Primary Cilium in Cancer Hallmarks



Abstract
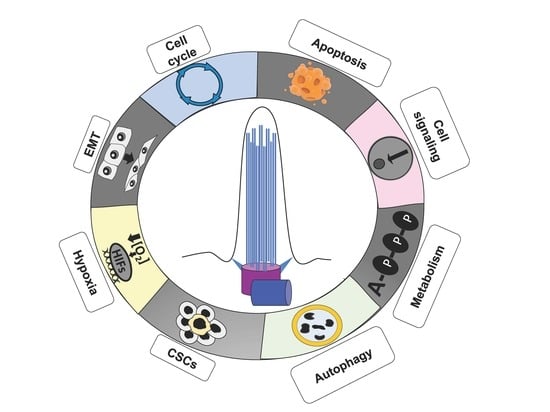
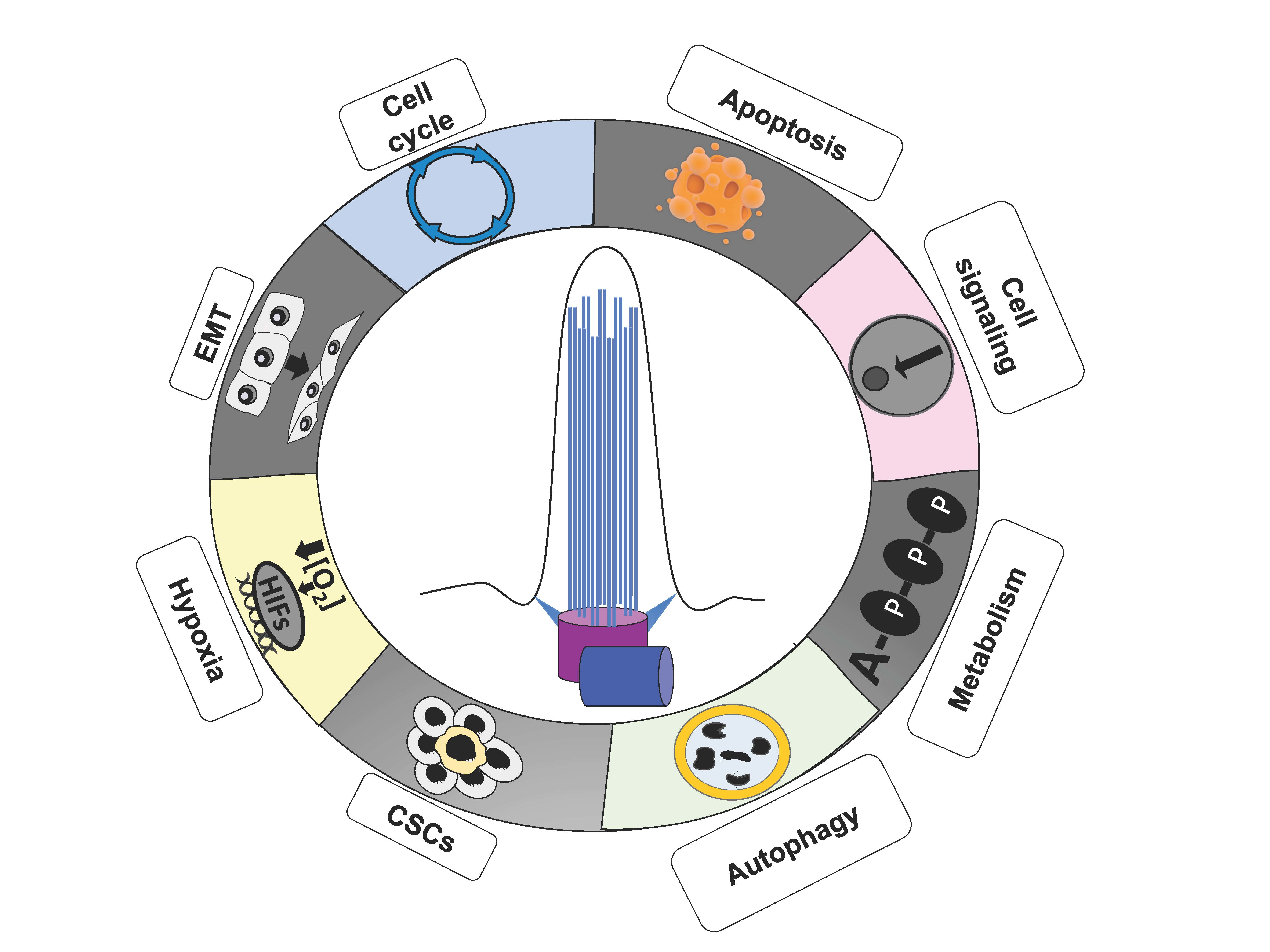{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Ciliogenesis as a Timeout for Cell Cycle Progression
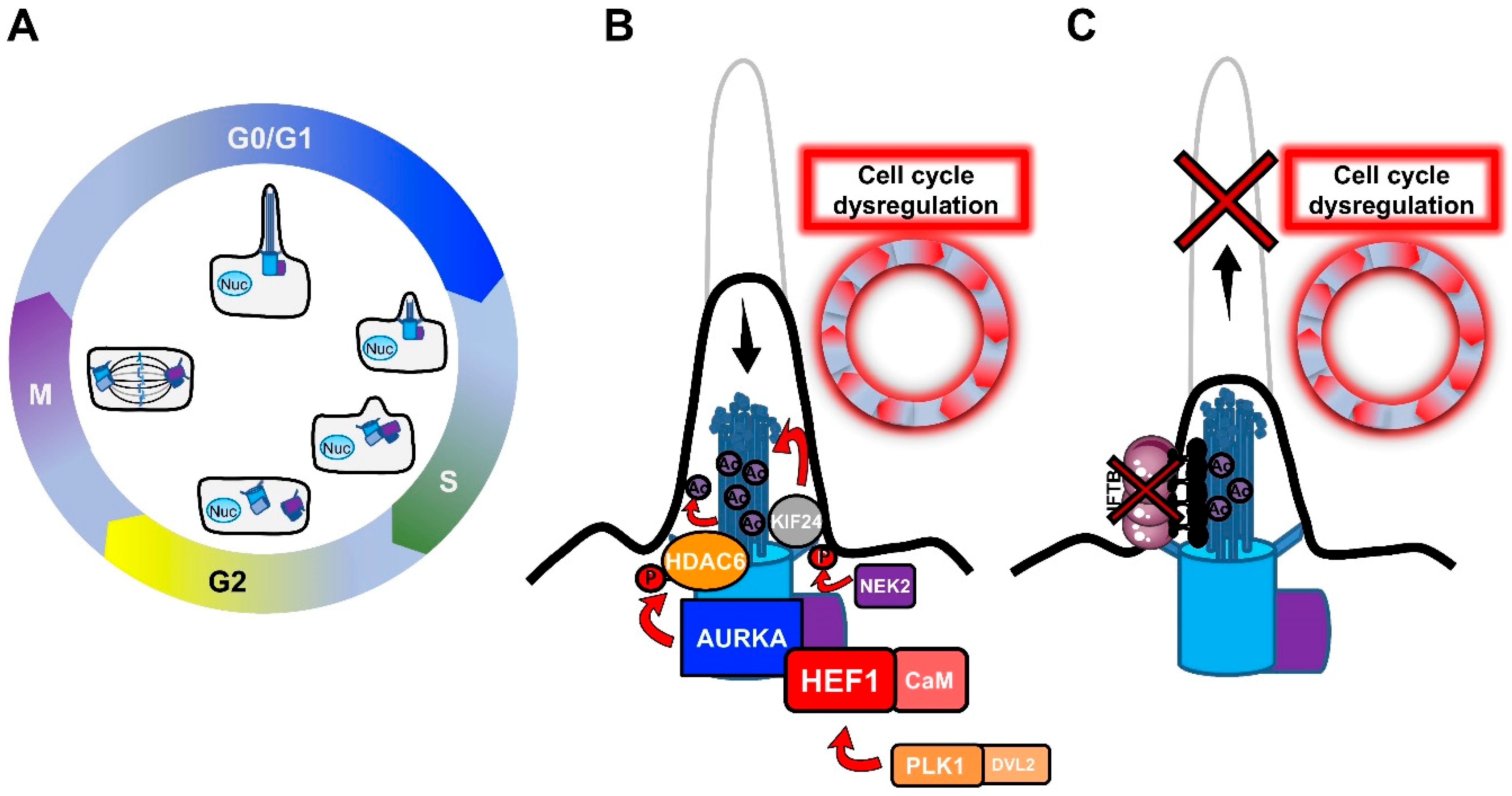
2.1. Primary Cilia and the Cell Cycle
- PLK1 is a mitotic kinase that regulates progression through the cell cycle by phosphorylating serine/threonine proteins on centrosomes, kinetochores, the mitotic spindle, and the midbody [49]. Activation of the non-canonical Wnt pathway induces the formation of the Plk1- disheveled segment polarity protein 2 (Dvl2) complex, which activates AurkA through the stabilization of Human Enhancer of Filamentation 1 (HEF1), thus inducing cilium disassembly [50].
- AURKA is a centrosomal mitotic kinase that regulates S phase entry. After mitosis, it localizes to the basal body, and is activated by the scaffold protein HEF1 and calmodulin (CaM) in the presence of calcium [51]. The HEF1–Ca2+/CaM–AURKA complex in turn activates the tubulin deacetylase histone deacetylase 6 (HDAC6), which destabilizes axonemal microtubules, inducing cilium disassembly [52].AURKA is the point of convergence between these two pathways, and cilium disassembly occurs downstream of its activation. AURKA was found to be upregulated in non-ciliated ovarian and clear cell renal cell carcinoma cancer cells [44,45], and HDAC6 inhibition restored primary cilia in chondrosarcoma and cholangiocarcinoma cancer cells, suppressing cell proliferation and their invasion capacity [53,54]. Similarly, HEF1 overexpression was associated with the metastasis of breast cancer and melanoma [55,56].
- NEK2 is another important regulator of both centrosome and basal body [57]. NEK2 exerts its role in the disassembly of the axonemal microtubules by phosphorylating the Kinesin Family Member 24 (KIF24), a member of the kinesin superfamily of microtubule-based motor proteins, which stimulates its microtubule-depolymerizing activity and prevents the formation of cilia in proliferating cells [58]. NEK2 and KIF24 were found to be overexpressed in breast cancer cells, and ablation of these proteins restored ciliation, thereby reducing proliferation.
2.2. Intraflagellar Transport (IFT)
3. Primary Cilium as a Mediator of Signaling Pathways
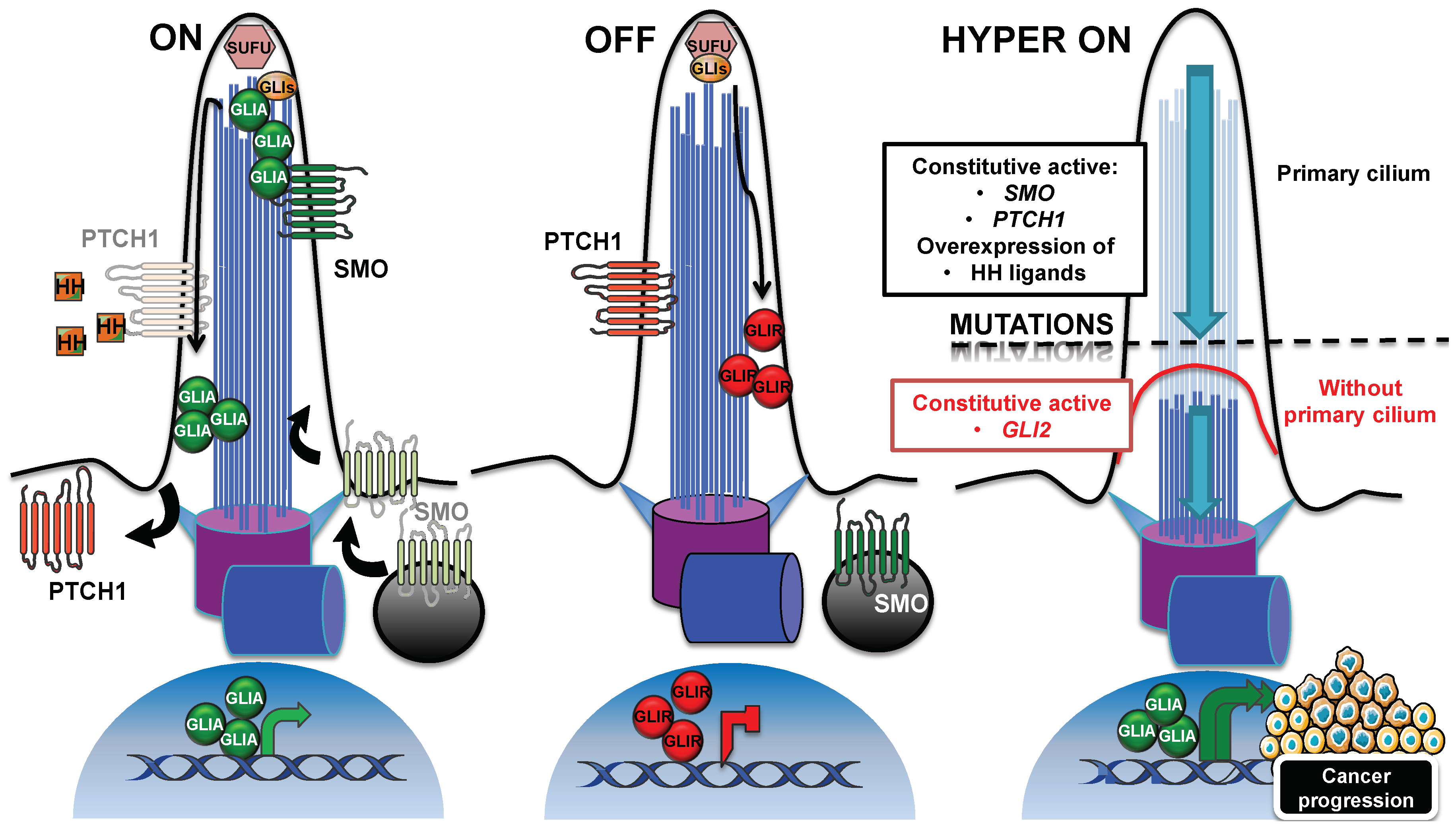
3.1. Hedgehog Signaling Pathway
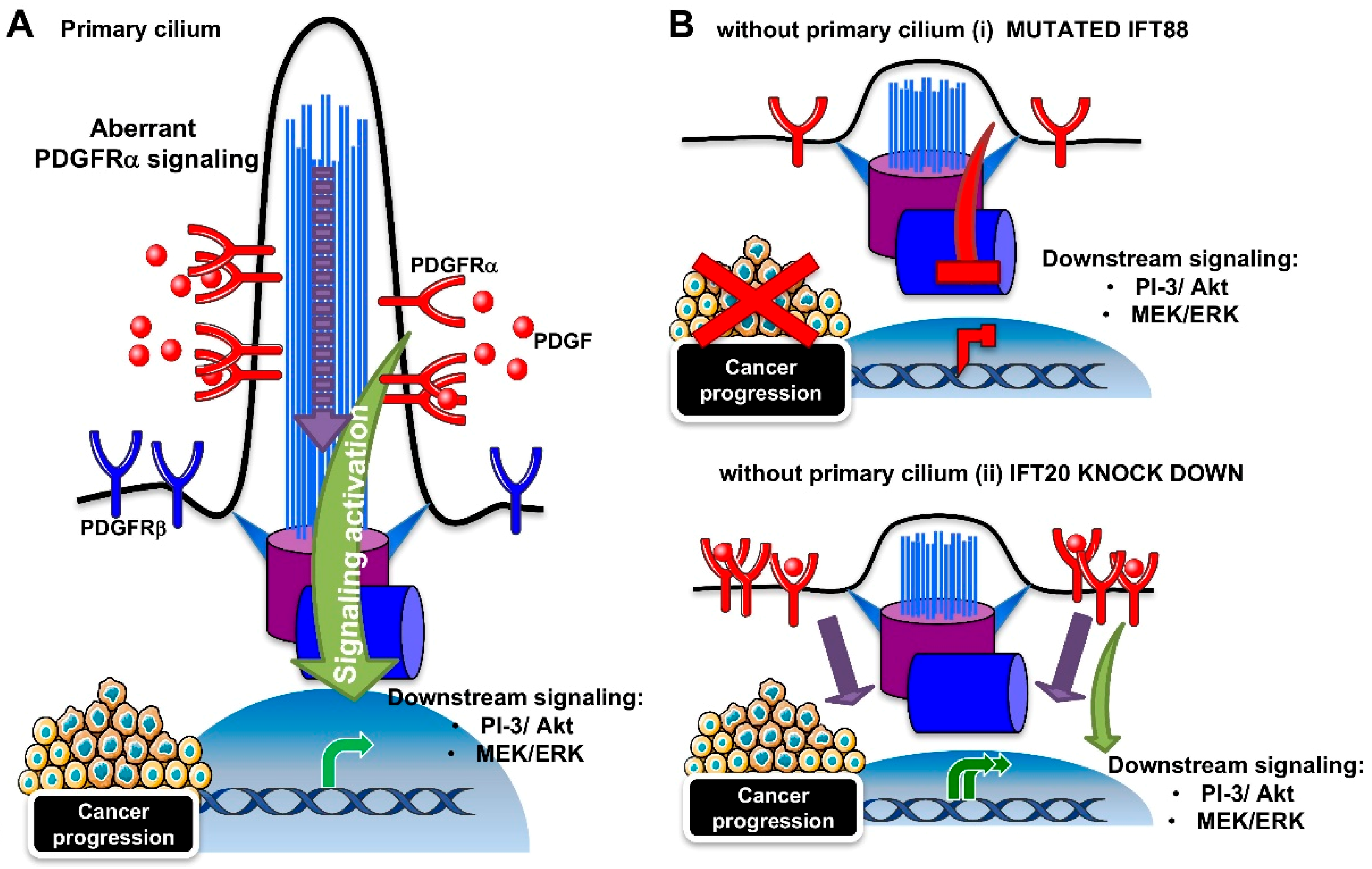
3.2. PDGFRα Signaling Pathway
3.3. Wnt Signaling Pathway
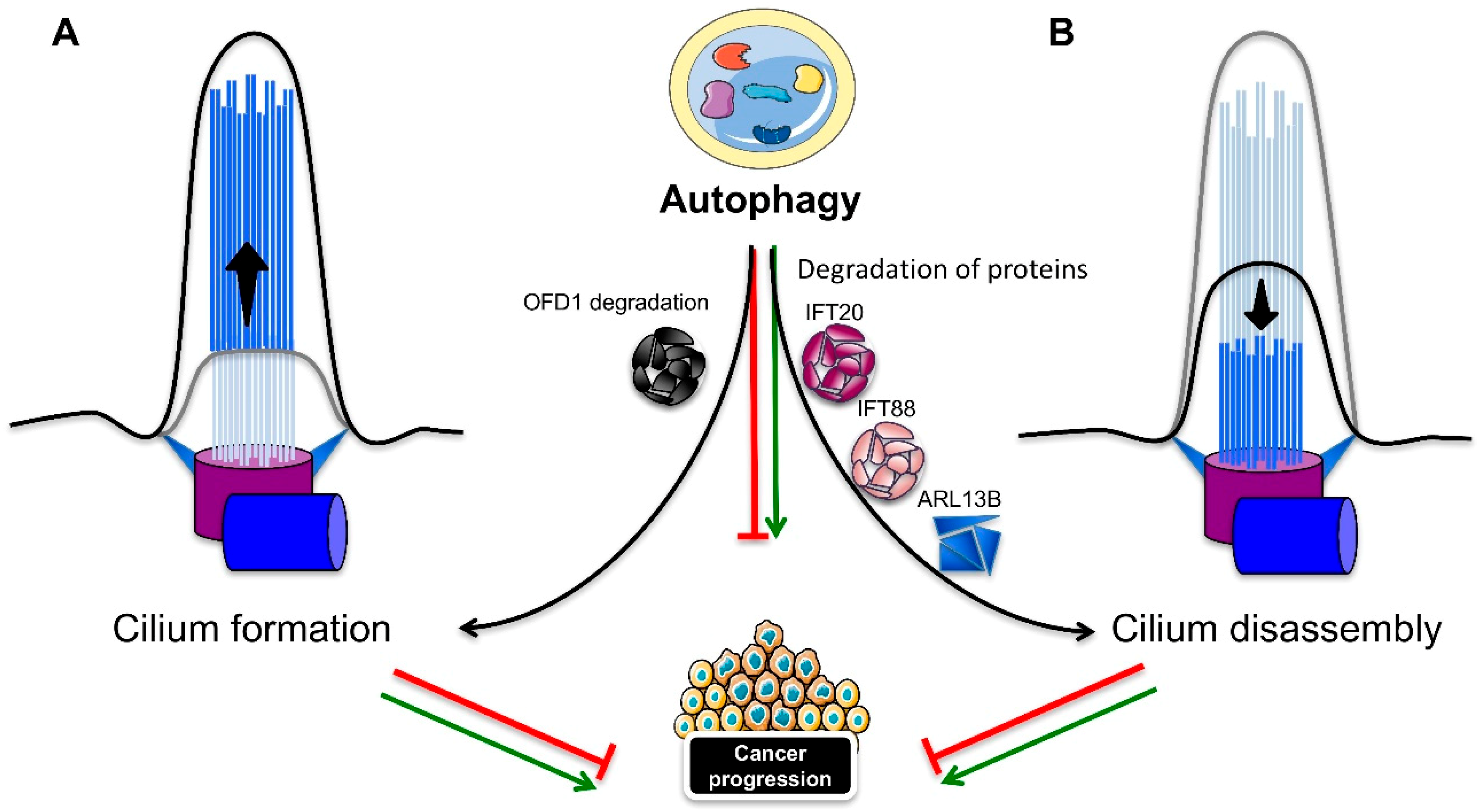
4. Primary Cilium and Autophagy
4.1. The Primary Cilium Regulates Autophagy
4.2. Autophagy Regulates Ciliogenesis
4.3. The Cilia–Autophagy Axis in Cancer Development
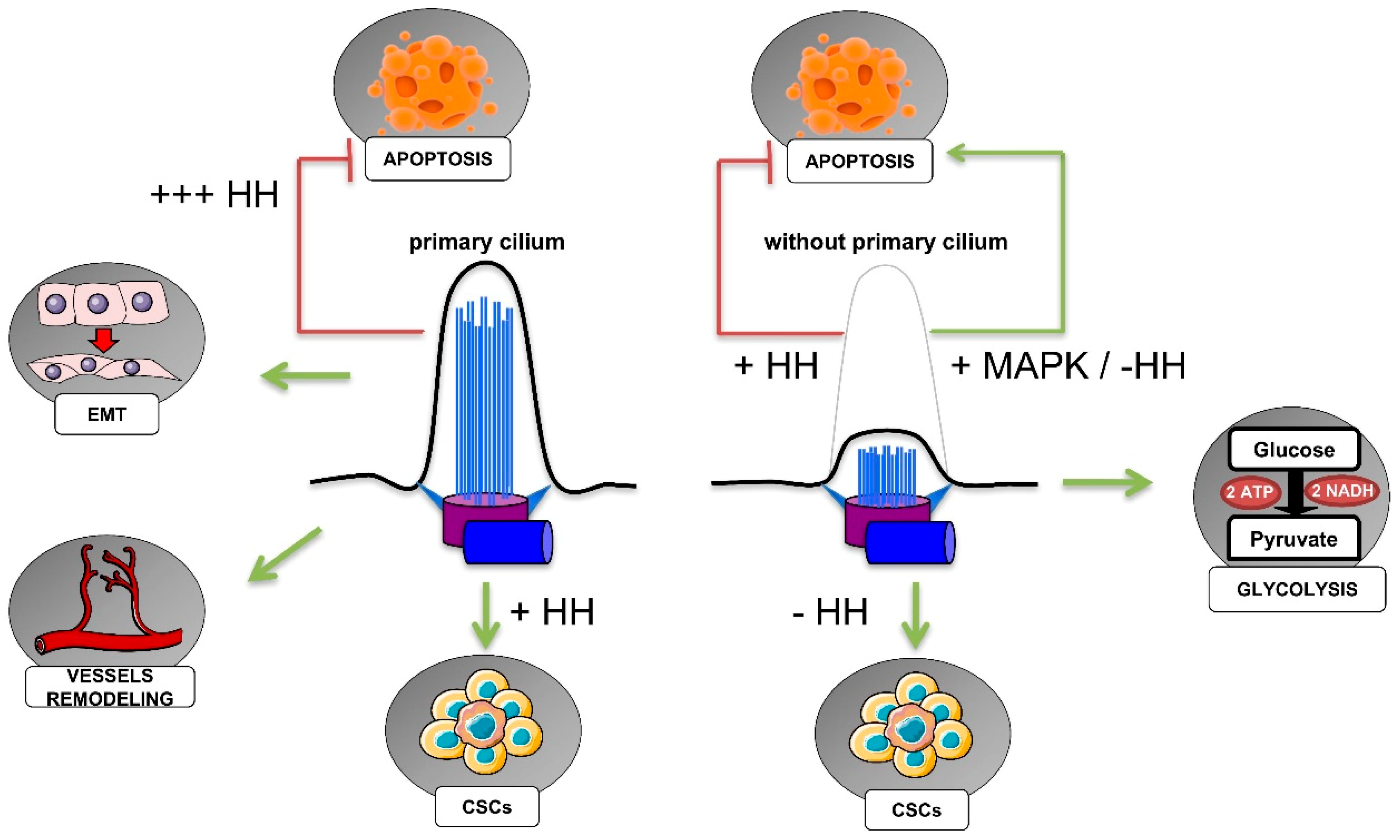
5. Primary Cilium, Hypoxia, and Cancer Hallmarks
5.1. HIF-Dependent Regulation of Ciliogenesis in Cancer
5.2. Primary Cilia in the Regulation of Cancer Cell Metabolism
5.3. Primary Cilia, Cancer Stem Cells, and the Epithelial–Mesenchymal Transition
5.4. Primary Cilia in Cell Death Resistance
5.5. Primary Cilia in Angiogenesis
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bloodgood, R.A. From central to rudimentary to primary: The history of an underappreciated organelle whose time has come. The primary cilium. Methods Cell Biol. 2009, 94, 3–52. [Google Scholar] [PubMed]
- Langerhans, P. Zur Anatomie des Amphioxus. Arch. Mikrosk. Anat. 1876, 12, 290–348. [Google Scholar] [CrossRef]
- Zimmermann, K.W. Beitrage zur kenntniss einiger drusen und epithelien. Arch. Mikrosk. Anat. 1898, 52, 552–706. [Google Scholar] [CrossRef]
- Henneguy, L.F. Sur les rapports des cils vibratiles avec les centrosomes. Arch. Anat. Microsc. 1898, 1, 481–496. [Google Scholar]
- Lenhossek, M.V. Ueber flimmerzellen. Verh. Anat. Ges. 1898, 12, 106–128. [Google Scholar]
- Cowdry, E.V. Flagellated thyroid cells in the dogfish (MUSTELUS CANIS). J. Exp. Med. 1921, 22, 289–299. [Google Scholar] [CrossRef]
- Allen, R.A. Isolated cilia in inner retinal neurons and in retinal pigment epithelium. J. Ultrastruct. Res. 1965, 12, 730–747. [Google Scholar] [CrossRef]
- Barnes, B.G. Ciliated secretory cells in the pars distalis of the mouse hypophysis. J. Ultrastruct. Res. 1961, 5, 453–467. [Google Scholar] [CrossRef]
- Latta, H.; Maunsbach, A.B.; Madden, S.C. Cilia in different segments of the rat nephron. J. Biophys. Biochem. Cytol. 1961, 11, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, W.; de Harven, E. L’ultrastructure du centriole et d’autres éléments de l’appariel achromatique. In Proceedings of the 4th International Congress Electron Microscopy; Bargmann, W., Peters, D., Wolpers, C., Eds.; Springer: Berlin, Germany, 1960; Volume 2, pp. 217–227. [Google Scholar]
- Scherft, J.P.; Daems, W.T. Single cilia in chondrocytes. J. Ultrastruct. Res. 1967, 19, 546–555. [Google Scholar] [CrossRef]
- Sorokin, S. Centrioles and the formation of rudimentary cilia by fibroblasts and smooth muscle cells. J. Cell. Biol. 1962, 15, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, D.W.; Porter, K.R. A study of the fine structure of ciliated epithelia. J. Morphol. 1954, 94, 221–281. [Google Scholar] [CrossRef]
- De Harven, E.; Bernhard, W. Etude au microscope de l’ultrastructure du centriole chez les vertébrés. Z. Zellforsch. Mikrosk. Anat. 1956, 45, 378–398. [Google Scholar] [CrossRef]
- Sorokin, S.P. Reconstructions of centriole formation and ciliogenesis in mammalian lungs. J. Cell. Sci. 1968, 3, 207–230. [Google Scholar] [PubMed]
- Sorokin, S.P. Centriole formation and ciliogenesis. Aspen Emphysema Conf. 1968, 11, 213–216. [Google Scholar] [PubMed]
- Sjostrand, F.S. The ultrastructure of the innersegments of the retinal rods of the guinea pig eye as revealed by electron microscopy. J. Cell. Comp. Physiol. 1953, 42, 45–70. [Google Scholar] [CrossRef] [PubMed]
- Praetorius, H.A.; Spring, K.R. Bending the MDCK cell primary cilium increases intracellular calcium. J. Membr. Biol. 2001, 184, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Kozminski, K.G.; Johnson, K.A.; Forscher, P.; Rosenbaum, J.L. A motility in the eukaryotic flagellum unrelated to flagellar beating. Proc. Natl. Acad. Sci. USA 1993, 90, 5519–5523. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, J.L.; Witman, G.B. Intraflagellar transport. Nat. Rev. Mol. Cell Biol. 2002, 3, 813–825. [Google Scholar] [CrossRef]
- Pazour, G.J.; Dickert, B.L.; Vucica, Y.; Seeley, E.S.; Rosenbaum, J.L.; Witman, G.B.; Cole, D.G. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J. Cell. Biol. 2000, 151, 709–718. [Google Scholar] [CrossRef]
- Pazour, G.J.; San Agustin, J.T.; Follit, J.A.; Rosenbaum, J.L.; Witman, G.B. Polycystin-2 localizes to kidney cilia and the ciliary level is elevated in orpk mice with polycystic kidney disease. Curr. Biol. 2002, 12, R378–R380. [Google Scholar] [CrossRef]
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Beales, P.L. Ciliopathies: An expanding disease spectrum. Pediatr. Nephrol. 2011, 26, 1039–1056. [Google Scholar] [CrossRef]
- Singla, V.; Reiter, J.F. The primary cilium as the cell’s antenna: Signaling at a sensory organelle. Science 2006, 313, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Mick, D.U.; Rodrigues, R.B.; Leib, R.D.; Adams, C.M.; Chien, A.S.; Gygi, S.P.; Nachury, M.V. Proteomics of Primary Cilia by Proximity Labeling. Dev. Cell 2015, 35, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Corbit, K.C.; Aanstad, P.; Singla, V.; Norman, A.R.; Stainier, D.Y.; Reiter, J.F. Vertebrate Smoothened functions at the primary cilium. Nature 2005, 437, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L.; Clement, C.A.; Teilmann, S.C.; Pazour, G.J.; Hoffmann, E.K.; Satir, P.; Christensen, S.T. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr. Biol. 2005, 15, 1861–1866. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.J.; May-Simera, H.; Eichers, E.R.; Kai, M.; Hill, J.; Jagger, D.J.; Leitch, C.C.; Chapple, J.P.; Munro, P.M.; Fisher, S.; et al. Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat. Genet. 2005, 37, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Fonte, V.G.; Searls, R.L.; Hilfer, S.R. The relationship of cilia with cell division and differentiation. J. Cell. Biol. 1971, 49, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar] [PubMed]
- Archer, F.L.; Wheatley, D.N. Cilia in cell-cultured fibroblasts. II. Incidence in mitotic and post-mitotic BHK 21-C13 fibroblasts. J. Anat. 1971, 109 Pt 2, 277–292. [Google Scholar] [PubMed]
- Ho, P.T.; Tucker, R.W. Centriole ciliation and cell cycle variability during G1 phase of BALB/c 3T3 cells. J. Cell Physiol. 1989, 139, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Chretien, D.; Buendia, B.; Fuller, S.D.; Karsenti, E. Reconstruction of the centrosome cycle from cryoelectron micrographs. J. Struct. Biol. 1997, 120, 117–133. [Google Scholar] [CrossRef]
- Ibrahim, R.; Messaoudi, C.; Chichon, F.J.; Celati, C.; Marco, S. Electron tomography study of isolated human centrioles. Microsc. Res. Tech. 2009, 72, 42–48. [Google Scholar] [CrossRef]
- Ringo, D.L. Flagellar motion and fine structure of the flagellar apparatus in Chlamydomonas. J. Cell. Biol. 1967, 33, 543–571. [Google Scholar] [CrossRef] [PubMed]
- Marshall, W.F. Basal bodies platforms for building cilia. Curr. Top. Dev. Biol. 2008, 85, 1–22. [Google Scholar] [PubMed]
- Joshi, H.C.; Palacios, M.J.; McNamara, L.; Cleveland, D.W. Gamma-tubulin is a centrosomal protein required for cell cycle-dependent microtubule nucleation. Nature 1992, 356, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Paz, J.; Luders, J. Microtubule-Organizing Centers: Towards a Minimal Parts List. Trends Cell Biol. 2018, 28, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K. Cytoplasmic microtubules and their functions. Prog. Biophys. Mol. Biol. 1974, 28, 371–420. [Google Scholar] [CrossRef]
- Nigg, E.A.; Stearns, T. The centrosome cycle: Centriole biogenesis, duplication and inherent asymmetries. Nat. Cell Biol. 2011, 13, 1154–1160. [Google Scholar] [CrossRef]
- Paridaen, J.T.; Wilsch-Brauninger, M.; Huttner, W.B. Asymmetric inheritance of centrosome-associated primary cilium membrane directs ciliogenesis after cell division. Cell 2013, 155, 333–344. [Google Scholar] [CrossRef]
- Basten, S.G.; Willekers, S.; Vermaat, J.S.; Slaats, G.G.; Voest, E.E.; van Diest, P.J.; Giles, R.H. Reduced cilia frequencies in human renal cell carcinomas versus neighboring parenchymal tissue. Cilia 2013, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Dere, R.; Perkins, A.L.; Bawa-Khalfe, T.; Jonasch, D.; Walker, C.L. beta-catenin links von Hippel-Lindau to aurora kinase A and loss of primary cilia in renal cell carcinoma. J. Am. Soc. Nephrol. 2015, 26, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Egeberg, D.L.; Lethan, M.; Manguso, R.; Schneider, L.; Awan, A.; Jorgensen, T.S.; Byskov, A.G.; Pedersen, L.B.; Christensen, S.T. Primary cilia and aberrant cell signaling in epithelial ovarian cancer. Cilia 2012, 1, 15. [Google Scholar] [CrossRef] [PubMed]
- Hassounah, N.B.; Nagle, R.; Saboda, K.; Roe, D.J.; Dalkin, B.L.; McDermott, K.M. Primary cilia are lost in preinvasive and invasive prostate cancer. PLoS ONE 2013, 8, e68521. [Google Scholar] [CrossRef]
- Menzl, I.; Lebeau, L.; Pandey, R.; Hassounah, N.B.; Li, F.W.; Nagle, R.; Weihs, K.; McDermott, K.M. Loss of primary cilia occurs early in breast cancer development. Cilia 2014, 3, 7. [Google Scholar] [CrossRef]
- Seeley, E.S.; Carriere, C.; Goetze, T.; Longnecker, D.S.; Korc, M. Pancreatic cancer and precursor pancreatic intraepithelial neoplasia lesions are devoid of primary cilia. Cancer Res. 2009, 69, 422–430. [Google Scholar] [CrossRef]
- Kishi, K.; van Vugt, M.A.; Okamoto, K.; Hayashi, Y.; Yaffe, M.B. Functional dynamics of Polo-like kinase 1 at the centrosome. Mol. Cell Biol. 2009, 29, 3134–3150. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Johmura, Y.; Yu, L.R.; Park, J.E.; Gao, Y.; Bang, J.K.; Zhou, M.; Veenstra, T.D.; Yeon Kim, B.; Lee, K.S. Identification of a novel Wnt5a-CK1varepsilon-Dvl2-Plk1-mediated primary cilia disassembly pathway. EMBO J. 2012, 31, 3104–3117. [Google Scholar] [CrossRef] [PubMed]
- Pugacheva, E.N.; Jablonski, S.A.; Hartman, T.R.; Henske, E.P.; Golemis, E.A. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell 2007, 129, 1351–1363. [Google Scholar] [CrossRef]
- Plotnikova, O.V.; Nikonova, A.S.; Loskutov, Y.V.; Kozyulina, P.Y.; Pugacheva, E.N.; Golemis, E.A. Calmodulin activation of Aurora-A kinase (AURKA) is required during ciliary disassembly and in mitosis. Mol. Biol. Cell 2012, 23, 2658–2670. [Google Scholar] [CrossRef]
- Gradilone, S.A.; Radtke, B.N.; Bogert, P.S.; Huang, B.Q.; Gajdos, G.B.; LaRusso, N.F. HDAC6 inhibition restores ciliary expression and decreases tumor growth. Cancer Res. 2013, 73, 2259–2270. [Google Scholar] [CrossRef]
- Xiang, W.; Guo, F.; Cheng, W.; Zhang, J.; Huang, J.; Wang, R.; Ma, Z.; Xu, K. HDAC6 inhibition suppresses chondrosarcoma by restoring the expression of primary cilia. Oncol. Rep. 2017, 38, 229–236. [Google Scholar] [CrossRef]
- Minn, A.J.; Gupta, G.P.; Siegel, P.M.; Bos, P.D.; Shu, W.; Giri, D.D.; Viale, A.; Olshen, A.B.; Gerald, W.L.; Massague, J. Genes that mediate breast cancer metastasis to lung. Nature 2005, 436, 518–524. [Google Scholar] [CrossRef]
- Kim, M.; Gans, J.D.; Nogueira, C.; Wang, A.; Paik, J.H.; Feng, B.; Brennan, C.; Hahn, W.C.; Cordon-Cardo, C.; Wagner, S.N.; et al. Comparative oncogenomics identifies NEDD9 as a melanoma metastasis gene. Cell 2006, 125, 1269–1281. [Google Scholar] [CrossRef]
- Spalluto, C.; Wilson, D.I.; Hearn, T. Nek2 localises to the distal portion of the mother centriole/basal body and is required for timely cilium disassembly at the G2/M transition. Eur. J. Cell. Biol. 2012, 91, 675–686. [Google Scholar] [CrossRef]
- Kim, S.; Lee, K.; Choi, J.H.; Ringstad, N.; Dynlacht, B.D. Nek2 activation of Kif24 ensures cilium disassembly during the cell cycle. Nat. Commun. 2015, 6, 8087. [Google Scholar] [CrossRef]
- Cole, D.G.; Diener, D.R.; Himelblau, A.L.; Beech, P.L.; Fuster, J.C.; Rosenbaum, J.L. Chlamydomonas kinesin-II-dependent intraflagellar transport (IFT): IFT particles contain proteins required for ciliary assembly in Caenorhabditis elegans sensory neurons. J. Cell. Biol. 1998, 141, 993–1008. [Google Scholar] [CrossRef]
- Piperno, G.; Mead, K. Transport of a novel complex in the cytoplasmic matrix of Chlamydomonas flagella. Proc. Natl. Acad. Sci. USA 1997, 94, 4457–4462. [Google Scholar] [CrossRef]
- Iomini, C.; Li, L.; Esparza, J.M.; Dutcher, S.K. Retrograde intraflagellar transport mutants identify complex A proteins with multiple genetic interactions in Chlamydomonas reinhardtii. Genetics 2009, 183, 885–896. [Google Scholar] [CrossRef]
- Kozminski, K.G.; Beech, P.L.; Rosenbaum, J.L. The Chlamydomonas kinesin-like protein FLA10 is involved in motility associated with the flagellar membrane. J. Cell. Biol. 1995, 131 Pt 1, 1517–1527. [Google Scholar] [CrossRef]
- Pazour, G.J.; Dickert, B.L.; Witman, G.B. The DHC1b (DHC2) isoform of cytoplasmic dynein is required for flagellar assembly. J. Cell. Biol. 1999, 144, 473–481. [Google Scholar] [CrossRef]
- Piperno, G.; Siuda, E.; Henderson, S.; Segil, M.; Vaananen, H.; Sassaroli, M. Distinct mutants of retrograde intraflagellar transport (IFT) share similar morphological and molecular defects. J. Cell. Biol. 1998, 143, 1591–1601. [Google Scholar] [CrossRef]
- Porter, M.E.; Bower, R.; Knott, J.A.; Byrd, P.; Dentler, W. Cytoplasmic dynein heavy chain 1b is required for flagellar assembly in Chlamydomonas. Mol. Biol. Cell 1999, 10, 693–712. [Google Scholar] [CrossRef]
- Tsao, C.C.; Gorovsky, M.A. Different effects of Tetrahymena IFT172 domains on anterograde and retrograde intraflagellar transport. Mol. Biol. Cell 2008, 19, 1450–1461. [Google Scholar] [CrossRef]
- Stepanek, L.; Pigino, G. Microtubule doublets are double-track railways for intraflagellar transport trains. Science 2016, 352, 721–724. [Google Scholar] [CrossRef]
- Jordan, M.A.; Diener, D.R.; Stepanek, L.; Pigino, G. The cryo-EM structure of intraflagellar transport trains reveals how dynein is inactivated to ensure unidirectional anterograde movement in cilia. Nat. Cell Biol. 2018, 20, 1250–1255. [Google Scholar] [CrossRef]
- Johnson, K.A.; Rosenbaum, J.L. Polarity of flagellar assembly in Chlamydomonas. J. Cell. Biol. 1992, 119, 1605–1611. [Google Scholar] [CrossRef]
- Bhogaraju, S.; Cajanek, L.; Fort, C.; Blisnick, T.; Weber, K.; Taschner, M.; Mizuno, N.; Lamla, S.; Bastin, P.; Nigg, E.A.; et al. Molecular basis of tubulin transport within the cilium by IFT74 and IFT81. Science 2013, 341, 1009–1012. [Google Scholar] [CrossRef]
- Taschner, M.; Weber, K.; Mourao, A.; Vetter, M.; Awasthi, M.; Stiegler, M.; Bhogaraju, S.; Lorentzen, E. Intraflagellar transport proteins 172, 80, 57, 54, 38, and 20 form a stable tubulin-binding IFT-B2 complex. EMBO J. 2016, 35, 773–790. [Google Scholar] [CrossRef]
- Craft, J.M.; Harris, J.A.; Hyman, S.; Kner, P.; Lechtreck, K.F. Tubulin transport by IFT is upregulated during ciliary growth by a cilium-autonomous mechanism. J. Cell. Biol. 2015, 208, 223–237. [Google Scholar] [CrossRef]
- Qin, H.; Wang, Z.; Diener, D.; Rosenbaum, J. Intraflagellar transport protein 27 is a small G protein involved in cell-cycle control. Curr. Biol. 2007, 17, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Inoko, A.; Matsuyama, M.; Goto, H.; Ohmuro-Matsuyama, Y.; Hayashi, Y.; Enomoto, M.; Ibi, M.; Urano, T.; Yonemura, S.; Kiyono, T.; et al. Trichoplein and Aurora A block aberrant primary cilia assembly in proliferating cells. J. Cell. Biol. 2012, 197, 391–405. [Google Scholar] [CrossRef]
- Lin, F.; Hiesberger, T.; Cordes, K.; Sinclair, A.M.; Goldstein, L.S.; Somlo, S.; Igarashi, P. Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2003, 100, 5286–5291. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Takebe, N.; Lorusso, P. Targeting the Hedgehog pathway in cancer. Ther. Adv. Med. Oncol. 2010, 2, 237–250. [Google Scholar] [CrossRef]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef]
- Zhang, J.; Lipinski, R.J.; Gipp, J.J.; Shaw, A.K.; Bushman, W. Hedgehog pathway responsiveness correlates with the presence of primary cilia on prostate stromal cells. BMC Dev. Biol. 2009, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hsia, E.Y.; Brigui, A.; Plessis, A.; Beachy, P.A.; Zheng, X. The role of ciliary trafficking in Hedgehog receptor signaling. Sci. Signal 2015, 8, ra55. [Google Scholar] [CrossRef]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Humke, E.W.; Dorn, K.V.; Milenkovic, L.; Scott, M.P.; Rohatgi, R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 2010, 24, 670–682. [Google Scholar] [CrossRef]
- Wen, X.; Lai, C.K.; Evangelista, M.; Hongo, J.A.; de Sauvage, F.J.; Scales, S.J. Kinetics of hedgehog-dependent full-length Gli3 accumulation in primary cilia and subsequent degradation. Mol. Cell Biol. 2010, 30, 1910–1922. [Google Scholar] [CrossRef]
- Haycraft, C.J.; Banizs, B.; Aydin-Son, Y.; Zhang, Q.; Michaud, E.J.; Yoder, B.K. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005, 1, e53. [Google Scholar] [CrossRef]
- Katoh, Y.; Katoh, M. Hedgehog target genes: Mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr. Mol. Med. 2009, 9, 873–886. [Google Scholar] [CrossRef]
- Huangfu, D.; Liu, A.; Rakeman, A.S.; Murcia, N.S.; Niswander, L.; Anderson, K.V. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 2003, 426, 83–87. [Google Scholar] [CrossRef]
- Liu, A.; Wang, B.; Niswander, L.A. Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development 2005, 132, 3103–3111. [Google Scholar] [CrossRef]
- Rana, A.A.; Barbera, J.P.; Rodriguez, T.A.; Lynch, D.; Hirst, E.; Smith, J.C.; Beddington, R.S. Targeted deletion of the novel cytoplasmic dynein mD2LIC disrupts the embryonic organiser, formation of the body axes and specification of ventral cell fates. Development 2004, 131, 4999–5007. [Google Scholar] [CrossRef]
- Huangfu, D.; Anderson, K.V. Cilia and Hedgehog responsiveness in the mouse. Proc. Natl. Acad. Sci. USA 2005, 102, 11325–11330. [Google Scholar] [CrossRef]
- Eguether, T.; Cordelieres, F.P.; Pazour, G.J. Intraflagellar transport is deeply integrated in hedgehog signaling. Mol. Biol. Cell 2018, 29, 1178–1189. [Google Scholar] [CrossRef]
- Milenkovic, L.; Weiss, L.E.; Yoon, J.; Roth, T.L.; Su, Y.S.; Sahl, S.J.; Scott, M.P.; Moerner, W.E. Single-molecule imaging of Hedgehog pathway protein Smoothened in primary cilia reveals binding events regulated by Patched1. Proc. Natl. Acad. Sci. USA 2015, 112, 8320–8325. [Google Scholar] [CrossRef]
- Larkins, C.E.; Aviles, G.D.; East, M.P.; Kahn, R.A.; Caspary, T. Arl13b regulates ciliogenesis and the dynamic localization of Shh signaling proteins. Mol. Biol. Cell 2011, 22, 4694–4703. [Google Scholar] [CrossRef]
- Raffel, C.; Jenkins, R.B.; Frederick, L.; Hebrink, D.; Alderete, B.; Fults, D.W.; James, C.D. Sporadic medulloblastomas contain PTCH mutations. Cancer Res. 1997, 57, 842–845. [Google Scholar]
- Taylor, M.D.; Liu, L.; Raffel, C.; Hui, C.C.; Mainprize, T.G.; Zhang, X.; Agatep, R.; Chiappa, S.; Gao, L.; Lowrance, A.; et al. Mutations in SUFU predispose to medulloblastoma. Nat. Genet. 2002, 31, 306–310. [Google Scholar] [CrossRef]
- Xie, J.; Murone, M.; Luoh, S.M.; Ryan, A.; Gu, Q.; Zhang, C.; Bonifas, J.M.; Lam, C.W.; Hynes, M.; Goddard, A.; et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 1998, 391, 90–92. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Bartolome, R.; Torres, D.; Marone, R.; Feng, X.; Martin, D.; Simaan, M.; Chen, M.; Weinstein, L.S.; Taylor, S.S.; Molinolo, A.A.; et al. Inactivation of a Galpha(s)-PKA tumour suppressor pathway in skin stem cells initiates basal-cell carcinogenesis. Nat. Cell Biol. 2015, 17, 793–803. [Google Scholar] [CrossRef]
- Bay, S.N.; Long, A.B.; Caspary, T. Disruption of the ciliary GTPase Arl13b suppresses Sonic hedgehog overactivation and inhibits medulloblastoma formation. Proc. Natl. Acad. Sci. USA 2018, 115, 1570–1575. [Google Scholar] [CrossRef]
- Wong, S.Y.; Seol, A.D.; So, P.L.; Ermilov, A.N.; Bichakjian, C.K.; Epstein, E.H., Jr.; Dlugosz, A.A.; Reiter, J.F. Primary cilia can both mediate and suppress Hedgehog pathway-dependent tumorigenesis. Nat. Med. 2009, 15, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.G.; Kim, H.J.; Dlugosz, A.A.; Ellison, D.W.; Gilbertson, R.J.; Alvarez-Buylla, A. Dual and opposing roles of primary cilia in medulloblastoma development. Nat. Med. 2009, 15, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Ali, S.A.; Al-Jazrawe, M.; Kandel, R.; Wunder, J.S.; Alman, B.A. Primary cilia attenuate hedgehog signalling in neoplastic chondrocytes. Oncogene 2013, 32, 5388–5396. [Google Scholar] [CrossRef]
- Hassounah, N.B.; Nunez, M.; Fordyce, C.; Roe, D.; Nagle, R.; Bunch, T.; McDermott, K.M. Inhibition of Ciliogenesis Promotes Hedgehog Signaling, Tumorigenesis, and Metastasis in Breast Cancer. Mol. Cancer Res. 2017, 15, 1421–1430. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Wen, X.; Ratti, N.; Loktev, A.; Rangell, L.; Scales, S.J.; Jackson, P.K. The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic hedgehog pathway via cAMP signaling. Cell 2013, 152, 210–223. [Google Scholar] [CrossRef]
- Shimada, I.S.; Hwang, S.H.; Somatilaka, B.N.; Wang, X.; Skowron, P.; Kim, J.; Kim, M.; Shelton, J.M.; Rajaram, V.; Xuan, Z.; et al. Basal Suppression of the Sonic Hedgehog Pathway by the G-Protein-Coupled Receptor Gpr161 Restricts Medulloblastoma Pathogenesis. Cell Rep. 2018, 22, 1169–1184. [Google Scholar] [CrossRef]
- Yu, J.; Ustach, C.; Kim, H.R. Platelet-derived growth factor signaling and human cancer. J. Biochem. Mol. Biol. 2003, 36, 49–59. [Google Scholar] [CrossRef]
- Schmid, F.M.; Schou, K.B.; Vilhelm, M.J.; Holm, M.S.; Breslin, L.; Farinelli, P.; Larsen, L.A.; Andersen, J.S.; Pedersen, L.B.; Christensen, S.T. IFT20 modulates ciliary PDGFRalpha signaling by regulating the stability of Cbl E3 ubiquitin ligases. J. Cell. Biol. 2018, 217, 151–161. [Google Scholar] [CrossRef]
- Papkoff, J.; Brown, A.M.; Varmus, H.E. The int-1 proto-oncogene products are glycoproteins that appear to enter the secretory pathway. Mol. Cell Biol. 1987, 7, 3978–3984. [Google Scholar] [CrossRef]
- Bhanot, P.; Brink, M.; Samos, C.H.; Hsieh, J.C.; Wang, Y.; Macke, J.P.; Andrew, D.; Nathans, J.; Nusse, R. A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature 1996, 382, 225–230. [Google Scholar] [CrossRef]
- Pinson, K.I.; Brennan, J.; Monkley, S.; Avery, B.J.; Skarnes, W.C. An LDL-receptor-related protein mediates Wnt signalling in mice. Nature 2000, 407, 535–538. [Google Scholar] [CrossRef]
- Wehrli, M.; Dougan, S.T.; Caldwell, K.; O’Keefe, L.; Schwartz, S.; Vaizel-Ohayon, D.; Schejter, E.; Tomlinson, A.; DiNardo, S. arrow encodes an LDL-receptor-related protein essential for Wingless signalling. Nature 2000, 407, 527–530. [Google Scholar] [CrossRef]
- Klingensmith, J.; Nusse, R.; Perrimon, N. The Drosophila segment polarity gene dishevelled encodes a novel protein required for response to the wingless signal. Genes Dev. 1994, 8, 118–130. [Google Scholar] [CrossRef]
- Rubinfeld, B.; Souza, B.; Albert, I.; Muller, O.; Chamberlain, S.H.; Masiarz, F.R.; Munemitsu, S.; Polakis, P. Association of the APC gene product with beta-catenin. Science 1993, 262, 1731–1734. [Google Scholar] [CrossRef]
- Salic, A.; Lee, E.; Mayer, L.; Kirschner, M.W. Control of beta-catenin stability: Reconstitution of the cytoplasmic steps of the wnt pathway in Xenopus egg extracts. Mol. Cell 2000, 5, 523–532. [Google Scholar] [CrossRef]
- Su, L.K.; Vogelstein, B.; Kinzler, K.W. Association of the APC tumor suppressor protein with catenins. Science 1993, 262, 1734–1737. [Google Scholar] [CrossRef]
- Behrens, J.; von Kries, J.P.; Kuhl, M.; Bruhn, L.; Wedlich, D.; Grosschedl, R.; Birchmeier, W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature 1996, 382, 638–642. [Google Scholar] [CrossRef]
- Sherwood, V. WNT signaling: An emerging mediator of cancer cell metabolism? Mol. Cell Biol. 2015, 35, 2–10. [Google Scholar] [CrossRef]
- Adler, P.N. Planar signaling and morphogenesis in Drosophila. Dev. Cell 2002, 2, 525–535. [Google Scholar] [CrossRef]
- Yang, Y.; Mlodzik, M. Wnt-Frizzled/planar cell polarity signaling: Cellular orientation by facing the wind (Wnt). Annu. Rev. Cell Dev. Biol. 2015, 31, 623–646. [Google Scholar] [CrossRef]
- Morgan, D.; Eley, L.; Sayer, J.; Strachan, T.; Yates, L.M.; Craighead, A.S.; Goodship, J.A. Expression analyses and interaction with the anaphase promoting complex protein Apc2 suggest a role for inversin in primary cilia and involvement in the cell cycle. Hum. Mol. Genet. 2002, 11, 3345–3350. [Google Scholar] [CrossRef]
- Simons, M.; Gloy, J.; Ganner, A.; Bullerkotte, A.; Bashkurov, M.; Kronig, C.; Schermer, B.; Benzing, T.; Cabello, O.A.; Jenny, A.; et al. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat. Genet. 2005, 37, 537–543. [Google Scholar] [CrossRef]
- Gerdes, J.M.; Liu, Y.; Zaghloul, N.A.; Leitch, C.C.; Lawson, S.S.; Kato, M.; Beachy, P.A.; Beales, P.L.; DeMartino, G.N.; Fisher, S.; et al. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat. Genet. 2007, 39, 1350–1360. [Google Scholar] [CrossRef]
- Gerdes, J.M.; Davis, E.E.; Katsanis, N. The vertebrate primary cilium in development, homeostasis, and disease. Cell 2009, 137, 32–45. [Google Scholar] [CrossRef]
- Corbit, K.C.; Shyer, A.E.; Dowdle, W.E.; Gaulden, J.; Singla, V.; Chen, M.H.; Chuang, P.T.; Reiter, J.F. Kif3a constrains beta-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat. Cell Biol. 2008, 10, 70–76. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Schroth, J.; Gleeson, J.G. Subcellular spatial regulation of canonical Wnt signalling at the primary cilium. Nat. Cell Biol. 2011, 13, 700–707. [Google Scholar] [CrossRef]
- Zingg, D.; Debbache, J.; Pena-Hernandez, R.; Antunes, A.T.; Schaefer, S.M.; Cheng, P.F.; Zimmerli, D.; Haeusel, J.; Calcada, R.R.; Tuncer, E.; et al. EZH2-Mediated Primary Cilium Deconstruction Drives Metastatic Melanoma Formation. Cancer Cell 2018, 34, 69–84.e14. [Google Scholar] [CrossRef]
- Moon, R.T.; Kohn, A.D.; De Ferrari, G.V.; Kaykas, A. WNT and beta-catenin signalling: Diseases and therapies. Nat. Rev. Genet. 2004, 5, 691–701. [Google Scholar] [CrossRef]
- Huang, P.; Schier, A.F. Dampened Hedgehog signaling but normal Wnt signaling in zebrafish without cilia. Development 2009, 136, 3089–3098. [Google Scholar] [CrossRef]
- Ocbina, P.J.; Tuson, M.; Anderson, K.V. Primary cilia are not required for normal canonical Wnt signaling in the mouse embryo. PLoS ONE 2009, 4, e6839. [Google Scholar] [CrossRef]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef]
- Mazure, N.M.; Pouyssegur, J. Hypoxia-induced autophagy: Cell death or cell survival? Curr. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef]
- Pampliega, O.; Orhon, I.; Patel, B.; Sridhar, S.; Diaz-Carretero, A.; Beau, I.; Codogno, P.; Satir, B.H.; Satir, P.; Cuervo, A.M. Functional interaction between autophagy and ciliogenesis. Nature 2013, 502, 194–200. [Google Scholar] [CrossRef]
- Follit, J.A.; Xu, F.; Keady, B.T.; Pazour, G.J. Characterization of mouse IFT complex B. Cell Motil. Cytoskelet. 2009, 66, 457–468. [Google Scholar] [CrossRef]
- Li, H.; Li, J.; Li, Y.; Singh, P.; Cao, L.; Xu, L.J.; Li, D.; Wang, Y.; Xie, Z.; Gui, Y.; et al. Sonic hedgehog promotes autophagy of vascular smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H1319–H1331. [Google Scholar] [CrossRef]
- Petralia, R.S.; Schwartz, C.M.; Wang, Y.X.; Kawamoto, E.M.; Mattson, M.P.; Yao, P.J. Sonic hedgehog promotes autophagy in hippocampal neurons. Biol. Open 2013, 2, 499–504. [Google Scholar] [CrossRef]
- Wang, S.; Livingston, M.J.; Su, Y.; Dong, Z. Reciprocal regulation of cilia and autophagy via the MTOR and proteasome pathways. Autophagy 2015, 11, 607–616. [Google Scholar] [CrossRef]
- Tang, Z.; Lin, M.G.; Stowe, T.R.; Chen, S.; Zhu, M.; Stearns, T.; Franco, B.; Zhong, Q. Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature 2013, 502, 254–257. [Google Scholar] [CrossRef]
- Singla, V.; Romaguera-Ros, M.; Garcia-Verdugo, J.M.; Reiter, J.F. Ofd1, a human disease gene, regulates the length and distal structure of centrioles. Dev. Cell 2010, 18, 410–424. [Google Scholar] [CrossRef]
- Maharjan, Y.; Lee, J.N.; Kwak, S.; Lim, H.; Dutta, R.K.; Liu, Z.Q.; So, H.S.; Park, R. Autophagy alteration prevents primary cilium disassembly in RPE1 cells. Biochem. Biophys. Res. Commun. 2018, 500, 242–248. [Google Scholar] [CrossRef]
- Takahashi, K.; Nagai, T.; Chiba, S.; Nakayama, K.; Mizuno, K. Glucose deprivation induces primary cilium formation through mTORC1 inactivation. J. Cell. Sci. 2018, 131, jcs208769. [Google Scholar] [CrossRef]
- Struchtrup, A.; Wiegering, A.; Stork, B.; Ruther, U.; Gerhardt, C. The ciliary protein RPGRIP1L governs autophagy independently of its proteasome-regulating function at the ciliary base in mouse embryonic fibroblasts. Autophagy 2018, 14, 567–583. [Google Scholar] [CrossRef]
- Liu, Z.Q.; Lee, J.N.; Son, M.; Lim, J.Y.; Dutta, R.K.; Maharjan, Y.; Kwak, S.; Oh, G.T.; Byun, K.; Choe, S.K.; et al. Ciliogenesis is reciprocally regulated by PPARA and NR1H4/FXR through controlling autophagy in vitro and in vivo. Autophagy 2018, 14, 1011–1027. [Google Scholar] [CrossRef]
- Xu, Q.; Liu, W.; Liu, X.; Otkur, W.; Hayashi, T.; Yamato, M.; Fujisaki, H.; Hattori, S.; Tashiro, S.I.; Ikejima, T. Type I collagen promotes primary cilia growth through down-regulating HDAC6-mediated autophagy in confluent mouse embryo fibroblast 3T3-L1 cells. J. Biosci. Bioeng. 2018, 125, 8–14. [Google Scholar] [CrossRef]
- Hsiao, C.J.; Chang, C.H.; Ibrahim, R.B.; Lin, I.H.; Wang, C.H.; Wang, W.J.; Tsai, J.W. Gli2 modulates cell cycle re-entry through autophagy-mediated regulation of the length of primary cilia. J. Cell. Sci. 2018, 131, jcs221218. [Google Scholar] [CrossRef]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes. Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Mancias, J.D.; Kimmelman, A.C. The Role of Autophagy in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 19–39. [Google Scholar] [CrossRef]
- Lee, J.; Yi, S.; Kang, Y.E.; Chang, J.Y.; Kim, J.T.; Sul, H.J.; Kim, J.O.; Kim, J.M.; Kim, J.; Porcelli, A.M.; et al. Defective ciliogenesis in thyroid hurthle cell tumors is associated with increased autophagy. Oncotarget 2016, 7, 79117–79130. [Google Scholar] [CrossRef]
- Choi, A.R.; Kim, J.H.; Yoon, S. Thioridazine specifically sensitizes drug-resistant cancer cells through highly increase in apoptosis and P-gp inhibition. Tumour Biol. 2014, 35, 9831–9838. [Google Scholar] [CrossRef]
- Bao, Z.; Huang, W. Thioridazine promotes primary ciliogenesis in lung cancer cells through enhancing cell autophagy. Int. J. Clin. Exp. Med. 2017, 10, 13960–13969. [Google Scholar]
- Pouyssegur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Wang, G.L.; Semenza, G.L. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 1995, 270, 1230–1237. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. The von Hippel-Lindau gene, kidney cancer, and oxygen sensing. J. Am. Soc. Nephrol. 2003, 14, 2703–2711. [Google Scholar] [CrossRef]
- Berra, E.; Richard, D.E.; Gothie, E.; Pouyssegur, J. HIF-1-dependent transcriptional activity is required for oxygen- mediated HIF-1alpha degradation. FEBS Lett. 2001, 491, 85–90. [Google Scholar] [CrossRef]
- Kallio, P.J.; Wilson, W.J.; O’Brien, S.; Makino, Y.; Poellinger, L. Regulation of the hypoxia-inducible transcription factor 1alpha by the ubiquitin-proteasome pathway. J. Biol. Chem. 1999, 274, 6519–6525. [Google Scholar] [CrossRef]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes. Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef]
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The impact of O2 availability on human cancer. Nat. Rev. Cancer 2008, 8, 967–975. [Google Scholar] [CrossRef]
- Ruan, K.; Song, G.; Ouyang, G. Role of hypoxia in the hallmarks of human cancer. J. Cell Biochem. 2009, 107, 1053–1062. [Google Scholar] [CrossRef]
- Decastro, G.J.; McKiernan, J.M. Epidemiology, clinical staging, and presentation of renal cell carcinoma. Urol. Clin. N. Am. 2008, 35, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Schraml, P.; Frew, I.J.; Thoma, C.R.; Boysen, G.; Struckmann, K.; Krek, W.; Moch, H. Sporadic clear cell renal cell carcinoma but not the papillary type is characterized by severely reduced frequency of primary cilia. Mod. Pathol. 2009, 22, 31–36. [Google Scholar] [CrossRef]
- Thoma, C.R.; Frew, I.J.; Hoerner, C.R.; Montani, M.; Moch, H.; Krek, W. pVHL and GSK3beta are components of a primary cilium-maintenance signalling network. Nat. Cell Biol. 2007, 9, 588–595. [Google Scholar] [CrossRef]
- Lutz, M.S.; Burk, R.D. Primary cilium formation requires von hippel-lindau gene function in renal-derived cells. Cancer Res. 2006, 66, 6903–6907. [Google Scholar] [CrossRef]
- Esteban, M.A.; Harten, S.K.; Tran, M.G.; Maxwell, P.H. Formation of primary cilia in the renal epithelium is regulated by the von Hippel-Lindau tumor suppressor protein. J. Am. Soc. Nephrol. 2006, 17, 1801–1806. [Google Scholar] [CrossRef]
- Ding, X.F.; Zhou, J.; Hu, Q.Y.; Liu, S.C.; Chen, G. The tumor suppressor pVHL down-regulates never-in-mitosis A-related kinase 8 via hypoxia-inducible factors to maintain cilia in human renal cancer cells. J. Biol. Chem. 2015, 290, 1389–1394. [Google Scholar] [CrossRef]
- Proulx-Bonneau, S.; Annabi, B. The primary cilium as a biomarker in the hypoxic adaptation of bone marrow-derived mesenchymal stromal cells: A role for the secreted frizzled-related proteins. Biomark. Insights 2011, 6, 107–118. [Google Scholar] [CrossRef]
- O’Toole, S.M.; Watson, D.S.; Novoselova, T.V.; Romano, L.E.L.; King, P.J.; Bradshaw, T.Y.; Thompson, C.L.; Knight, M.M.; Sharp, T.V.; Barnes, M.R.; et al. Oncometabolite induced primary cilia loss in pheochromocytoma. Endocr.-Relat. Cancer 2019, 26, 165–180. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef]
- Xu, J.; Li, H.; Wang, B.; Xu, Y.; Yang, J.; Zhang, X.; Harten, S.K.; Shukla, D.; Maxwell, P.H.; Pei, D.; et al. VHL inactivation induces HEF1 and Aurora kinase A. J. Am. Soc. Nephrol. 2010, 21, 2041–2046. [Google Scholar] [CrossRef]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Lee, J.; Yi, S.; Won, M.; Song, Y.S.; Yi, H.S.; Park, Y.J.; Park, K.C.; Kim, J.T.; Chang, J.Y.; Lee, M.J.; et al. Loss-of-function of IFT88 determines metabolic phenotypes in thyroid cancer. Oncogene 2018, 37, 4455–4474. [Google Scholar] [CrossRef]
- Jeong, A.L.; Ka, H.I.; Han, S.; Lee, S.; Lee, E.W.; Soh, S.J.; Joo, H.J.; Sumiyasuren, B.; Park, J.Y.; Lim, J.S.; et al. Oncoprotein CIP2A promotes the disassembly of primary cilia and inhibits glycolytic metabolism. EMBO Rep. 2018, 19, 5. [Google Scholar] [CrossRef]
- Lee, H.; Song, J.; Jung, J.H.; Ko, H.W. Primary cilia in energy balance signaling and metabolic disorder. BMB Rep. 2015, 48, 647–654. [Google Scholar] [CrossRef]
- Volta, F.; Gerdes, J.M. The role of primary cilia in obesity and diabetes. Ann. N. Y. Acad. Sci. 2017, 1391, 71–84. [Google Scholar] [CrossRef]
- Kim, H.; Lin, Q.; Glazer, P.M.; Yun, Z. The hypoxic tumor microenvironment in vivo selects the cancer stem cell fate of breast cancer cells. Breast Cancer Res. 2018, 20, 16. [Google Scholar] [CrossRef]
- Li, Z.; Rich, J.N. Hypoxia and hypoxia inducible factors in cancer stem cell maintenance. Curr. Top. Microbiol. Immunol. 2010, 345, 21–30. [Google Scholar]
- Peng, G.; Liu, Y. Hypoxia-inducible factors in cancer stem cells and inflammation. Trends Pharmacol. Sci. 2015, 36, 374–383. [Google Scholar] [CrossRef]
- Gate, D.; Danielpour, M.; Bannykh, S.; Town, T. Characterization of cancer stem cells and primary cilia in medulloblastoma. CNS Neurol. Disord. Drug Targ. 2015, 14, 600–611. [Google Scholar] [CrossRef]
- Guen, V.J.; Chavarria, T.E.; Kroger, C.; Ye, X.; Weinberg, R.A.; Lees, J.A. EMT programs promote basal mammary stem cell and tumor-initiating cell stemness by inducing primary ciliogenesis and Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2017, 114, E10532–E10539. [Google Scholar] [CrossRef]
- Han, S.J.; Jung, J.K.; Im, S.S.; Lee, S.R.; Jang, B.C.; Park, K.M.; Kim, J.I. Deficiency of primary cilia in kidney epithelial cells induces epithelial to mesenchymal transition. Biochem. Biophys. Res. Commun. 2018, 496, 450–454. [Google Scholar] [CrossRef]
- Jenks, A.D.; Vyse, S.; Wong, J.P.; Kostaras, E.; Keller, D.; Burgoyne, T.; Shoemark, A.; Tsalikis, A.; de la Roche, M.; Michaelis, M.; et al. Primary Cilia Mediate Diverse Kinase Inhibitor Resistance Mechanisms in Cancer. Cell Rep. 2018, 23, 3042–3055. [Google Scholar] [CrossRef]
- Wang, S.; Wei, Q.; Dong, G.; Dong, Z. ERK-mediated suppression of cilia in cisplatin-induced tubular cell apoptosis and acute kidney injury. Biochim. Biophys. Acta 2013, 1832, 1582–1590. [Google Scholar] [CrossRef]
- Hoang-Minh, L.B.; Deleyrolle, L.P.; Nakamura, N.S.; Parker, A.K.; Martuscello, R.T.; Reynolds, B.A.; Sarkisian, M.R. PCM1 Depletion Inhibits Glioblastoma Cell Ciliogenesis and Increases Cell Death and Sensitivity to Temozolomide. Transl. Oncol. 2016, 9, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Pak, E.; Ornell, K.J.; Pazyra-Murphy, M.F.; MacKenzie, E.L.; Chadwick, E.J.; Ponomaryov, T.; Kelleher, J.F.; Segal, R.A. A Transposon Screen Identifies Loss of Primary Cilia as a Mechanism of Resistance to SMO Inhibitors. Cancer Discov. 2017, 7, 1436–1449. [Google Scholar] [CrossRef]
- Nauli, S.M.; Kawanabe, Y.; Kaminski, J.J.; Pearce, W.J.; Ingber, D.E.; Zhou, J. Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation 2008, 117, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Dinsmore, C.; Reiter, J.F. Endothelial primary cilia inhibit atherosclerosis. EMBO Rep. 2016, 17, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Eisa-Beygi, S.; Benslimane, F.M.; El-Rass, S.; Prabhudesai, S.; Abdelrasoul, M.K.A.; Simpson, P.M.; Yalcin, H.C.; Burrows, P.E.; Ramchandran, R. Characterization of Endothelial Cilia Distribution During Cerebral-Vascular Development in Zebrafish (Danio rerio). Arterioscler. Thrombosis Vasc. Biol. 2018, 38, 2806–2818. [Google Scholar] [CrossRef]
- Goetz, J.G.; Steed, E.; Ferreira, R.R.; Roth, S.; Ramspacher, C.; Boselli, F.; Charvin, G.; Liebling, M.; Wyart, C.; Schwab, Y.; et al. Endothelial cilia mediate low flow sensing during zebrafish vascular development. Cell Rep. 2014, 6, 799–808. [Google Scholar] [CrossRef]
- Vion, A.C.; Alt, S.; Klaus-Bergmann, A.; Szymborska, A.; Zheng, T.; Perovic, T.; Hammoutene, A.; Oliveira, M.B.; Bartels-Klein, E.; Hollfinger, I.; et al. Primary cilia sensitize endothelial cells to BMP and prevent excessive vascular regression. J. Cell. Biol. 2018, 217, 1651–1665. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fabbri, L.; Bost, F.; Mazure, N.M. Primary Cilium in Cancer Hallmarks. Int. J. Mol. Sci. 2019, 20, 1336. https://doi.org/10.3390/ijms20061336
Fabbri L, Bost F, Mazure NM. Primary Cilium in Cancer Hallmarks. International Journal of Molecular Sciences. 2019; 20(6):1336. https://doi.org/10.3390/ijms20061336
Chicago/Turabian StyleFabbri, Lucilla, Frédéric Bost, and Nathalie M. Mazure. 2019. "Primary Cilium in Cancer Hallmarks" International Journal of Molecular Sciences 20, no. 6: 1336. https://doi.org/10.3390/ijms20061336
APA StyleFabbri, L., Bost, F., & Mazure, N. M. (2019). Primary Cilium in Cancer Hallmarks. International Journal of Molecular Sciences, 20(6), 1336. https://doi.org/10.3390/ijms20061336