brenner, M.K.; Dotti, G.; Savoldo, B. T Lymphocytes Coexpressing CCR4 and a Chimeric Antigen Receptor Targeting CD30 Have Improved Homing and Antitumor Activity in a Hodgkin Tumor Model. Blood 2009, 113, 6392–6402. [Google Scholar] [CrossRef]
- Craddock, J.A.; Lu, A.; Bear, A.; Pule, M.; Brenner, M.K.; Rooney, C.M.; Foster, A.E. Enhanced Tumor Trafficking of GD2 Chimeric Antigen Receptor T Cells by Expression of the Chemokine Receptor CCR2b. J. Immunother. Cancer 2010, 33, 780–788. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aim | Modulation | Approaches |
|---|---|---|
| Enhancing selectivity | Simultaneous targeting of multiple antigens | Tandem CAR [130] |
| iCAR (inhibitory CAR) [129] | ||
| scFv modulation | Fine-tune scFv affinities [113] | |
| Inducible CARs | synNotch CAR [124] | |
| Enhancing killing potential | Co-stimulatory domains | JAK-STAT CAR [82] |
| 3rd generation ICOS CAR [128] | ||
| Cytokine production | TRUCK system [86] | |
| Checkpoint Blockade | Secretion of PD-1 scFv [125] | |
| Targeted delivery of CAR cDNA to disrupt a locus | CRIPSR guided to Fas, endogenous TCR, PD-1, and LAG-3 [131,132,133] | |
| TALEN-mediated multi-drug resistant CARs [134] | ||
| Immune cell recruitment | 7 × 19 CAR (co-expressing IL-7 and CCL 19) [126] | |
| Regulating activity | Suicide Gene | Inducible Caspase9 [135] |
| Antibody-mediated depletion via marker antigen [43] | ||
| Switchable CAR | Tumor targeting anti- or nanobody (UniCAR, SUPRA CAR) [85,136] | |
| Dimerization through small molecules [137] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benmebarek, M.-R.; Karches, C.H.; Cadilha, B.L.; Lesch, S.; Endres, S.; Kobold, S. Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. Int. J. Mol. Sci. 2019, 20, 1283. https://doi.org/10.3390/ijms20061283
Benmebarek M-R, Karches CH, Cadilha BL, Lesch S, Endres S, Kobold S. Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. International Journal of Molecular Sciences. 2019; 20(6):1283. https://doi.org/10.3390/ijms20061283
Chicago/Turabian StyleBenmebarek, Mohamed-Reda, Clara Helke Karches, Bruno Loureiro Cadilha, Stefanie Lesch, Stefan Endres, and Sebastian Kobold. 2019. "Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells" International Journal of Molecular Sciences 20, no. 6: 1283. https://doi.org/10.3390/ijms20061283
APA StyleBenmebarek, M.-R., Karches, C. H., Cadilha, B. L., Lesch, S., Endres, S., & Kobold, S. (2019). Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. International Journal of Molecular Sciences, 20(6), 1283. https://doi.org/10.3390/ijms20061283