Fatty Acid Synthesis and Degradation Interplay to Regulate the Oxidative Stress in Cancer Cells

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
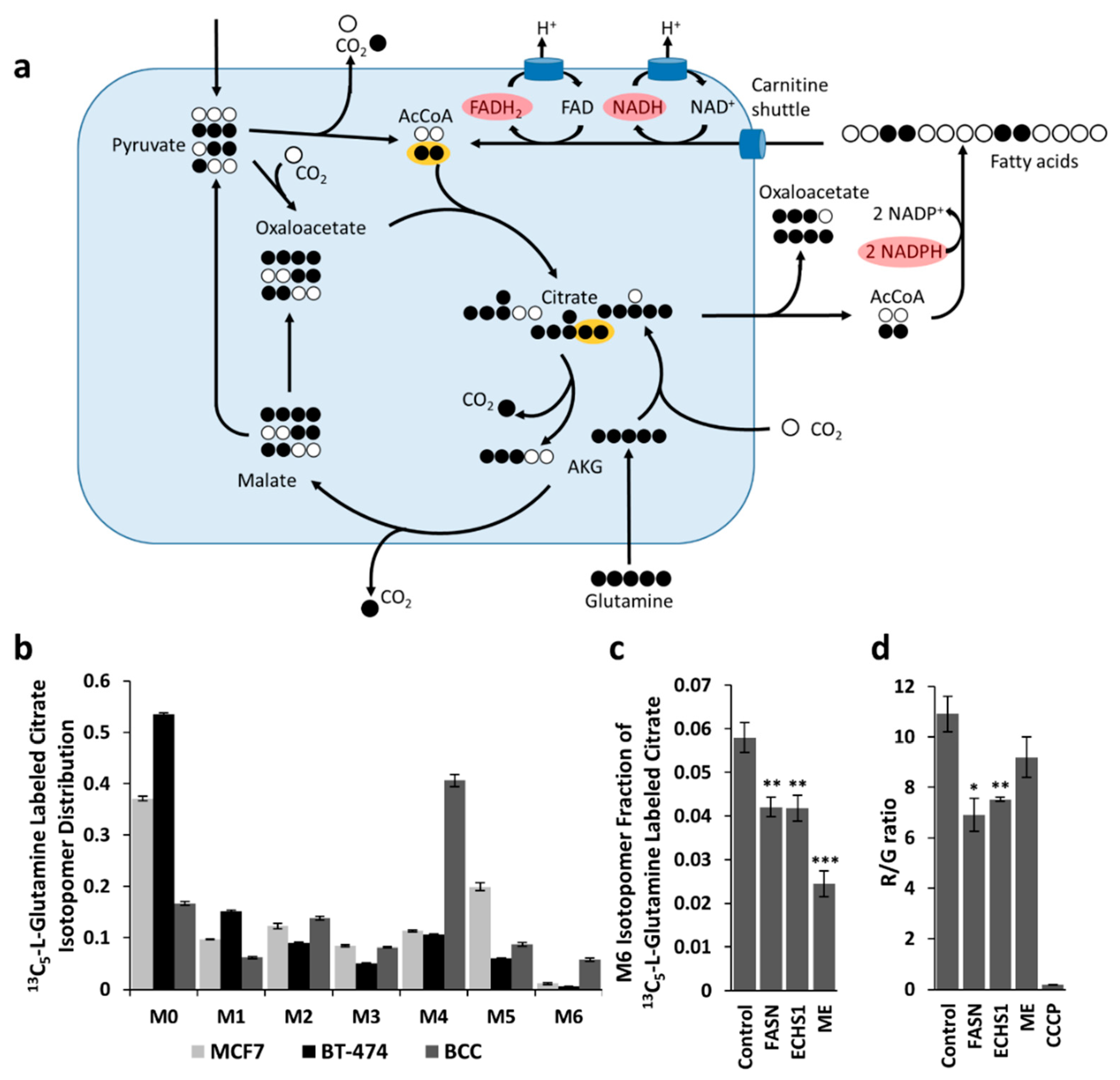
2.1. Assessment of Simultaneous FAS and FAO Using 13C Labeling
2.2. Assessment of Simultaneous FAS and FAO Using Measurements of Mitochondrial Membrane Potential
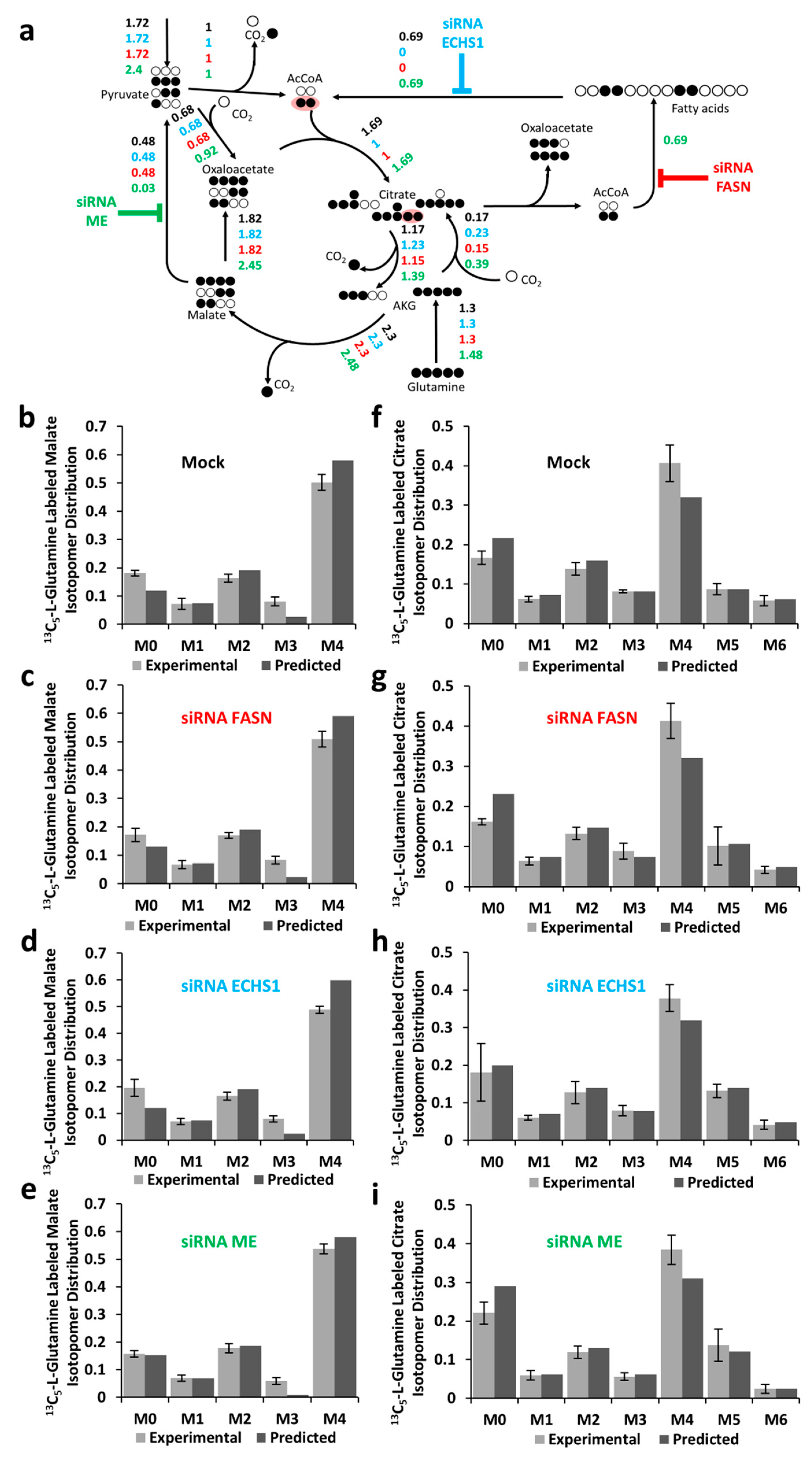
2.3. Metabolic Flux Analysis of BCC Cells
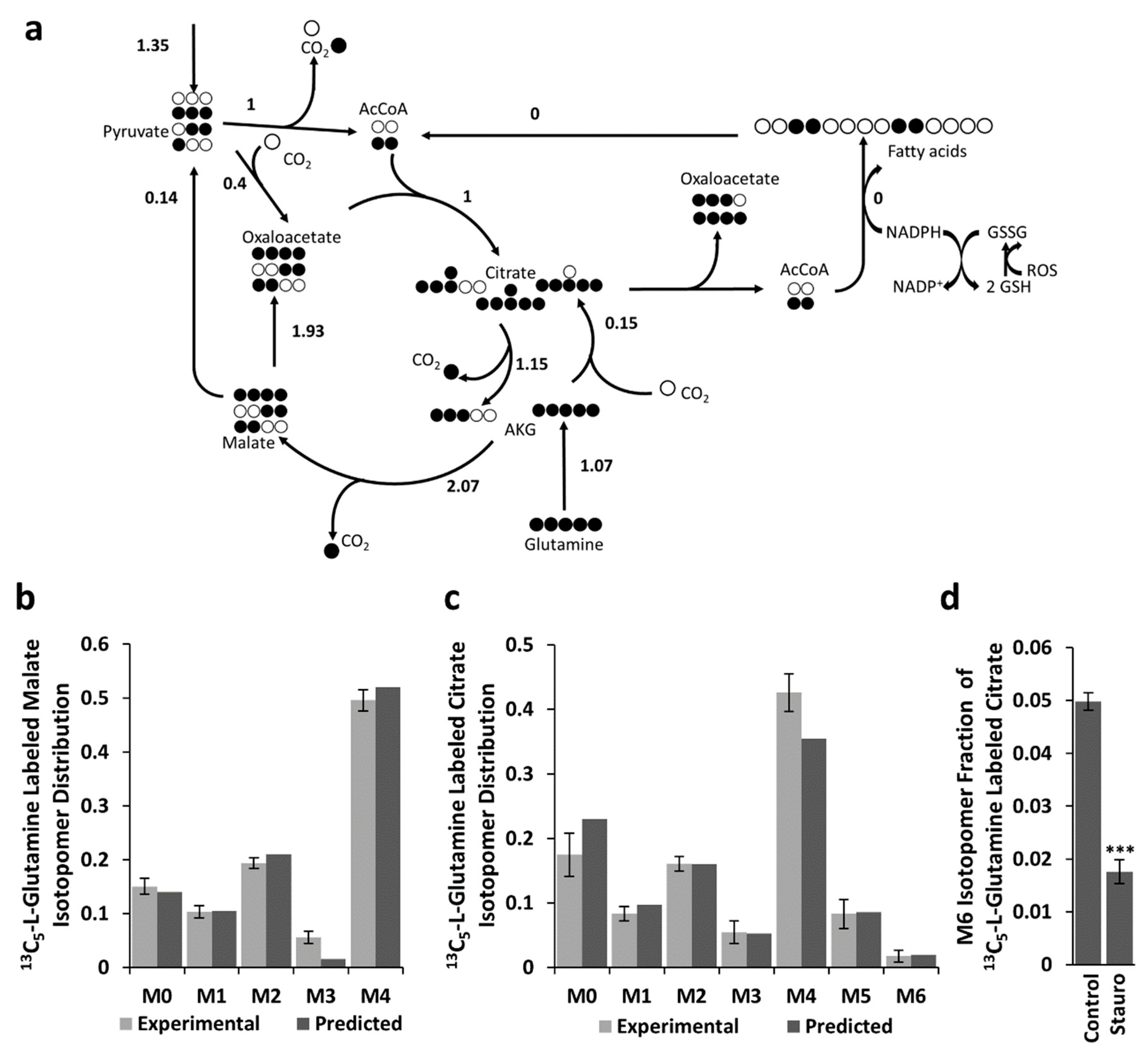
2.4. Effects of Oxidative Stress on Metabolic Fluxes
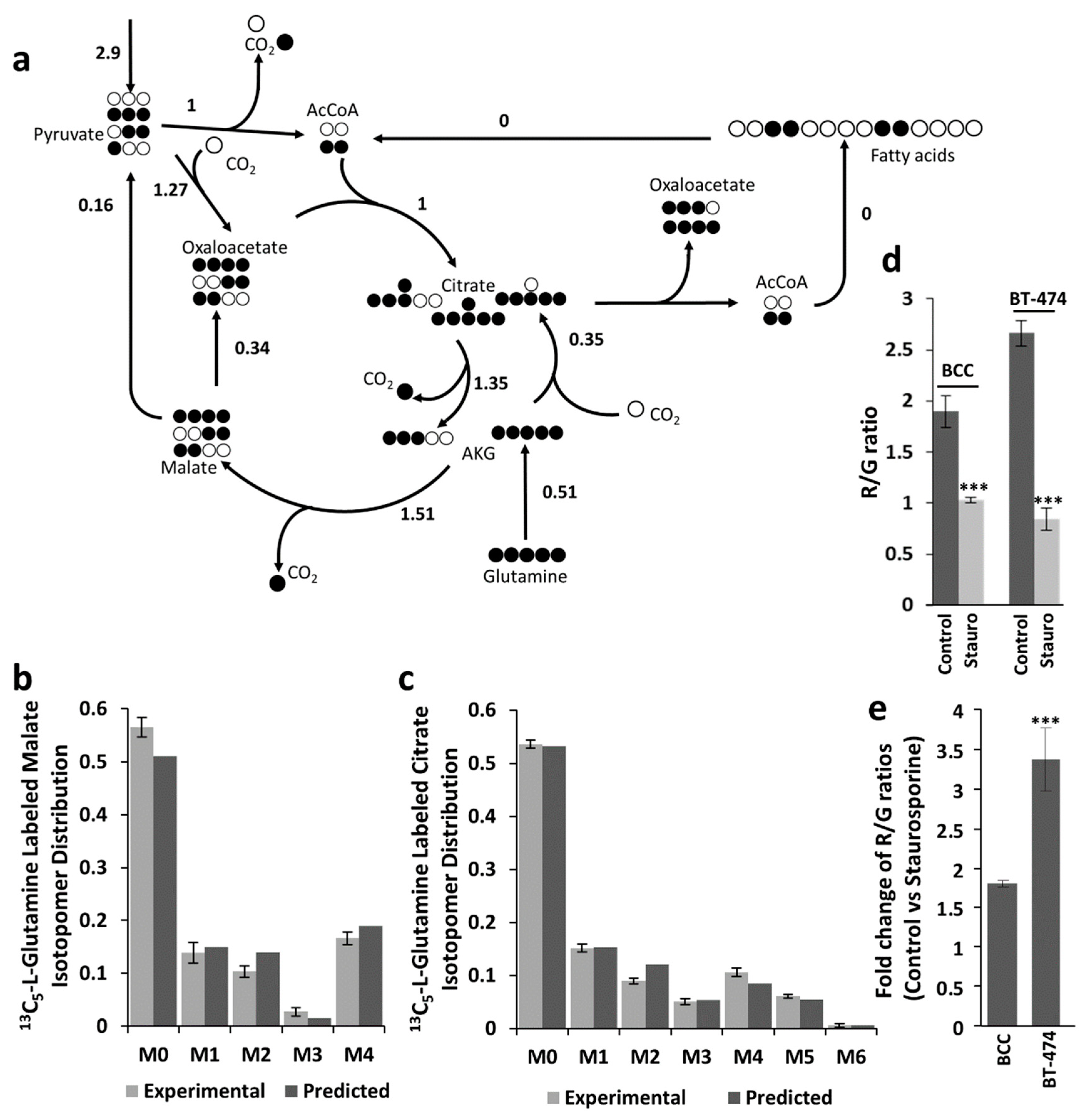
2.5. Resistance of BCC and BT-474 Cells to Oxidative Stress
3. Discussion
4. Methods
4.1. Cell Lines and Culture Medium
4.2. siRNA Transfection
4.3. 13C Labeling
4.4. UPLC-ESI-MS Conditions
4.5. Fitting of Metabolic Flux Distributions
4.6. Western Blot Analysis
4.7. Flow Cytometry Assay
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Medes, G.; Thomas, A.; Weinhouse, S. Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neo-plastic tissue slices in vitro. Cancer Res. 1953, 13, 27–29. [Google Scholar] [PubMed]
- Kuhajda, F.P.; Jenner, K.; Wood, F.D.; Hennigar, R.A.; Jacobs, L.B.; Dick, J.D.; Pasternack, G.R. Fatty acid synthesis: A potential selective target for antineoplastic therapy. Proc. Natl. Acad. Sci. USA 1994, 91, 6379–6383. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, J.V.; Vanderhoydonc, F.; Elgamal, A.A.; Eelen, M.; Vercaeren, I.; Joniau, S.; Heyns, W.; Verhoeven, G. Selective activation of the fatty acid synthesis pathway in human prostate cancer. Int. J. Cancer 2000, 88, 176–179. [Google Scholar] [CrossRef] [Green Version]
- Bauer, D.E.; Hatzivasssiliou, G.; Zhao, F.; Andreadis, C.; Thompson, C.B. ATP citrate lyase is an important component of cell growth and transformation. Oncogene 2005, 24, 6314–6322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckers, A.; Organe, S.; Timmermans, L.; Scheys, K.; Peeters, A.; Brusselmans, K.; Verhoeven, G.; Swinnen, J.V. Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res. 2007, 67, 8180–8187. [Google Scholar] [CrossRef] [PubMed]
- Quijano, C.; Cao, L.; Fergusson, M.M.; Romero, H.; Liu, J.; Gutkind, S.; Rovira, I.I.; Mohney, R.P.; Karoly, E.D.; Finkel, T. Oncogene-induced senescence results in marked metabolic and bioenergetics alterations. Cell Cycle 2012, 11, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Marien, E.; Meister, M.; Muley, T.; Fieuws, S.; Bordel, S.; Derua, R.; Spraggins, J.; Van de Plas, R.; Dehairs, J.; Wouters, J.; et al. Non-small cell lung cancer (NSCLC) is characterized by dramatic changes in phospholipid profiles. Int. J. Cancer 2015, 137, 1539–1548. [Google Scholar] [CrossRef]
- Feizi, A.; Bordel, S. Metabolic and protein interaction sub-networks controlling the proliferation rate of cancer cells and their impact on patient survival. Sci. Rep. 2013, 3, 3041. [Google Scholar] [CrossRef]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- Qu, Q.; Zeng, F.; Liu, X.; Wang, Q.J.; Deng, F. Fatty acid oxidation and carnitine palmitoyltransferase I: Emerging therapeutic targets in cancer. Cell Death Dis. 2016, 4, e532. [Google Scholar] [CrossRef]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarijan, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Invest. 2010, 120, 142–156. [Google Scholar] [CrossRef]
- Carracedo, A.; Weiss, D.; Leliaert, A.K.; Bhasin, M.; de Boer, V.C.; Laurent, G.; Adams, A.C.; Sundvall, M.; Song, S.J.; Ito, K.; et al. A metabolic prosurvival role for PML in breast cancer. J. Clin. Invest. 2012, 122, 3088–3100. [Google Scholar] [CrossRef] [Green Version]
- Zaugg, K.; Yao, Y.; Reilly, P.T.; Kannan, K.; Kiarash, R.; Mason, J.; Huang, P.; Sawyer, S.K.; Fuerth, B.; Faubert, B.; et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011, 25, 1041–1051. [Google Scholar] [CrossRef] [Green Version]
- Abu-Elheiga, L.; Matzuk, M.M.; Abo-Hashema, K.A.; Wakil, S.J. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science 2001, 291, 2613–2616. [Google Scholar] [CrossRef]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009, 461, 109–113. [Google Scholar] [CrossRef]
- Jeon, S.M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumor cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef]
- Caro, P.; Kishan, A.U.; Norberg, E.; Stanley, I.A.; Chapuy, B.; Ficarro, S.B.; Polak, K.; Tondera, D.; Gounarides, J.; Yin, H.; et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell 2012, 22, 547–560. [Google Scholar] [CrossRef]
- Schlaepfer, I.R.; Rider, L.; Rodrigues, L.U.; Gijón, M.A.; Pac, C.T.; Romero, L.; Cimic, A.; Sirintrapun, S.J.; Glodé, L.M.; Eckel, R.H.; et al. Lipid catabolism via CPT1 as a therapeutic target for prostate cáncer. Mol. Cancer Ther. 2014, 13, 2361–2371. [Google Scholar] [CrossRef]
- Tirado-Velez, J.M.; Joumady, I.; Saez-Benito, A.; Cozar-Castellano, I.; Perdomo, G. Inhibition of fatty acid metabolism reduces human mieloma cells proliferation. PLoS ONE 2012, 7, e46484. [Google Scholar] [CrossRef]
- Fendt, S.M.; Bell, E.L.; Keibler, M.A.; Olenchock, B.A.; Mayers, J.R.; Wasylenko, T.M.; Vokes, N.I.; Guarente, L.; Vander Heiden, M.G.; Stephanopoulos, G. Reductive glutamine metabolism is a function of α-ketoglutarate to citrate ratio in cells. Nat. Commun. 2013, 4, 2236. [Google Scholar] [CrossRef]
- Antanavičiutė, I.; Mikalayeva, V.; Ceslevičienė, I.; Milašiūtė, G.; Skeberdis, V.A.; Bordel, S. Transcriptional hallmarks of cancer cell lines reveal and emerging role of branched chain amino acid catabolism. Sci. Rep. 2017, 7, 7820. [Google Scholar] [CrossRef]
- Antoniewicz, M.R.; Kelleher, J.K.; Stephanopoulos, G. Elementary metabolite units (EMU): A novel framework for modeling isotopic distributions. Metab. Eng. 2007, 9, 68–86. [Google Scholar] [CrossRef]
- Kruman, I.; Guo, Q.; Mattson, M.P. Calcium and reactive oxygen species mediate staurosporine-induced mitochondrial disfunction and apoptosis in PC12 cells. J. Neurosci. Res. 1998, 51, 293–308. [Google Scholar] [CrossRef]
- Borgos, S.E.F.; Bordel, S.; Sletta, H.; Ertesvag, H.; Jakobsen, O.; Bruheim, P.; Ellingsen, T.E.; Nielsen, J.; Valla, S. Mapping global effects of the anti-sigma factor MucA in Pseudomonas fluorescensSBW25 through genome-scale metabolic modeling. BMC Syst. Biol. 2013, 7, 19. [Google Scholar] [CrossRef]
- Sellick, C.A.; Hansen, R.; Stephens, G.M.; Goodacre, R.; Dickson, A.J. Metabolite extraction from suspension-cultured mammalian cells for global metabolite profiling. Nat. Protoc. 2011, 6, 1241–1249. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikalayeva, V.; Ceslevičienė, I.; Sarapinienė, I.; Žvikas, V.; Skeberdis, V.A.; Jakštas, V.; Bordel, S. Fatty Acid Synthesis and Degradation Interplay to Regulate the Oxidative Stress in Cancer Cells. Int. J. Mol. Sci. 2019, 20, 1348. https://doi.org/10.3390/ijms20061348
Mikalayeva V, Ceslevičienė I, Sarapinienė I, Žvikas V, Skeberdis VA, Jakštas V, Bordel S. Fatty Acid Synthesis and Degradation Interplay to Regulate the Oxidative Stress in Cancer Cells. International Journal of Molecular Sciences. 2019; 20(6):1348. https://doi.org/10.3390/ijms20061348
Chicago/Turabian StyleMikalayeva, Valeryia, Ieva Ceslevičienė, Ieva Sarapinienė, Vaidotas Žvikas, Vytenis Arvydas Skeberdis, Valdas Jakštas, and Sergio Bordel. 2019. "Fatty Acid Synthesis and Degradation Interplay to Regulate the Oxidative Stress in Cancer Cells" International Journal of Molecular Sciences 20, no. 6: 1348. https://doi.org/10.3390/ijms20061348
APA StyleMikalayeva, V., Ceslevičienė, I., Sarapinienė, I., Žvikas, V., Skeberdis, V. A., Jakštas, V., & Bordel, S. (2019). Fatty Acid Synthesis and Degradation Interplay to Regulate the Oxidative Stress in Cancer Cells. International Journal of Molecular Sciences, 20(6), 1348. https://doi.org/10.3390/ijms20061348