QT Assessment in Early Drug Development: The Long and the Short of It

Abstract
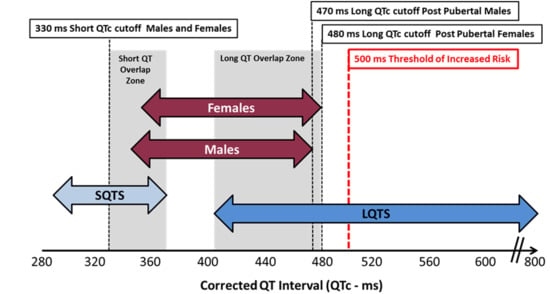
1. History of The QT Interval and Its Importance in Early Drug Development
2. Overview of Ventricular Repolarization Electrophysiology
- ➢
- Phase 0: The sharp upstroke of the action potential is primarily the result of a transient and rapid influx of Na+ (INa) through opening of Na+ channels;
- ➢
- Phase 1: The termination of the action potential upstroke and initiation of the early repolarization phase are mediated by inactivation of Na+ channels and the transient outward movement of K+ (Ito) through K+ channels and chloride (Cl−) ions;
- ➢
- Phase 2: The action potential plateau is ascribed to the balance between the influx of Ca2+ (ICa) through long opening L-type Ca2+ channels and the efflux of repolarizing K+ currents;
- ➢
- Phase 3: The sustained downward slope of the action potential and the late repolarization phase are due to the egress of K+ (IKr and IKs) through delayed rectifier K+ channels;
- ➢
- Phase 4: The resting membrane potential is supported by the inward rectifier K+ current (IK1), the sodium potassium ATPase pump, and the Na+/Ca2+ exchanger.
3. How Is the QTc Calculated: Popular Correction Formulae for QT Values
4. What Is a Normal QTc Value
5. Measurement of the QT Interval
6. Problematic and Challenging Issues in QT Assessment
- ➢
- Artifact: Measurement of the QT interval should be performed in tracings without any artifact that may obscure the intervals and lead to erroneous values. As such, a segment of the extracted ECG devoid of artifact should be used for measurement or additional “clean” ECGs as close to the nominal timepoint specified in the protocol time and events schedule should be secured and used for interval assessment. To aid in this regard there are automated computer algorithms designed to ensure that extracted cardiodynamic ECGs are obtained without significant artifact, dysrhythmias or heart rate instability.
- ➢
- U waves: U waves are common especially in young individuals with relatively slow heart rates and often are distinct positive waves after the T wave and best seen in leads V2 and V3 (Figure 2). They are thought to represent a final phase of ventricular repolarization involving the summation of early afterdepolarizations or repolarization of mid myocardial M cells, papillary muscles or purkinje fibers [51]. U waves may be attenuated with filtering or indistinct when there is significant tachycardia. The U wave should be clearly identified as distinct from the T wave and should not be included in QT measurement as normal QU values have not been established and inclusion of the U wave would lead to gross over-measurement of the QT interval. When it is unclear if U waves are present, inspection of neighboring leads on the ECG may be helpful in separating a discrete U wave from the T wave.
- ➢
- Bifid T waves: T waves may be notched or bifid in appearance and the end of the T in these cases should be measured after the second peak. Also, careful inspection of notched T wave morphology and the distance between notches may be useful in distinguishing the T wave from a superimposed U wave. While differentiating a notched T wave from a superimposed U wave can be difficult, viewing alternate leads in the tracing should be performed to help make this distinction.
- ➢
- Flat T waves: When the T waves are flat, measurement of the QT interval should be carried out in a lead(s) where the T waves are positive, monophasic and best defined. In the absence of any positive unidirectional T waves, a clearly visible monophasic negative T wave would also suffice for this assessment. When a single lead median beat approach is being used and the designated lead is not suitable for measurement, the alternate lead utilized should be identified on the report and subsequent ECGs from that subject should all be measured in that same lead in order to provide procedural consistency and reduce variability that may be misconstrued as drug mediated.
- ➢
- T-U fusion: U waves may be fused with the T wave thereby artificially prolonging the QT interval by casual visual inspection. In this setting, as initially recommended by Lepeschkin and Surawicz [52], a tangent line should be drawn through the steepest portion of the T wave downslope until it intersects the isoelectric line (defined by the T-P segment) and that crossing point is to be designated as the end of the T wave.
- ➢
- Arrhythmias: Whenever the RR interval shows significant variability as in the case of sinus arrhythmia or atrial fibrillation, multiple evaluable QT complexes should be measured and the QT value averaged for all complexes so as to avoid over or under estimation of the QT interval. In the case of premature supraventricular or ventricular beats, measurement in the complex immediately following the premature beat should be avoided as ventricular repolarization is altered in the complex after a premature beat.
- ➢
- Asymptotic prolonged downsloping T waves: This is a finding in which the QT interval can easily be overmeasured due to a prolonged T wave “tail”. As such, when the end of the T wave approaches the isoelectric line asymptotically, a tangent function utilizing the steepest portion of the downslope of the T wave should be employed as described for T-U fusion.
- ➢
- Wide QRS complexes: The presence of a widened QRS such as with bundle branch block, ventricular pacing, pre-excitation, or intraventricular conduction delays, may contribute to a prolonged QTc interval which may not be a consequence of significantly altered ventricular repolarization. In these circumstances, the formula QTc = measured QTc-(QRS-100 ms) has been suggested to provide a clinically useful determination of the true QTc interval [41]. This approach has been advocated by those involved in assessing individuals with suspected or known congenital long QTc syndromes.
- ➢
- Misconnected limb leads: In cases where there is limb lead misconnection involving only reversed arm leads, measurement can still be affected in lead II if a single lead median beat approach is used. In cases where lead II is effected by misconnected limb leads, an alternative precordial lead such as V5 is suggested. Misconnected limb leads should not significantly alter the QT measurement when a representative 12 lead median beat is utilized.
7. QTc Syndromes
8. Long QTc Syndromes (LQTS)
8.1. Acquired LQTS
8.2. Congenital LQTS
9. Short QTc Syndrome (SQTS)
9.1. Acquired SQTS
9.2. Congenital SQTS
9.3. Criteria for Diagnosis SQTS
10. The Evolution of Regulatory Guidance Regarding Ventricular Repolarization
11. Current FDA Guidance for Assessing QT Liability
11.1. Comprehensive In Vitro Proarrhythmia Assay (CiPA)
11.1.1. Preclinical
11.1.2. Clinical
11.2. Concentration QT Modelling
12. Commentary
13. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| APD | action potential duration |
| CiPA | comprehensive in vitro proarrhythmia assay |
| Cmax | maximal concentration |
| cQT | concentration QT modelling |
| ECG | electrocardiogram |
| FDA | Food and Drug Administration |
| hERG | human ether a-go-go related gene |
| ICH | International Conference on Harmonization |
| LQTS | long QT syndrome |
| MAD | multiple ascending dose |
| NCE | new chemical entity |
| QTc | corrected QT |
| SAD | single ascending dose |
| SCD | sudden cardiac death |
| SQTS | short QT syndrome |
| TdP | Torsades de Pointes |
| TQT | thorough QT |
References
- Einthoven, W. Ueber die Form des menschlichen Electrocardiogramms. Pflügers Arch. Eur. J. Physiol. 1895, 60, 101–123. [Google Scholar] [CrossRef]
- Jervell, A.; Lange-Nielsen, F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am. Heart J. 1957, 54, 59–68. [Google Scholar] [CrossRef]
- Romano, C.; Gemme, G.; Pongiglione, R. Rare Cardiac Arrythmias of the Pediatric Age. Ii. Syncopal Attacks Due to Paroxysmal Ventricular Fibrillation. (Presentation of 1st Case in Italian Pediatric Literature). Clin. Pediatr. (Bologna) 1963, 45, 656–683. [Google Scholar] [PubMed]
- Ward, O.C. A New Familial Cardiac Syndrome in Children. J. Ir. Med. Assoc. 1964, 54, 103–106. [Google Scholar] [PubMed]
- Selzer, A.; Wray, H.W. Quinidine Syncope. Paroxysmal Ventricular Fibrillation Occurring during Treatment of Chronic Atrial Arrhythmias. Circulation 1964, 30, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Dessertenne, F. Ventricular tachycardia with 2 variable opposing foci. Arch. Mal. Coeur Vaiss. 1966, 59, 263–272. [Google Scholar] [PubMed]
- Meanock, C.I.; Noble, M.I. A case of prenylamine toxicity showing the torsade de pointes phenomenon in sinus rhythm? Postgrad. Med. J. 1981, 57, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Monahan, B.P.; Ferguson, C.L.; Killeavy, E.S.; Lloyd, B.K.; Troy, J.; Cantilena, L.R., Jr. Torsades de pointes occurring in association with terfenadine use. JAMA 1990, 264, 2788–2790. [Google Scholar] [CrossRef] [PubMed]
- Fung, M.; Thornton, A.; Mybeck, K.; Wu, J.H.-H.; Hornbuckle, K.; Muniz, E. Evaluation of the Characteristics of Safety Withdrawal of Prescription Drugs from Worldwide Pharmaceutical Markets-1960 to 1999. Drug Inf. J. 2001, 35, 293–317. [Google Scholar] [CrossRef]
- Points to Consider: The Assessment of the Potential for QT Interval Prolongation by Non Cardiovascular Medicinal Products; Committee for Proprietary Medicinal Products, Ed.; The European Agency for the Evaluation of Medicinal Products: London, UK, 1997. [Google Scholar]
- Draft Guidance: Assessment of the QT Prolongation Potential of Non Anti-Arrhythmic Drugs; Health Canada: Ottawa, ON, Canada, 2001.
- Implementation Working Group ICH E14 Guideline. The Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs. Q&A R3. Available online: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E14/E14_Q_As_R3__Step4.pdf (accessed on 26 September 2017).
- The Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs E14. Available online: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E14/E14_Guideline.pdf (accessed on 26 September 2017).
- Kwon, C.H.; Kim, S.H. Intraoperative management of critical arrhythmia. Korean J. Anesthesiol. 2017, 70, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, N.; Grunnet, M.; Olesen, S.P. Cardiac potassium channel subtypes: New roles in repolarization and arrhythmia. Physiol. Rev. 2014, 94, 609–653. [Google Scholar] [CrossRef] [PubMed]
- Grant, A.O. Cardiac ion channels. Circ. Arrhythm. Electrophysiol. 2009, 2, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Jeevaratnam, K.; Chadda, K.R.; Huang, C.L.; Camm, A.J. Cardiac Potassium Channels: Physiological Insights for Targeted Therapy. J. Cardiovasc. Pharmacol. Ther. 2018, 23, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Q.; Purhonen, P.; Hebert, H. Structure of potassium channels. Cell. Mol. Life Sci. 2015, 72, 3677–3693. [Google Scholar] [CrossRef] [PubMed]
- Roden, D.M. Cellular basis of drug-induced torsades de pointes. Br. J. Pharmacol. 2008, 154, 1502–1507. [Google Scholar] [CrossRef] [PubMed]
- Bazett, H.C. An analysis of the time-relations of the electrocardiograms. Heart 1920, 7, 353–370. [Google Scholar] [CrossRef]
- Fridericia, L.S. Die systolendauer im elektrokardiogramm bei normalen menschen und bei herzkranken. Acta Med. Scand. 1920, 53, 469–486. [Google Scholar] [CrossRef]
- Sagie, A.; Larson, M.G.; Goldberg, R.J.; Bengtson, J.R.; Levy, D. An improved method for adjusting the QT interval for heart rate (the Framingham Heart Study). Am. J. Cardiol. 1992, 70, 797–801. [Google Scholar] [CrossRef]
- Hodges, M.; Salerno, D.; Erlien, D. Bazett’s QT correction reviewed. Evidence that a linear QT correction for heart is better. J. Am. Coll. Cardiol. 1983, 1, 694. [Google Scholar]
- Rautaharju, P.M.; Mason, J.W.; Akiyama, T. New age- and sex-specific criteria for QT prolongation based on rate correction formulas that minimize bias at the upper normal limits. Int. J. Cardiol. 2014, 174, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Dmitrienke, A.A.; Sides, G.D.; Winters, K.J.; Kovacs, R.J.; Rebhun, D.M.; Bloom, J.C.; Groh, W.; Eisenberg, P.R. Electrocardiogram Reference Ranges Derived from a Standardized Clinical Trial Population. Drug Inf. J. 2005, 39, 395–405. [Google Scholar] [CrossRef]
- Van de Water, A.; Verheyen, J.; Xhonneux, R.; Reneman, R.S. An improved method to correct the QT interval of the electrocardiogram for changes in heart rate. J. Pharmacol. Methods 1989, 22, 207–217. [Google Scholar] [CrossRef]
- Rabkin, S.W.; Szefer, E.; Thompson, D.J.S. A New QT Interval Correction Formulae to Adjust for Increases in Heart Rate. JACC Clin. Electrophysiol. 2017, 3, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Vandenberk, B.; Vandael, E.; Robyns, T.; Vandenberghe, J.; Garweg, C.; Foulon, V.; Ector, J.; Willems, R. Which QT Correction Formulae to Use for QT Monitoring? J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- FDA Guidance for Industry E14 (2017) Clinical Evaluation of QT/QTc Interval Prolongation for Non-Antiarrhythmic Drugs—Questions and Answers (R3). Available online: https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm073161.pdf (accessed on 26 September 2017).
- Phan, D.Q.; Silka, M.J.; Lan, Y.T.; Chang, R.K. Comparison of formulas for calculation of the corrected QT interval in infants and young children. J. Pediatr. 2015, 166, 960–964. [Google Scholar] [CrossRef] [PubMed]
- Morganroth, J. Cardiac Safety of Noncardiac Drugs: Practical Guidelines for Clinical Research and Drug Development; Humana Press: New York, NY, USA, 2005. [Google Scholar] [CrossRef]
- Goldenberg, I.; Moss, A.J.; Zareba, W. QT interval: How to measure it and what is “normal”. J. Cardiovasc. Electrophysiol. 2006, 17, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Malik, M. QTc evaluation for drugs with a substantial heart rate effect. In Cardiac Safety Research Consortium Think Tank; Cardiac Safety Research Consortium: Washington, DC, USA, 2018. [Google Scholar]
- Malik, M. Methods of Subject-Specific Heart Rate Corrections. J. Clin. Pharmacol. 2018, 58, 1020–1024. [Google Scholar] [CrossRef] [PubMed]
- Garnett, C.E.; Zhu, H.; Malik, M.; Fossa, A.A.; Zhang, J.; Badilini, F.; Li, J.; Darpo, B.; Sager, P.; Rodriguez, I. Methodologies to characterize the QT/corrected QT interval in the presence of drug-induced heart rate changes or other autonomic effects. Am. Heart J. 2012, 163, 912–930. [Google Scholar] [CrossRef] [PubMed]
- Rautaharju, P.M.; Surawicz, B.; Gettes, L.S.; Bailey, J.J.; Childers, R.; Deal, B.J.; Gorgels, A.; Hancock, E.W.; Josephson, M.; Kligfield, P.; et al. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: Part IV: The ST segment, T and U waves, and the QT interval: A scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J. Am. Coll. Cardiol. 2009, 53, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Drew, B.J.; Ackerman, M.J.; Funk, M.; Gibler, W.B.; Kligfield, P.; Menon, V.; Philippides, G.J.; Roden, D.M.; Zareba, W.; American Heart Association Acute Cardiac Care Committee of the Council on Clinical Cardiology; et al. Prevention of torsade de pointes in hospital settings: A scientific statement from the American Heart Association and the American College of Cardiology Foundation. J. Am. Coll. Cardiol. 2010, 55, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Rijnbeek, P.R.; van Herpen, G.; Bots, M.L.; Man, S.; Verweij, N.; Hofman, A.; Hillege, H.; Numans, M.E.; Swenne, C.A.; Witteman, J.C.; et al. Normal values of the electrocardiogram for ages 16-90 years. J. Electrocardiol. 2014, 47, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.W.; Ramseth, D.J.; Chanter, D.O.; Moon, T.E.; Goodman, D.B.; Mendzelevski, B. Electrocardiographic reference ranges derived from 79,743 ambulatory subjects. J. Electrocardiol. 2007, 40, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Olbertz, J.; Lester, R.M.; Combs, M. Establishing Normal Ranges for ECG Intervals in a Normal Healthy Population. Available online: https://www.celerion.com/wp-content/uploads/2015/06/Celerion_DIA-2015_Establishing-Normal-Ranges-for-ECG-Intervals-in-a-Normal-Healthy-Population.pdf (accessed on 3 December,2018).
- Ackerman, M. Long QTs, Brugada, CPVT: From Genetics to Clinical Practice. In Proceedings of the Arrhythmias and the Heart Symposium, Kauai, HA, USA, 28 February 2018. [Google Scholar]
- Postema, P.G.; Wilde, A.A. The measurement of the QT interval. Curr. Cardiol. Rev. 2014, 10, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Lester, R.M.; Olbertz, J. Early drug development: Assessment of proarrhythmic risk and cardiovascular safety. Expert Rev. Clin. Pharmacol. 2016, 9, 1611–1618. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.K.; Soliman, E.Z. ECG abnormalities and stroke incidence. Expert Rev. Cardiovasc. Ther. 2013, 11, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Salvi, V.; Karnad, D.R.; Kerkar, V.; Panicker, G.K.; Manohar, D.; Natekar, M.; Kothari, S.; Narula, D.; Lokhandwala, Y. Choice of an alternative lead for QT interval measurement in serial ECGs when Lead II is not suitable for analysis. Indian Heart J. 2012, 64, 535–540. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Molnar, J.; Zhang, F.; Weiss, J.; Ehlert, F.A.; Rosenthal, J.E. Diurnal pattern of QTc interval: How long is prolonged? Possible relation to circadian triggers of cardiovascular events. J. Am. Coll. Cardiol. 1996, 27, 76–83. [Google Scholar] [CrossRef]
- Viskin, S.; Rosovski, U.; Sands, A.J.; Chen, E.; Kistler, P.M.; Kalman, J.M.; Rodriguez Chavez, L.; Iturralde Torres, P.; Cruz, F.F.; Centurion, O.A.; et al. Inaccurate electrocardiographic interpretation of long QT: The majority of physicians cannot recognize a long QT when they see one. Heart Rhythm 2005, 2, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Lester, R.M.; Azzam, S.M.; Erskine, C.; Clark, K.; Olbertz, J. Triplicate ECGs are Sufficient in Obtaining Precise Estimates of QTcF. Available online: https://www.celerion.com/wp-content/uploads/2016/03/Celerion_2016-ASCPT_Triplicate-ECGs-are-Sufficient-in-Obtaining-Precise-Estimates-of-QTcF.pdf (accessed on 3 December 2018).
- Natekar, M.; Hingorani, P.; Gupta, P.; Karnad, D.R.; Kothari, S.; de Vries, M.; Zumbrunnen, T.; Narula, D. Effect of number of replicate electrocardiograms recorded at each time point in a thorough QT study on sample size and study cost. J. Clin. Pharmacol. 2011, 51, 908–914. [Google Scholar] [CrossRef] [PubMed]
- Darpo, B. The Expert Precision QT Approach: Driving Earlier Assessments of Cardiac Safety and Supporting Regulatory Change. Available online: https://www.ert.com/wp-content/uploads/2018/09/WhitePaper_EPQT_030518.pdf (accessed on 3 December 2018).
- Hopenfeld, B.; Ashikaga, H. Origin of the electrocardiographic U wave: Effects of M cells and dynamic gap junction coupling. Ann. Biomed. Eng. 2010, 38, 1060–1070. [Google Scholar] [CrossRef] [PubMed]
- Lepeschkin, E.; Surawicz, B. The measurement of the Q-T interval of the electrocardiogram. Circulation 1952, 6, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Fossa, A.A. Beat-to-beat ECG restitution: A review and proposal for a new biomarker to assess cardiac stress and ventricular tachyarrhythmia vulnerability. Ann. Noninvasive Electrocardiol. 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Page, A.; McNitt, S.; Xia, X.; Zareba, W.; Couderc, J.P. Population-based beat-to-beat QT analysis from Holter recordings in the long QT syndrome. J. Electrocardiol. 2017, 50, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Lux, R.L.; Sower, C.T.; Allen, N.; Etheridge, S.P.; Tristani-Firouzi, M.; Saarel, E.V. The application of root mean square electrocardiography (RMS ECG) for the detection of acquired and congenital long QT syndrome. PLoS ONE 2014, 9, e85689. [Google Scholar] [CrossRef] [PubMed]
- Lux, R.L.; Fuller, M.S.; MacLeod, R.S.; Ershler, P.R.; Punske, B.B.; Taccardi, B. Noninvasive indices of repolarization and its dispersion. J. Electrocardiol. 1999, 32 (Suppl. 1), 153–157. [Google Scholar] [CrossRef]
- Shah, R.R. The significance of QT interval in drug development. Br. J. Clin. Pharmacol. 2002, 54, 188–202. [Google Scholar] [CrossRef] [PubMed]
- Zeltser, D.; Justo, D.; Halkin, A.; Prokhorov, V.; Heller, K.; Viskin, S. Torsade de pointes due to noncardiac drugs: Most patients have easily identifiable risk factors. Medicine (Baltimore) 2003, 82, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Khan, Q.; Ismail, M.; Khan, S. Frequency, characteristics and risk factors of QT interval prolonging drugs and drug-drug interactions in cancer patients: A multicenter study. BMC Pharmacol. Toxicol. 2017, 18, 75. [Google Scholar] [CrossRef] [PubMed]
- Roden, D.M.; Abraham, R.L. Refining repolarization reserve. Heart Rhythm 2011, 8, 1756–1757. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Ackerman, M.J. The long QT syndrome: A transatlantic clinical approach to diagnosis and therapy. Eur. Heart J. 2013, 34, 3109–3116. [Google Scholar] [CrossRef] [PubMed]
- Brenyo, A.J.; Huang, D.T.; Aktas, M.K. Congenital long and short QT syndromes. Cardiology 2012, 122, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Wilde, A.A.; Horie, M.; Cho, Y.; Behr, E.R.; Berul, C.; Blom, N.; Brugada, J.; Chiang, C.E.; Huikuri, H.; et al. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Europace 2013, 15, 1389–1406. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, M. Channelopathies and Cardiomyopathies: Indications and Pitfalls of Genetic Testing. In Proceedings of the Arrhythmias and the Heart, Kauai, HA, USA, 28 February 2018. [Google Scholar]
- Gussak, I.; Brugada, P.; Brugada, J.; Wright, R.S.; Kopecky, S.L.; Chaitman, B.R.; Bjerregaard, P. Idiopathic short QT interval: A new clinical syndrome? Cardiology 2000, 94, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Rudic, B.; Schimpf, R.; Borggrefe, M. Short QT Syndrome—Review of Diagnosis and Treatment. Arrhythm. Electrophysiol. Rev. 2014, 3, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Dhutia, H.; Malhotra, A.; Parpia, S.; Gabus, V.; Finocchiaro, G.; Mellor, G.; Merghani, A.; Millar, L.; Narain, R.; Sheikh, N.; et al. The prevalence and significance of a short QT interval in 18,825 low-risk individuals including athletes. Br. J. Sports Med. 2016, 50, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.; Yan, G.X.; Antzelevitch, C. Short QT syndrome: From bench to bedside. Circ. Arrhythm. Electrophysiol. 2010, 3, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Comelli, I.; Lippi, G.; Mossini, G.; Gonzi, G.; Meschi, T.; Borghi, L.; Cervellin, G. The dark side of the QT interval. The Short QT Syndrome: Pathophysiology, clinical presentation and management. Emerg. Care J. 2012, 8, 6. [Google Scholar] [CrossRef][Green Version]
- Gollob, M.H.; Redpath, C.J.; Roberts, J.D. The short QT syndrome: Proposed diagnostic criteria. J. Am. Coll. Cardiol. 2011, 57, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Gintant, G.A.; Bi, D.; Kozeli, D.; Pettit, S.D.; Pierson, J.B.; Skinner, M.; Willard, J.; Wisialowski, T.; Koerner, J.; et al. Can non-clinical repolarization assays predict the results of clinical thorough QT studies? Results from a research consortium. Br. J. Pharmacol. 2018, 175, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Kleiman, R.B.; Shah, R.R.; Morganroth, J. Replacing the thorough QT study: Reflections of a baby in the bath water. Br. J. Clin. Pharmacol. 2014, 78, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.R. Drug-induced QT interval prolongation: Does ethnicity of the thorough QT study population matter? Br. J. Clin. Pharmacol. 2013, 75, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Collection of Race and Ethnicity Data in Clinical Trials. Available online: https://www.fda.gov/downloads/regulatoryinformation/guidances/ucm126396.pdf (accessed on 3 December 2018).
- Bibbins-Domingo, K.; Fernandez, A. BiDil for heart failure in black patients: Implications of the U.S. Food and Drug Administration approval. Ann. Intern. Med. 2007, 146, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Sager, P.T.; Gintant, G.; Turner, J.R.; Pettit, S.; Stockbridge, N. Rechanneling the cardiac proarrhythmia safety paradigm: A meeting report from the Cardiac Safety Research Consortium. Am. Heart J. 2014, 167, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Vicente, J.; Johannesen, L.; Galeotti, L.; Strauss, D.G. Mechanisms of sex and age differences in ventricular repolarization in humans. Am. Heart J. 2014, 168, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Vicente, J.; Johannesen, L.; Hosseini, M.; Mason, J.W.; Sager, P.T.; Pueyo, E.; Strauss, D.G. Electrocardiographic Biomarkers for Detection of Drug-Induced Late Sodium Current Block. PLoS ONE 2016, 11, e0163619. [Google Scholar] [CrossRef] [PubMed]
- Johannesen, L.; Vicente, J.; Mason, J.W.; Sanabria, C.; Waite-Labott, K.; Hong, M.; Guo, P.; Lin, J.; Sorensen, J.S.; Galeotti, L.; et al. Differentiating drug-induced multichannel block on the electrocardiogram: Randomized study of dofetilide, quinidine, ranolazine, and verapamil. Clin. Pharmacol. Ther. 2014, 96, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Johannesen, L.; Vicente, J.; Mason, J.W.; Erato, C.; Sanabria, C.; Waite-Labott, K.; Hong, M.; Lin, J.; Guo, P.; Mutlib, A.; et al. Late sodium current block for drug-induced long QT syndrome: Results from a prospective clinical trial. Clin. Pharmacol. Ther. 2016, 99, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Vicente, J. New ECG Biomarkers and their Potential Role in CiPA: Results and Implications. Available online: http://www.cardiac-safety.org/wp-content/uploads/2018/05/08-Jose-Ruiz-Vicente-CSRC-CiPA-ECG-component-May-21-2018.pdf (accessed on 3 December 2018).
- Badilini, F.; Libretti, G.; Vaglio, M. Automated JTpeak analysis by BRAVO. J. Electrocardiol. 2017, 50, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Couderc, J.P.; Ma, S.; Page, A.; Besaw, C.; Xia, J.; Chiu, W.B.; de Bie, J.; Vicente, J.; Vaglio, M.; Badilini, F.; et al. An evaluation of multiple algorithms for the measurement of the heart rate corrected JTpeak interval. J. Electrocardiol. 2017, 50, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Marathe, D. Recent Insights from the FDA QT-IRT on Concentration-QTc Analysis and Requirements for TQT Study (‘waiver’) Substitution. Available online: http://www.cardiac-safety.org/wp-content/uploads/2018/05/01-Dhananjay_Marathe_-CSRC_May2018_Final.pdf (accessed on 3 December 2018).
- Strauss, D. The Potential Role of CiPA on Drug Discovery, Development, and Regulatory Pathways. Available online: http://www.cardiac-safety.org/wp-content/uploads/2018/05/03-David-Strauss-Talk-1-CiPA-Potential-Role-CSRC-5-20-2018-v2.pdf (accessed on 3 December 2018).
- Garnett, C.; Bonate, P.L.; Dang, Q.; Ferber, G.; Huang, D.; Liu, J.; Mehrotra, D.; Riley, S.; Sager, P.; Tornoe, C.; et al. Scientific white paper on concentration-QTc modeling. J. Pharmacokinet. Pharmacodyn. 2018, 45, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Grenier, J.; Paglialunga, S.; Morimoto, B.H.; Lester, R.M. Evaluating cardiac risk: Exposure response analysis in early clinical drug development. Drug Healthc. Patient Saf. 2018, 10, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Garnett, C.E.; Beasley, N.; Bhattaram, V.A.; Jadhav, P.R.; Madabushi, R.; Stockbridge, N.; Tornoe, C.W.; Wang, Y.; Zhu, H.; Gobburu, J.V. Concentration-QT relationships play a key role in the evaluation of proarrhythmic risk during regulatory review. J. Clin. Pharmacol. 2008, 48, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Darpo, B.; Garnett, C.; Keirns, J.; Stockbridge, N. Implications of the IQ-CSRC Prospective Study: Time to Revise ICH E14. Drug Saf. 2015, 38, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Ferber, G.; Zhou, M.; Dota, C.; Garnett, C.; Keirns, J.; Malik, M.; Stockbridge, N.; Darpo, B. Can Bias Evaluation Provide Protection Against False-Negative Results in QT Studies Without a Positive Control Using Exposure-Response Analysis? J. Clin. Pharmacol. 2017, 57, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Darpo, B.; Sarapa, N.; Garnett, C.; Benson, C.; Dota, C.; Ferber, G.; Jarugula, V.; Johannesen, L.; Keirns, J.; Krudys, K.; et al. The IQ-CSRC prospective clinical Phase 1 study: “Can early QT assessment using exposure response analysis replace the thorough QT study?”. Ann. Noninvasive Electrocardiol. 2014, 19, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Ferber, G.; Fernandes, S.; Taubel, J. Estimation of the Power of the Food Effect on QTc to Show Assay Sensitivity. J. Clin. Pharmacol. 2018, 58, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, B. QTcF postural changes as positive control for TQT studies: Eliminating the moxifloxacin group. In Proceedings of the Drug Information Association (DIA), Chicago, IL, USA, 11–13 June 2011. [Google Scholar]
- Marathe, D. Regulatroy perspective for using C-QTc as the primary analysis: Trial design, ECG quality evaluation, evaluation of modeling/simulation results, and decision making. In Proceedings of the Cardiac Safety Research Consortium Meeting, Washington, DC, USA, 6 April 2016. [Google Scholar]
- Choo, W.K.; Turpie, D.; Milne, K.; Davidson, L.; Elofuke, P.; Whitfield, J.; Broadhurst, P. Prescribers’ practice of assessing arrhythmia risk with QT-prolonging medications. Cardiovasc. Ther. 2014, 32, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Broszko, M.; Stanciu, C.N. Survey of EKG Monitoring Practices: A Necessity or Prolonged Nuisance? Available online: htttp://doi.org/10.1176/appi.ajp-rj.2017.120303 (accessed on 18 December 2018).
- Curtis, L.H.; Ostbye, T.; Sendersky, V.; Hutchison, S.; Allen LaPointe, N.M.; Al-Khatib, S.M.; Usdin Yasuda, S.; Dans, P.E.; Wright, A.; Califf, R.M.; et al. Prescription of QT-prolonging drugs in a cohort of about 5 million outpatients. Am. J. Med. 2003, 114, 135–141. [Google Scholar] [CrossRef]
- Tisdale, J.E.; Jaynes, H.A.; Kingery, J.R.; Overholser, B.R.; Mourad, N.A.; Trujillo, T.N.; Kovacs, R.J. Effectiveness of a clinical decision support system for reducing the risk of QT interval prolongation in hospitalized patients. Circ. Cardiovasc. Qual. Outcomes 2014, 7, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Malik, M. Drug-Induced QT/QTc Interval Shortening: Lessons from Drug-Induced QT/QTc Prolongation. Drug Saf. 2016, 39, 647–659. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula Name | Equation | Reference |
|---|---|---|
| Bazett | QTcB = QT/RR1/2 | [20] |
| Fridericia | QTcFri = QT/RR1/3 | [21] |
| Framingham | QTcFra = QT + 0.154 (1 − RR) | [22] |
| Hodges | QTcH = QT + 0.00175 ([60/RR] − 60) | [23] |
| Rautaharju | QTcR = QT − 0.185 (RR − 1) + k (k = + 0.006 s for men and + 0 s for women) | [24] |
| Individual | QTci = QTi/RRibi multiple mathematical formulae have been proposed (see below) | |
| Dmitrienko | QTcDMT: mixed effects modeling formula | [25] |
| Population based | QTcP = QT/RRb off treatment baseline ECGs | |
| Van de Water | QTc = QT − 0.087{(60/HR) − 1} | [26] |
| Other | Cross validated spline correction factor which is independent of HR | [27] |
| Drug | Therapeutic Class | Year Withdrawn from Market |
|---|---|---|
| Prenylamine | Angina | 1988 |
| Terodiline | Urinary Incontinence | 1991 |
| Sparfloxacin | Antibiotic | 1996 |
| Terfenadine | Antihistamine | 1998 |
| Sertindole | Antipsychotic | 1998 |
| Astemizole | Antihistamine | 1999 |
| Grepafloxacin | Antibiotic | 1999 |
| Cisapride | Prokinetic | 2000 |
| Droperidol | Antipsychotic | 2001 |
| Levomethadyl | Opiate Dependence | 2003 |
| Propoxyphene | Analgesic | 2015 |
| Risk Factor |
|---|
| QTc > 500 ms |
| Use of QT prolonging drug(s) |
| Abnormal repolarization morphology on ECG: notching of T waves, long Tpeak-Tend |
| Underlying heart disease: heart failure or myocardial infarction |
| Female gender |
| Hypokalemia |
| Hypomagnesemia |
| Hypocalcemia |
| Hypothyroidism |
| Advanced age |
| Bradycardia |
| Premature contractions producing short-long-short cycles |
| Impaired hepatic clearance of drugs |
| Diuretic use |
| Renal failure |
| Latent congenital LQTS polymorphisms |
| Abnormal repolarization reserve |
| Combinations of 2 or more risk factors |
| Category. | Criteria | Score |
|---|---|---|
| Electrocardiogram | QTcB interval: | |
| ≥480 ms | 3 | |
| 460–479 ms | 2 | |
| 450–459 (male) ms | 1 | |
| QTcB 4th minute of recovery from exercise stress test ≥480 ms | 1 | |
| Torsade de Pointes | 2 | |
| T-wave alternans | 1 | |
| Notched T-wave in three leads | 1 | |
| Low heart rate for age (below the 2nd percentile) | 0.5 | |
| Clinical History | Syncope: | |
| With stressful activity | 2 | |
| Without stressful activity | 1 | |
| Congenital deafness | 0.5 | |
| Family History | Family members with definite LQTS | 1 |
| Unexplained sudden cardiac death below age 30 among immediate family members | 0.5 | |
| Diagnosis | Probability of LQTS | Sum of Score |
| Low | ≤1 | |
| Intermediate | 1.5 to 3 | |
| High | ≥3.5 |
| SQTS Subtype. | Gene Name | Protein Name | Function | SQTS Mechanism |
|---|---|---|---|---|
| SQT-1 | KCNH2 | Kv11.1 | α-subunit IKr | Gain-of-function |
| SQT-2 | KCNQ1 | Kv7.1 | α-subunit IKs | Gain-of-function |
| SQT-3 | KCNJ1 | Kir2.1 | α-subunit IK1 | Gain-of-function |
| SQT-4 | CACNA1C | Cav1.2 | α-subunit IL,Ca | Loss-of-function |
| SQT-5 | CACNB2 | Cavβ2 | β2-subunit IL,Ca | Loss-of-function |
| SQT-6 | CACNA2D1 | Cavδ1 | δ1-subunit IL,Ca | Loss-of-function |
| Category | Criteria | Score |
|---|---|---|
| Electrocardiogram | QTc interval: | |
| <370 ms | 1 | |
| <350 ms | 2 | |
| <330 ms | 3 | |
| Jpoint-Tpeak interval <120 ms | 1 | |
| Clinical History | History of sudden cardiac arrest | 2 |
| Documented polymorphic VT or VF | 2 | |
| Unexplained syncope | 1 | |
| Atrial fibrillation | 1 | |
| Family History | Family member with high-probability SQTS | 2 |
| Family member with autopsy-negative sudden cardiac death | 1 | |
| Sudden infant death syndrome | 1 | |
| Genotype | Genotype positive | 2 |
| Mutation of undetermined significance in a culprit gene | 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lester, R.M.; Paglialunga, S.; Johnson, I.A. QT Assessment in Early Drug Development: The Long and the Short of It. Int. J. Mol. Sci. 2019, 20, 1324. https://doi.org/10.3390/ijms20061324
Lester RM, Paglialunga S, Johnson IA. QT Assessment in Early Drug Development: The Long and the Short of It. International Journal of Molecular Sciences. 2019; 20(6):1324. https://doi.org/10.3390/ijms20061324
Chicago/Turabian StyleLester, Robert M., Sabina Paglialunga, and Ian A. Johnson. 2019. "QT Assessment in Early Drug Development: The Long and the Short of It" International Journal of Molecular Sciences 20, no. 6: 1324. https://doi.org/10.3390/ijms20061324
APA StyleLester, R. M., Paglialunga, S., & Johnson, I. A. (2019). QT Assessment in Early Drug Development: The Long and the Short of It. International Journal of Molecular Sciences, 20(6), 1324. https://doi.org/10.3390/ijms20061324