Transcriptomic Impact of IMA-08401, a Novel AHR Agonist Resembling Laquinimod, on Rat Liver

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
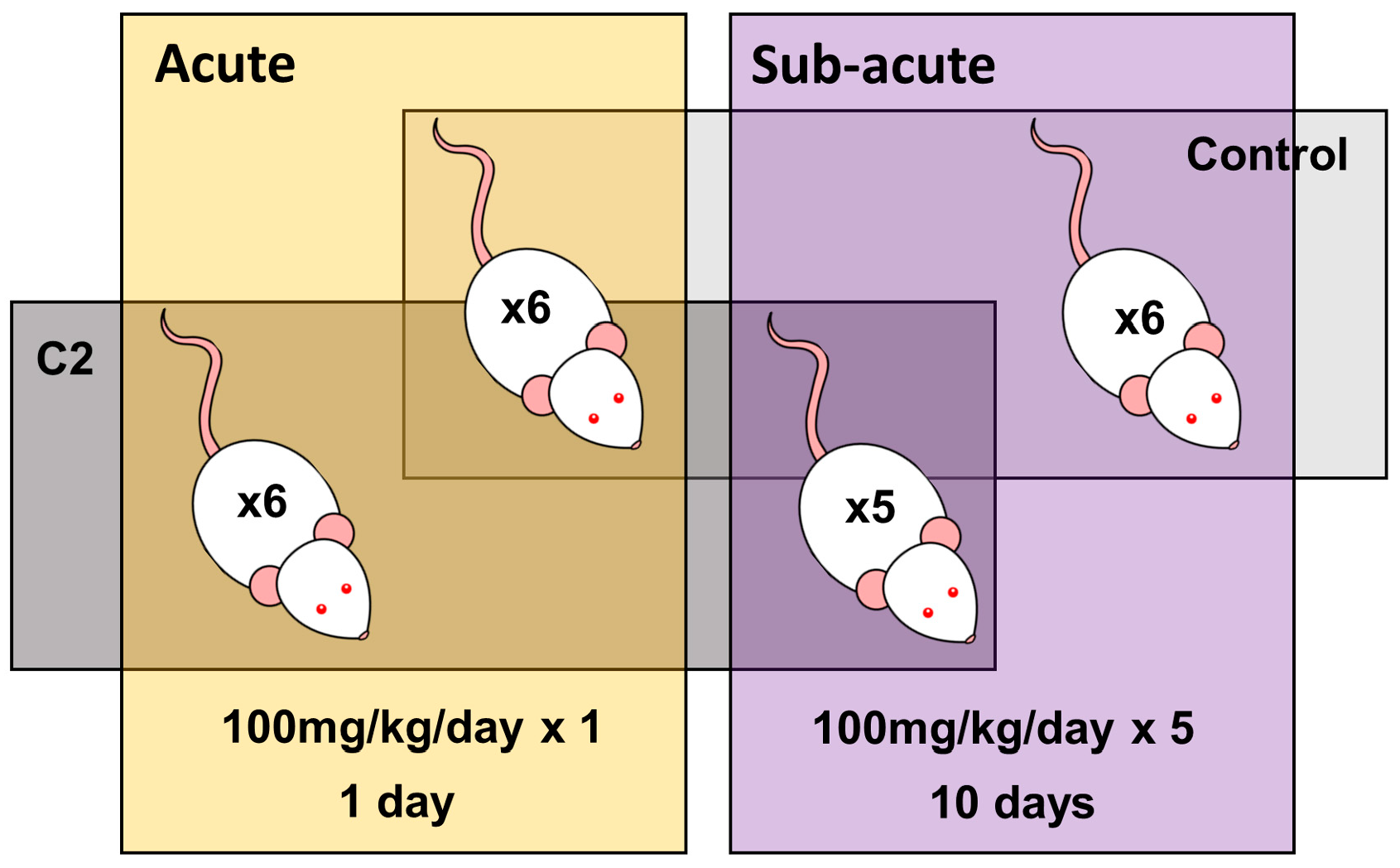
2.1. Experimental Design
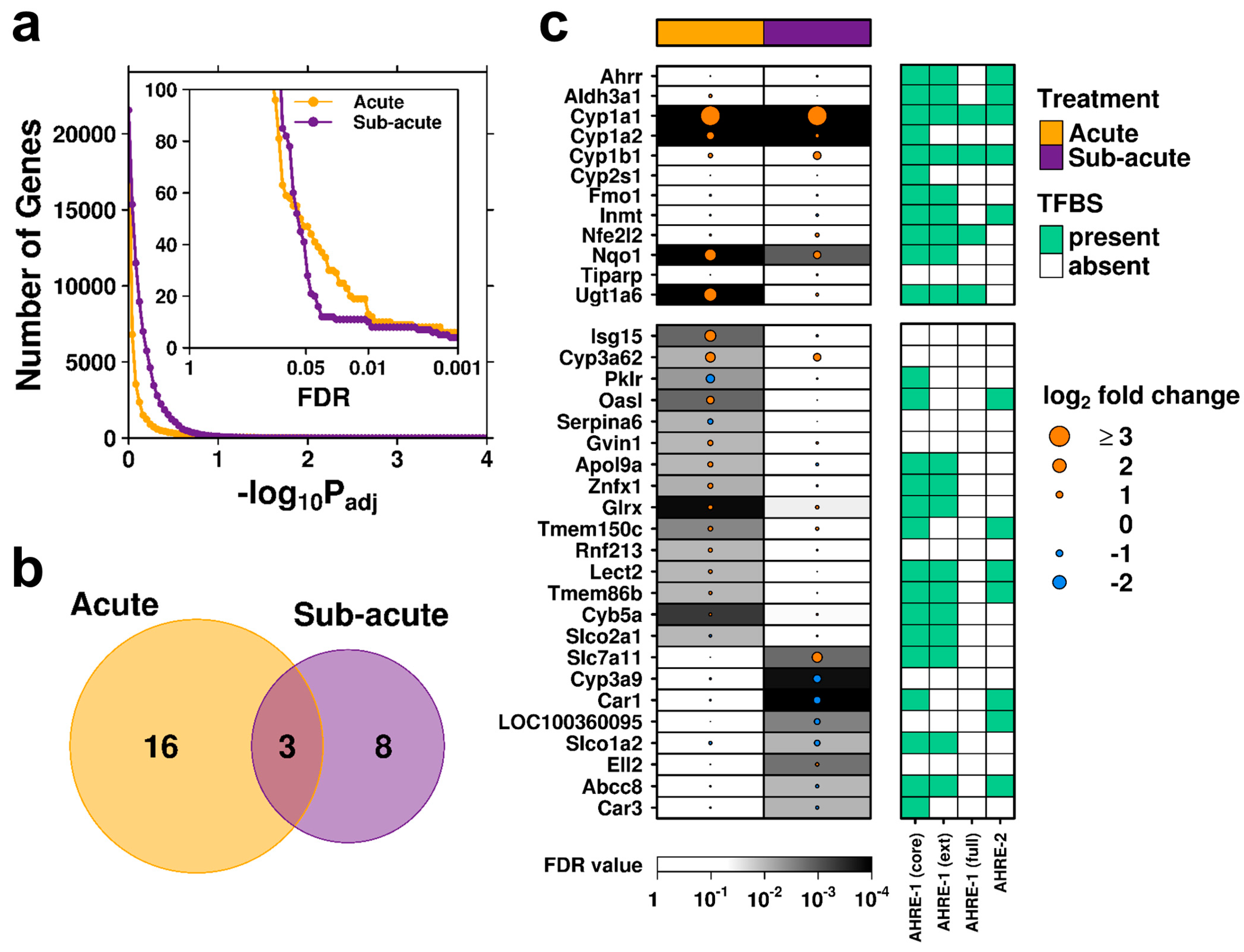
2.2. Transcriptomic Profiles of Acute and Subacute Treatments Differ Considerably
2.3. Rapid Recovery Observed Following Subacute Exposure to C2
2.4. Acute Exposure to C2 May Stimulate an Immune Response
2.5. Subacute Exposure to C2 Leads to Oxidative Stress
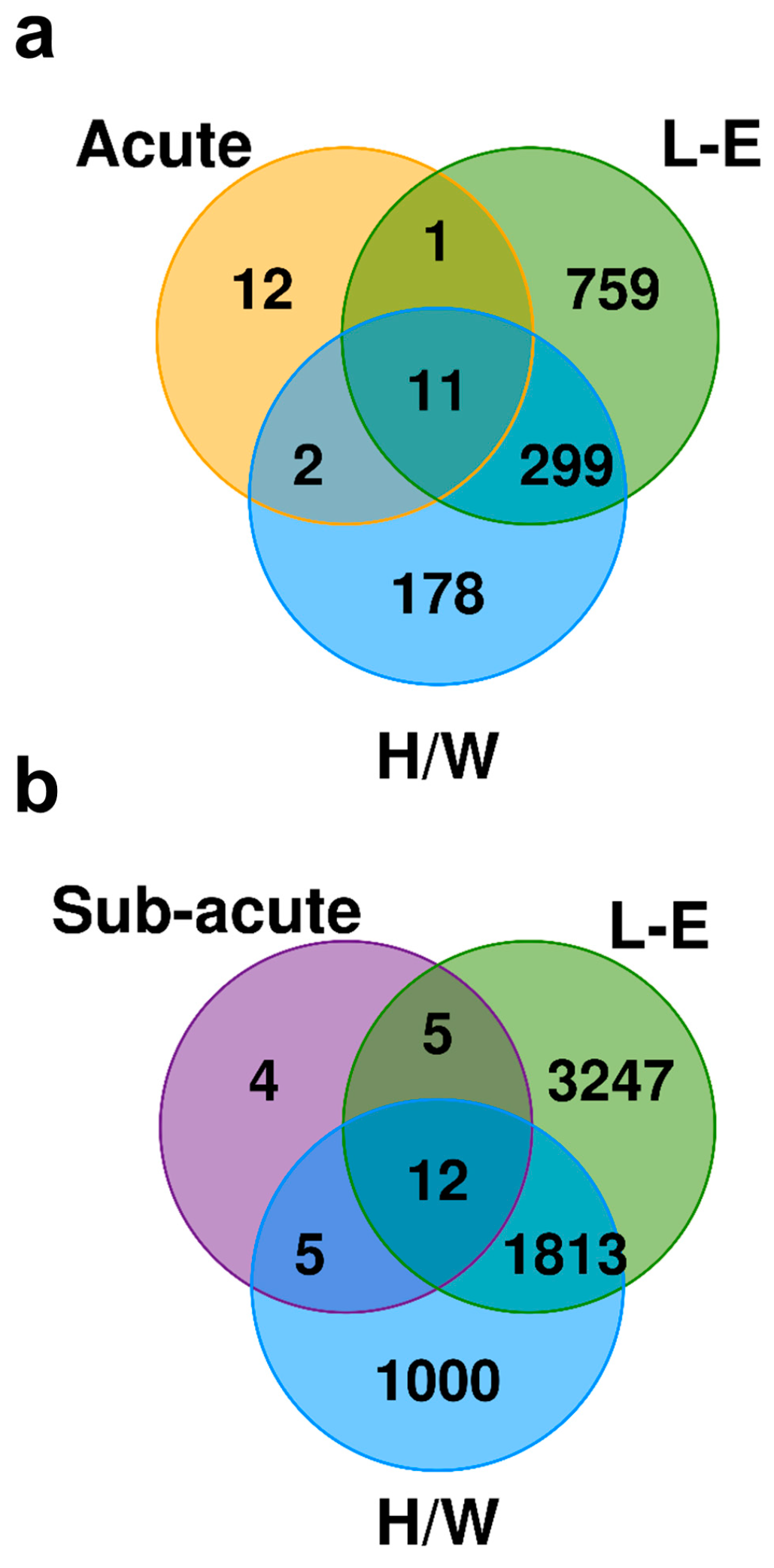
2.6. Exposure to C2 Leads to a Drastically Different Transcriptomic Profile From the Prototypical AHR Agonist TCDD
3. Discussion
4. Materials and Methods
4.1. Animal Handling and Sample Preparation
4.2. Data Generation and Preprocessing
4.3. Statistical Analysis and Visualizations
4.4. Transcription Factor Binding Site (TFBS) Analysis
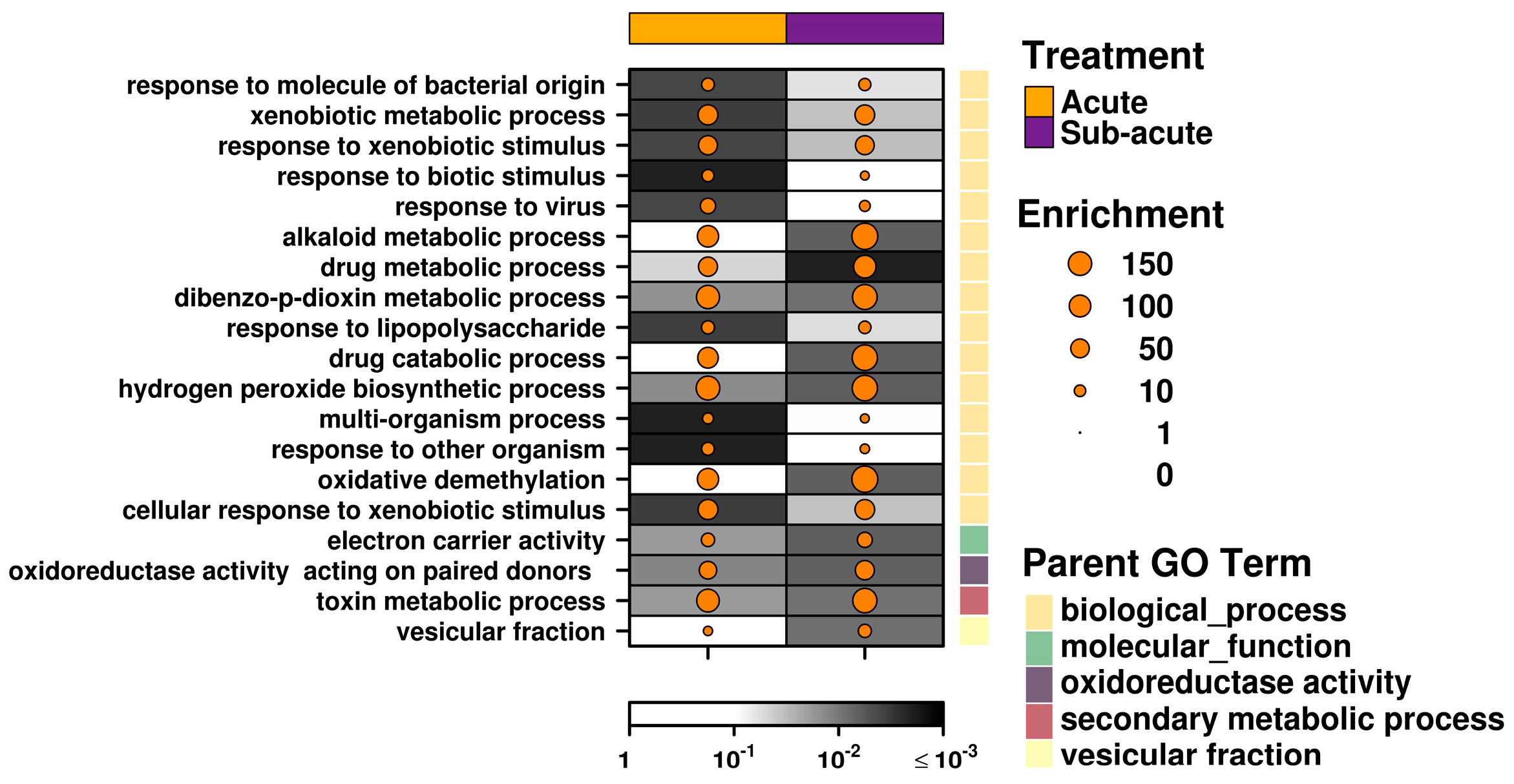
4.5. Pathway Analysis
4.6. Comparison to TCDD
4.7. Data Availability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AHR | Aryl hydrocarbon receptor |
| SAHRM | selective AHR modulator |
| LAQ | Laquinimod |
| DELAQ | Deethylated laquinimod |
| TCDD | 2,3,7,8-tetrachlorodibenzo-p-dioxin |
| C2 | IMA-08401; N-acetyl-N-phenyl-4-acetoxy-5-chloro-1,2-dihydro-1-methyl-2-oxo-quinoline-3-carboxamide |
| AHRE | AHR response elements |
| H/W | Han/Wistar (Kuopio) |
| L-E | Long–Evans (Turku/AB) |
References
- Vaz, F.; Silva, M.R.; Ascensao, J.L. Enhanced lymphokine-activated killer cell activity by an immunomodulator, Roquinimex. Br. J. Cancer 1995, 72, 1498–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirkey, B.L.; Slavin, S.; Vistica, B.P.; Podgor, M.J.; Gery, I. Immunomodulatory effects of linomide in animals immunized with immunopathogenic retinal antigens: Dissociation between different immune functions. Clin. Exp. Immunol. 1997, 108, 539–544. [Google Scholar] [CrossRef]
- Xiao, Z.Y.; Zhou, W.X.; Zhang, Y.X.; Cheng, J.P.; He, J.F.; Yang, R.F.; Yun, L.H. Inhibitory effect of linomide on lipopolysaccharide-induced proinflammatory cytokine tumor necrosis factor-alpha production in RAW264.7 macrophages through suppression of NF-kappaB, p38, and JNK activation. Immunol. Lett. 2007, 114, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.T.; Pili, R.; Qian, D.Z.; Dalrymple, S.L.; Garrison, J.B.; Kyprianou, N.; Bjork, A.; Olsson, A.; Leanderson, T. Identification of ABR-215050 as lead second generation quinoline-3-carboxamide anti-angiogenic agent for the treatment of prostate cancer. Prostate 2006, 66, 1768–1778. [Google Scholar] [CrossRef] [PubMed]
- Brunmark, C.; Runstrom, A.; Ohlsson, L.; Sparre, B.; Brodin, T.; Astrom, M.; Hedlund, G. The new orally active immunoregulator laquinimod (ABR-215062) effectively inhibits development and relapses of experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2002, 130, 163–172. [Google Scholar] [CrossRef]
- Yang, J.S.; Xu, L.Y.; Xiao, B.G.; Hedlund, G.; Link, H. Laquinimod (ABR-215062) suppresses the development of experimental autoimmune encephalomyelitis, modulates the Th1/Th2 balance and induces the Th3 cytokine TGF-beta in Lewis rats. J. Neuroimmunol. 2004, 156, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Polman, C.; Barkhof, F.; Sandberg-Wollheim, M.; Linde, A.; Nordle, O.; Nederman, T. Treatment with laquinimod reduces development of active MRI lesions in relapsing MS. Neurology 2005, 64, 987–991. [Google Scholar] [CrossRef] [PubMed]
- D’Haens, G.; Sandborn, W.J.; Colombel, J.F.; Rutgeerts, P.; Brown, K.; Barkay, H.; Sakov, A.; Haviv, A.; Feagan, B.G. A phase II study of laquinimod in Crohn’s disease. Gut 2015, 64, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Kaye, J.; Piryatinsky, V.; Birnberg, T.; Hingaly, T.; Raymond, E.; Kashi, R.; Amit-Romach, E.; Caballero, I.S.; Towfic, F.; Ator, M.A.; et al. Laquinimod arrests experimental autoimmune encephalomyelitis by activating the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2016, 113, E6145–E6152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comi, G.; Abramsky, O.; Arbizu, T.; Boyko, A.; Gold, R.; Havrdova, E.; Komoly, S.; Selmaj, K.; Sharrack, B.; Filippi, M. Oral laquinimod in patients with relapsing-remitting multiple sclerosis: 36-week double-blind active extension of the multi-centre, randomized, double-blind, parallel-group placebo-controlled study. Mult. Scler. 2010, 16, 1360–1366. [Google Scholar] [CrossRef]
- Filippi, M.; Rocca, M.A.; Pagani, E.; De Stefano, N.; Jeffery, D.; Kappos, L.; Montalban, X.; Boyko, A.N.; Comi, G. Placebo-controlled trial of oral laquinimod in multiple sclerosis: MRI evidence of an effect on brain tissue damage. J. Neurol. Neurosurg. Psychiatry 2014, 85, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, T.L.; Sorensen, P.S.; Selmaj, K.; Zipp, F.; Havrdova, E.; Cohen, J.A.; Sasson, N.; Gilgun-Sherki, Y.; Arnold, D.L. A randomized placebo-controlled phase III trial of oral laquinimod for multiple sclerosis. J. Neurol. 2014, 261, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, P.S.; Comi, G.; Vollmer, T.L.; Montalban, X.; Kappos, L.; Dadon, Y.; Gorfine, T.; Margalit, M.; Sasson, N.; Rubinchick, S.; et al. Laquinimod Safety Profile: Pooled Analyses from the ALLEGRO and BRAVO Trials. Int. J. MS Care 2017, 19, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.; Mahmoudjanlou, Y.; Duscha, A.; Massa, M.G.; Thone, J.; Esser, C.; Gold, R.; Haghikia, A. The immunomodulatory effect of laquinimod in CNS autoimmunity is mediated by the aryl hydrocarbon receptor. J. Neuroimmunol. 2016, 298, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Boutros, P.C.; Moffat, I.D.; Franc, M.A.; Tijet, N.; Tuomisto, J.; Pohjanvirta, R.; Okey, A.B. Dioxin-responsive AHRE-II gene battery: Identification by phylogenetic footprinting. Biochem. Biophys. Res. Commun. 2004, 321, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.D.; Prokopec, S.D.; Smith, A.B.; Okey, A.B.; Pohjanvirta, R.; Boutros, P.C. TCDD dysregulation of 13 AHR-target genes in rat liver. Toxicol. Appl. Pharmacol. 2014, 274, 445–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrulis, J.R.; Perdew, G.H. The role of chaperone proteins in the aryl hydrocarbon receptor core complex. Chem. Biol. Interact. 2002, 141, 25–40. [Google Scholar] [CrossRef] [Green Version]
- Reyes, H.; Reisz-Porszasz, S.; Hankinson, O. Identification of the Ah receptor nuclear translocator protein (Arnt) as a component of the DNA binding form of the Ah receptor. Science 1992, 256, 1193–1195. [Google Scholar] [CrossRef]
- Tijet, N.; Boutros, P.C.; Moffat, I.D.; Okey, A.B.; Tuomisto, J.; Pohjanvirta, R. Aryl hydrocarbon receptor regulates distinct dioxin-dependent and dioxin-independent gene batteries. Mol. Pharmacol. 2006, 69, 140–153. [Google Scholar] [CrossRef]
- Yeager, R.L.; Reisman, S.A.; Aleksunes, L.M.; Klaassen, C.D. Introducing the “TCDD-inducible AhR-Nrf2 gene battery”. Toxicol. Sci. 2009, 111, 238–246. [Google Scholar] [CrossRef]
- Prokopec, S.D.; Watson, J.D.; Lee, J.; Pohjanvirta, R.; Boutros, P.C. Sex-related differences in murine hepatic transcriptional and proteomic responses to TCDD. Toxicol. Appl. Pharmacol. 2015, 284, 188–196. [Google Scholar] [CrossRef] [Green Version]
- Prokopec, S.D.; Houlahan, K.E.; Sun, R.X.; Watson, J.D.; Yao, C.Q.; Lee, J.; P’ng, C.; Pang, R.; Wu, A.H.; Chong, L.C.; et al. Compendium of TCDD-mediated transcriptomic response datasets in mammalian model systems. BMC Genom. 2017, 18, 78. [Google Scholar] [CrossRef]
- Hou, L.; Zhang, X.; Wang, D.; Baccarelli, A. Environmental chemical exposures and human epigenetics. Int. J. Epidemiol. 2012, 41, 79–105. [Google Scholar] [CrossRef]
- Patrizi, B.; Siciliani de Cumis, M. TCDD Toxicity Mediated by Epigenetic Mechanisms. Int. J. Mol. Sci. 2018, 19, 4101. [Google Scholar] [CrossRef]
- Willett, K.L. Considering Epigenetics in Adverse Outcome Pathways. In A Systems Biology Approach to Advancing Adverse Outcome Pathways for Risk Assessment; Natàlia Garcia-Reyero, C.A.M., Ed.; Springer International Publishing: Cham, Switzerland, 2011. [Google Scholar]
- Singh, N.P.; Singh, U.P.; Singh, B.; Price, R.L.; Nagarkatti, M.; Nagarkatti, P.S. Activation of aryl hydrocarbon receptor (AhR) leads to reciprocal epigenetic regulation of FoxP3 and IL-17 expression and amelioration of experimental colitis. PLoS ONE 2011, 6, e23522. [Google Scholar] [CrossRef]
- Brunnberg, S.; Elin Swedenborg, E.; Gustafsson, J.-Å. Functional Interactions of AHR with other Receptors. In The AH Receptor in Biology and Toxicology; Pohjanvirta, R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011. [Google Scholar]
- Matsumura, F. Nongenomic Route of Action of TCDD: Identity, Characteristics, and Toxicological Significance. In The AH Receptor in Biology and Toxicology; Pohjanvirta, R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011. [Google Scholar]
- Ohtake, F.; Kato, S. The E3 Ubiquitin Ligase Activity of Transcription Factor AHR Permits Nongenomic Regulation of Biological Pathways. In The AH Receptor in Biology and Toxicology; Pohjanvirta, R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011. [Google Scholar]
- Pohjanvirta, R.; Tuomisto, J. Short-term toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in laboratory animals: Effects, mechanisms, and animal models. Pharmacol. Rev. 1994, 46, 483–549. [Google Scholar]
- Xu, J.; Ye, Y.; Huang, F.; Chen, H.; Wu, H.; Huang, J.; Hu, J.; Xia, D.; Wu, Y. Association between dioxin and cancer incidence and mortality: A meta-analysis. Sci. Rep. 2016, 6, 38012. [Google Scholar] [CrossRef]
- Huang, T.; Jiang, W.; Ling, Z.; Zhao, Y.; Gao, H.; Ma, J. Trend of cancer risk of Chinese inhabitants to dioxins due to changes in dietary patterns: 1980–2009. Sci. Rep. 2016, 6, 21997. [Google Scholar] [CrossRef] [Green Version]
- Ansbaugh, N.; Shannon, J.; Mori, M.; Farris, P.E.; Garzotto, M. Agent Orange as a risk factor for high-grade prostate cancer. Cancer 2013, 119, 2399–2404. [Google Scholar] [CrossRef]
- Warner, M.; Mocarelli, P.; Samuels, S.; Needham, L.; Brambilla, P.; Eskenazi, B. Dioxin exposure and cancer risk in the Seveso Women’s Health Study. Environ. Health Perspect. 2011, 119, 1700–1705. [Google Scholar] [CrossRef]
- Pelcl, T.; Skrha, J., Jr.; Prazny, M.; Vlckova, S.; Fenclova, Z.; Navratil, T.; Malik, J.; Diblik, P.; Zikan, V.; Pelclova, D. Diabetes, Cardiovascular Disorders and 2,3,7,8-Tetrachlorodibenzo-p-Dioxin Body Burden in Czech Patients 50 Years After the Intoxication. Basic Clin. Pharmacol. Toxicol. 2018, 123, 356–359. [Google Scholar] [CrossRef]
- Mannetje, A.T.; Eng, A.; Walls, C.; Dryson, E.; Douwes, J.; Bertazzi, P.; Ryder-Lewis, S.; Scott, D.; Brooks, C.; McLean, D.; et al. Morbidity in New Zealand pesticide producers exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Environ. Int. 2018, 110, 22–31. [Google Scholar] [CrossRef]
- Warner, M.; Mocarelli, P.; Brambilla, P.; Wesselink, A.; Samuels, S.; Signorini, S.; Eskenazi, B. Diabetes, metabolic syndrome, and obesity in relation to serum dioxin concentrations: The Seveso women’s health study. Environ. Health Perspect. 2013, 121, 906–911. [Google Scholar] [CrossRef]
- Fernandez-Salguero, P.M.; Hilbert, D.M.; Rudikoff, S.; Ward, J.M.; Gonzalez, F.J. Aryl-hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol. Appl. Pharmacol. 1996, 140, 173–179. [Google Scholar] [CrossRef]
- Mimura, J.; Yamashita, K.; Nakamura, K.; Morita, M.; Takagi, T.N.; Nakao, K.; Ema, M.; Sogawa, K.; Yasuda, M.; Katsuki, M.; et al. Loss of teratogenic response to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice lacking the Ah (dioxin) receptor. Genes Cells 1997, 2, 645–654. [Google Scholar] [CrossRef]
- Okey, A.B.; Franc, M.A.; Moffat, I.D.; Tijet, N.; Boutros, P.C.; Korkalainen, M.; Tuomisto, J.; Pohjanvirta, R. Toxicological implications of polymorphisms in receptors for xenobiotic chemicals: The case of the aryl hydrocarbon receptor. Toxicol. Appl. Pharmacol. 2005, 207, 43–51. [Google Scholar] [CrossRef]
- Tuvesson, H.; Hallin, I.; Ellman, M.; Sparre, B.; Gunnarsson, P.O.; Seidegard, J. In vitro metabolism and in vivo pharmacokinetics of quinoline 3-carboxamide derivatives in various species. Xenobiotica 2005, 35, 293–304. [Google Scholar] [CrossRef]
- Mahiout, S.; Linden, J.; Esteban, J.; Sanchez-Perez, I.; Sankari, S.; Pettersson, L.; Hakansson, H.; Pohjanvirta, R. Toxicological characterisation of two novel selective aryl hydrocarbon receptor modulators in Sprague-Dawley rats. Toxicol. Appl. Pharmacol. 2017, 326, 54–65. [Google Scholar] [CrossRef]
- Lensu, S.; Tiittanen, P.; Linden, J.; Tuomisto, J.; Pohjanvirta, R. Effects of a single exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on macro- and microstructures of feeding and drinking in two differently TCDD-sensitive rat strains. Pharmacol. Biochem. Behav. 2011, 99, 487–499. [Google Scholar] [CrossRef]
- Yao, C.Q.; Prokopec, S.D.; Watson, J.D.; Pang, R.; P’ng, C.; Chong, L.C.; Harding, N.J.; Pohjanvirta, R.; Okey, A.B.; Boutros, P.C. Inter-strain heterogeneity in rat hepatic transcriptomic responses to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Toxicol. Appl. Pharmacol. 2012, 260, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Boutros, P.C.; Yao, C.Q.; Watson, J.D.; Wu, A.H.; Moffat, I.D.; Prokopec, S.D.; Smith, A.B.; Okey, A.B.; Pohjanvirta, R. Hepatic transcriptomic responses to TCDD in dioxin-sensitive and dioxin-resistant rats during the onset of toxicity. Toxicol Appl. Pharmacol. 2011, 251, 119–129. [Google Scholar] [CrossRef]
- Lenschow, D.J.; Lai, C.; Frias-Staheli, N.; Giannakopoulos, N.V.; Lutz, A.; Wolff, T.; Osiak, A.; Levine, B.; Schmidt, R.E.; Garcia-Sastre, A.; et al. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc. Natl. Acad. Sci. USA 2007, 104, 1371–1376. [Google Scholar] [CrossRef] [Green Version]
- Hsiang, T.Y.; Zhao, C.; Krug, R.M. Interferon-induced ISG15 conjugation inhibits influenza A virus gene expression and replication in human cells. J. Virol. 2009, 83, 5971–5977. [Google Scholar] [CrossRef]
- Kuang, Z.; Seo, E.J.; Leis, J. Mechanism of inhibition of retrovirus release from cells by interferon-induced gene ISG15. J. Virol. 2011, 85, 7153–7161. [Google Scholar] [CrossRef]
- Bogunovic, D.; Byun, M.; Durfee, L.A.; Abhyankar, A.; Sanal, O.; Mansouri, D.; Salem, S.; Radovanovic, I.; Grant, A.V.; Adimi, P.; et al. Mycobacterial disease and impaired IFN-gamma immunity in humans with inherited ISG15 deficiency. Science 2012, 337, 1684–1688. [Google Scholar] [CrossRef]
- Ishibashi, M.; Wakita, T.; Esumi, M. 2’,5’-Oligoadenylate synthetase-like gene highly induced by hepatitis C virus infection in human liver is inhibitory to viral replication in vitro. Biochem. Biophys. Res. Commun. 2010, 392, 397–402. [Google Scholar] [CrossRef]
- Guo, X.; Li, X.; Xu, Y.; Sun, T.; Yang, G.; Wu, Z.; Li, E. Identification of OASL d, a splice variant of human OASL, with antiviral activity. Int. J. Biochem. Cell Biol. 2012, 44, 1133–1138. [Google Scholar] [CrossRef]
- Klamp, T.; Boehm, U.; Schenk, D.; Pfeffer, K.; Howard, J.C. A giant GTPase, very large inducible GTPase-1, is inducible by IFNs. J. Immunol. 2003, 171, 1255–1265. [Google Scholar] [CrossRef]
- Machado, P.; Pereira, R.; Rocha, A.M.; Manco, L.; Fernandes, N.; Miranda, J.; Ribeiro, L.; do Rosario, V.E.; Amorim, A.; Gusmao, L.; et al. Malaria: Looking for selection signatures in the human PKLR gene region. Br. J. Haematol. 2010, 149, 775–784. [Google Scholar] [CrossRef]
- Hill, L.A.; Bodnar, T.S.; Weinberg, J.; Hammond, G.L. Corticosteroid-binding globulin is a biomarker of inflammation onset and severity in female rats. J. Endocrinol. 2016, 230, 215–225. [Google Scholar] [CrossRef] [Green Version]
- Bannai, S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J. Biol. Chem. 1986, 261, 2256–2263. [Google Scholar]
- Kaleeba, J.A.; Berger, E.A. Kaposi’s sarcoma-associated herpesvirus fusion-entry receptor: Cystine transporter xCT. Science 2006, 311, 1921–1924. [Google Scholar] [CrossRef]
- Pampliega, O.; Domercq, M.; Soria, F.N.; Villoslada, P.; Rodriguez-Antiguedad, A.; Matute, C. Increased expression of cystine/glutamate antiporter in multiple sclerosis. J. Neuroinflamm. 2011, 8, 63. [Google Scholar] [CrossRef]
- Kuhara, M.; Wang, J.; Flores, M.J.; Qiao, Z.; Koizumi, Y.; Koyota, S.; Taniguchi, N.; Sugiyama, T. Sexual dimorphism in LEC rat liver: Suppression of carbonic anhydrase III by copper accumulation during hepatocarcinogenesis. Biomed. Res. 2011, 32, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Moffat, I.D.; Boutros, P.C.; Chen, H.; Okey, A.B.; Pohjanvirta, R. Aryl hydrocarbon receptor (AHR)-regulated transcriptomic changes in rats sensitive or resistant to major dioxin toxicities. BMC Genom. 2010, 11, 263. [Google Scholar] [CrossRef]
- Watson, J.D.; Prokopec, S.D.; Smith, A.B.; Okey, A.B.; Pohjanvirta, R.; Boutros, P.C. 2,3,7,8 Tetrachlorodibenzo-p-dioxin-induced RNA abundance changes identify Ackr3, Col18a1, Cyb5a and Glud1 as candidate mediators of toxicity. Arch. Toxicol. 2017, 91, 325–338. [Google Scholar] [CrossRef]
- Mahiout, S.; Tagliabue, S.G.; Nasri, A.; Omoruyi, I.M.; Pettersson, L.; Bonati, L.; Pohjanvirta, R. In vitro toxicity and in silico docking analysis of two novel selective AH-receptor modulators. Toxicol. In Vitro 2018, 52, 178–188. [Google Scholar] [CrossRef]
- Pohjanvirta, R.; Vartiainen, T.; Uusi-Rauva, A.; Monkkonen, J.; Tuomisto, J. Tissue distribution, metabolism, and excretion of 14C-TCDD in a TCDD-susceptible and a TCDD-resistant rat strain. Pharmacol. Toxicol. 1990, 66, 93–100. [Google Scholar] [CrossRef]
- Kopec, A.K.; Burgoon, L.D.; Ibrahim-Aibo, D.; Burg, A.R.; Lee, A.W.; Tashiro, C.; Potter, D.; Sharratt, B.; Harkema, J.R.; Rowlands, J.C.; et al. Automated dose-response analysis and comparative toxicogenomic evaluation of the hepatic effects elicited by TCDD, TCDF, and PCB126 in C57BL/6 mice. Toxicol. Sci. 2010, 118, 286–297. [Google Scholar] [CrossRef]
- Nault, R.; Forgacs, A.L.; Dere, E.; Zacharewski, T.R. Comparisons of differential gene expression elicited by TCDD, PCB126, betaNF, or ICZ in mouse hepatoma Hepa1c1c7 cells and C57BL/6 mouse liver. Toxicol. Lett. 2013, 223, 52–59. [Google Scholar] [CrossRef]
- Wang, T.; Wyrick, K.L.; Pecka, M.R.; Wills, T.B.; Vorderstrasse, B.A. Mechanistic exploration of AhR-mediated host protection against Streptococcus pneumoniae infection. Int. Immunopharmacol. 2012, 13, 490–498. [Google Scholar] [CrossRef] [Green Version]
- DeKrey, G.K.; Teagarden, R.E.; Lenberg, J.L.; Titus, R.G. 2,3,7,8-tetrachlorodibenzo-p-dioxin slows the progression of experimental cutaneous Leishmaniasis in susceptible BALB/c and SCID mice. PLoS ONE 2013, 8, e76259. [Google Scholar] [CrossRef]
- Rannug, A.; Rannug, U. The tryptophan derivative 6-formylindolo[3,2-b]carbazole, FICZ, a dynamic mediator of endogenous aryl hydrocarbon receptor signaling, balances cell growth and differentiation. Crit. Rev. Toxicol. 2018, 48, 555–574. [Google Scholar] [CrossRef]
- Veldhoen, M.; Hirota, K.; Westendorf, A.M.; Buer, J.; Dumoutier, L.; Renauld, J.C.; Stockinger, B. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 2008, 453, 106–109. [Google Scholar] [CrossRef]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef]
- Duarte, J.H.; Di Meglio, P.; Hirota, K.; Ahlfors, H.; Stockinger, B. Differential influences of the aryl hydrocarbon receptor on Th17 mediated responses in vitro and in vivo. PLoS ONE 2013, 8, e79819. [Google Scholar] [CrossRef]
- Stohs, S.J.; Hassoun, E.A. Dioxin-Activated AHR: Toxic Responses and the Induction of Oxidative Stress. In The AH Receptor in Biology and Toxicology; Pohjanvirta, R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011. [Google Scholar]
- Park, S.L.; Justiniano, R.; Williams, J.D.; Cabello, C.M.; Qiao, S.; Wondrak, G.T. The Tryptophan-Derived Endogenous Aryl Hydrocarbon Receptor Ligand 6-Formylindolo[3,2-b]Carbazole Is a Nanomolar UVA Photosensitizer in Epidermal Keratinocytes. J. Investig. Dermatol. 2015, 135, 1649–1658. [Google Scholar] [CrossRef]
- Brem, R.; Macpherson, P.; Guven, M.; Karran, P. Oxidative stress induced by UVA photoactivation of the tryptophan UVB photoproduct 6-formylindolo[3,2-b]carbazole (FICZ) inhibits nucleotide excision repair in human cells. Sci. Rep. 2017, 7, 4310. [Google Scholar] [CrossRef]
- Pohjanvirta, R.; Kulju, T.; Morselt, A.F.; Tuominen, R.; Juvonen, R.; Rozman, K.; Mannisto, P.; Collan, Y.; Sainio, E.L.; Tuomisto, J. Target tissue morphology and serum biochemistry following 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) exposure in a TCDD-susceptible and a TCDD-resistant rat strain. Fundam. Appl. Toxicol. 1989, 12, 698–712. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [Green Version]
- Dai, M.; Wang, P.; Boyd, A.D.; Kostov, G.; Athey, B.; Jones, E.G.; Bunney, W.E.; Myers, R.M.; Speed, T.P.; Akil, H.; et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005, 33, e175. [Google Scholar] [CrossRef]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3. [Google Scholar] [CrossRef]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [Green Version]
- P’ng, C.; Green, J.; Chong, L.C.; Waggott, D.; Prokopec, S.D.; Shamsi, M.; Nguyen, F.; Mak, D.Y.F.; Lam, F.; Albuquerque, M.A.; et al. BPG: Seamless, automated and interactive visualization of scientific data. BMC Bioinform. 2019, 20, 42. [Google Scholar] [CrossRef]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef]
- Denison, M.S.; Fisher, J.M.; Whitlock, J.P., Jr. The DNA recognition site for the dioxin-Ah receptor complex. Nucleotide sequence and functional analysis. J. Biol. Chem. 1988, 263, 17221–17224. [Google Scholar]
- Sogawa, K.; Numayama-Tsuruta, K.; Takahashi, T.; Matsushita, N.; Miura, C.; Nikawa, J.; Gotoh, O.; Kikuchi, Y.; Fujii-Kuriyama, Y. A novel induction mechanism of the rat CYP1A2 gene mediated by Ah receptor-Arnt heterodimer. Biochem. Biophys. Res. Commun. 2004, 318, 746–755. [Google Scholar] [CrossRef]
- Zeeberg, B.R.; Qin, H.; Narasimhan, S.; Sunshine, M.; Cao, H.; Kane, D.W.; Reimers, M.; Stephens, R.M.; Bryant, D.; Burt, S.K.; et al. High-Throughput GoMiner, an ‘industrial-strength’ integrative gene ontology tool for interpretation of multiple-microarray experiments, with application to studies of Common Variable Immune Deficiency (CVID). BMC Bioinform. 2005, 6, 168. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Sprague-Dawley (C2) | Time Point | H/W (TCDD) | L-E (TCDD) | |
|---|---|---|---|---|---|
| Car1 | Acute | −0.25 | 19 h | −0.29 * | −0.19 |
| Subacute | −1.18 *** | 4 days | −0.87 *** | −0.05 | |
| 10 days | −2.24 *** | −0.16 | |||
| Car3 | Acute | −0.22 | 19 h | −0.64 | −1.38 ** |
| Subacute | −0.48 ** | 4 days | 0.04 | −5.08 *** | |
| 10 days | −0.68 | −5.13 *** |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prokopec, S.D.; Pohjanvirta, R.; Mahiout, S.; Pettersson, L.; Boutros, P.C. Transcriptomic Impact of IMA-08401, a Novel AHR Agonist Resembling Laquinimod, on Rat Liver. Int. J. Mol. Sci. 2019, 20, 1370. https://doi.org/10.3390/ijms20061370
Prokopec SD, Pohjanvirta R, Mahiout S, Pettersson L, Boutros PC. Transcriptomic Impact of IMA-08401, a Novel AHR Agonist Resembling Laquinimod, on Rat Liver. International Journal of Molecular Sciences. 2019; 20(6):1370. https://doi.org/10.3390/ijms20061370
Chicago/Turabian StyleProkopec, Stephenie D., Raimo Pohjanvirta, Selma Mahiout, Lars Pettersson, and Paul C. Boutros. 2019. "Transcriptomic Impact of IMA-08401, a Novel AHR Agonist Resembling Laquinimod, on Rat Liver" International Journal of Molecular Sciences 20, no. 6: 1370. https://doi.org/10.3390/ijms20061370
APA StyleProkopec, S. D., Pohjanvirta, R., Mahiout, S., Pettersson, L., & Boutros, P. C. (2019). Transcriptomic Impact of IMA-08401, a Novel AHR Agonist Resembling Laquinimod, on Rat Liver. International Journal of Molecular Sciences, 20(6), 1370. https://doi.org/10.3390/ijms20061370