Multifactorial Contribution of Notch Signaling in Head and Neck Squamous Cell Carcinoma



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
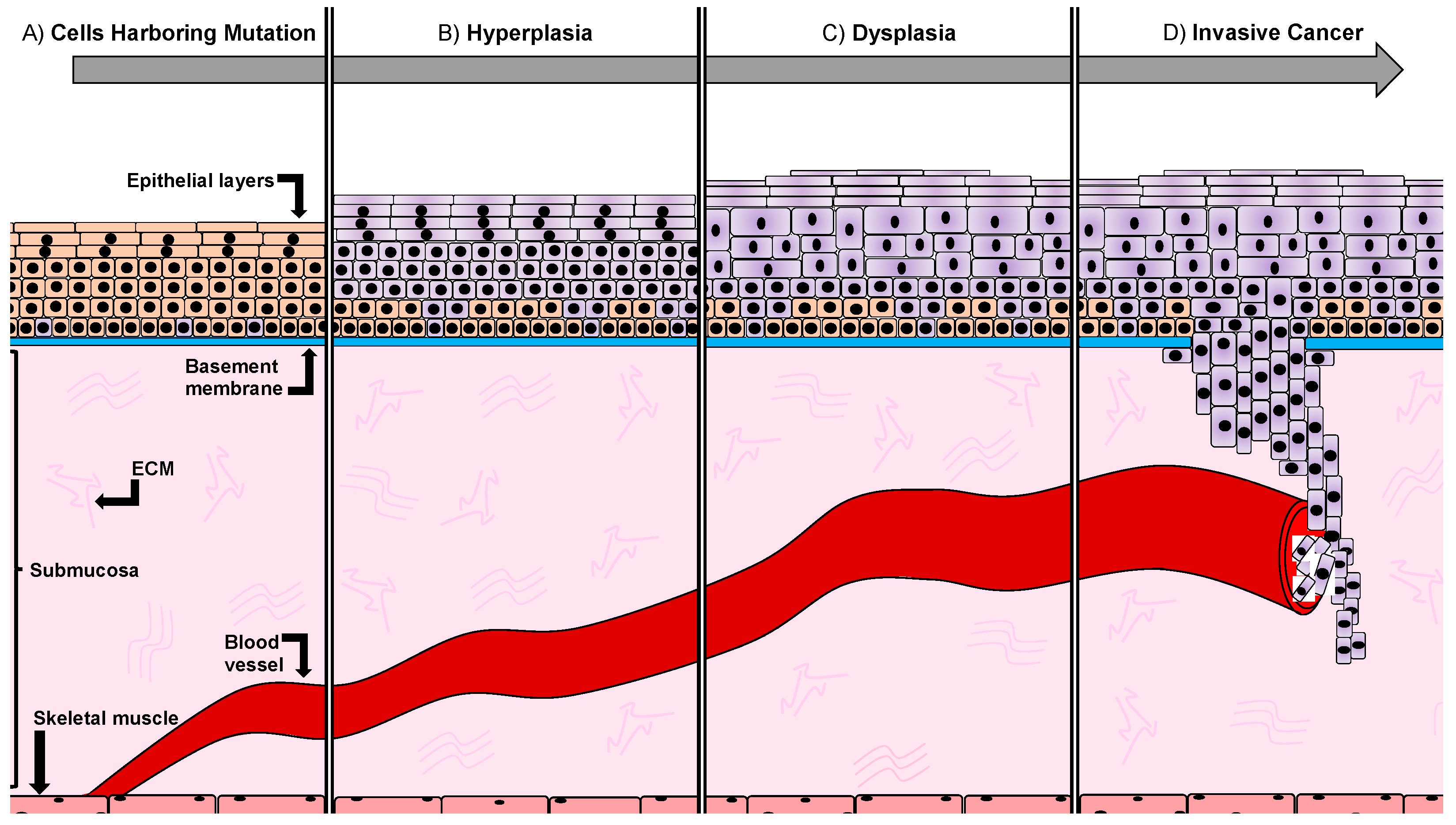
1.1. Developmental Stages of Oral Squamous Carcinoma
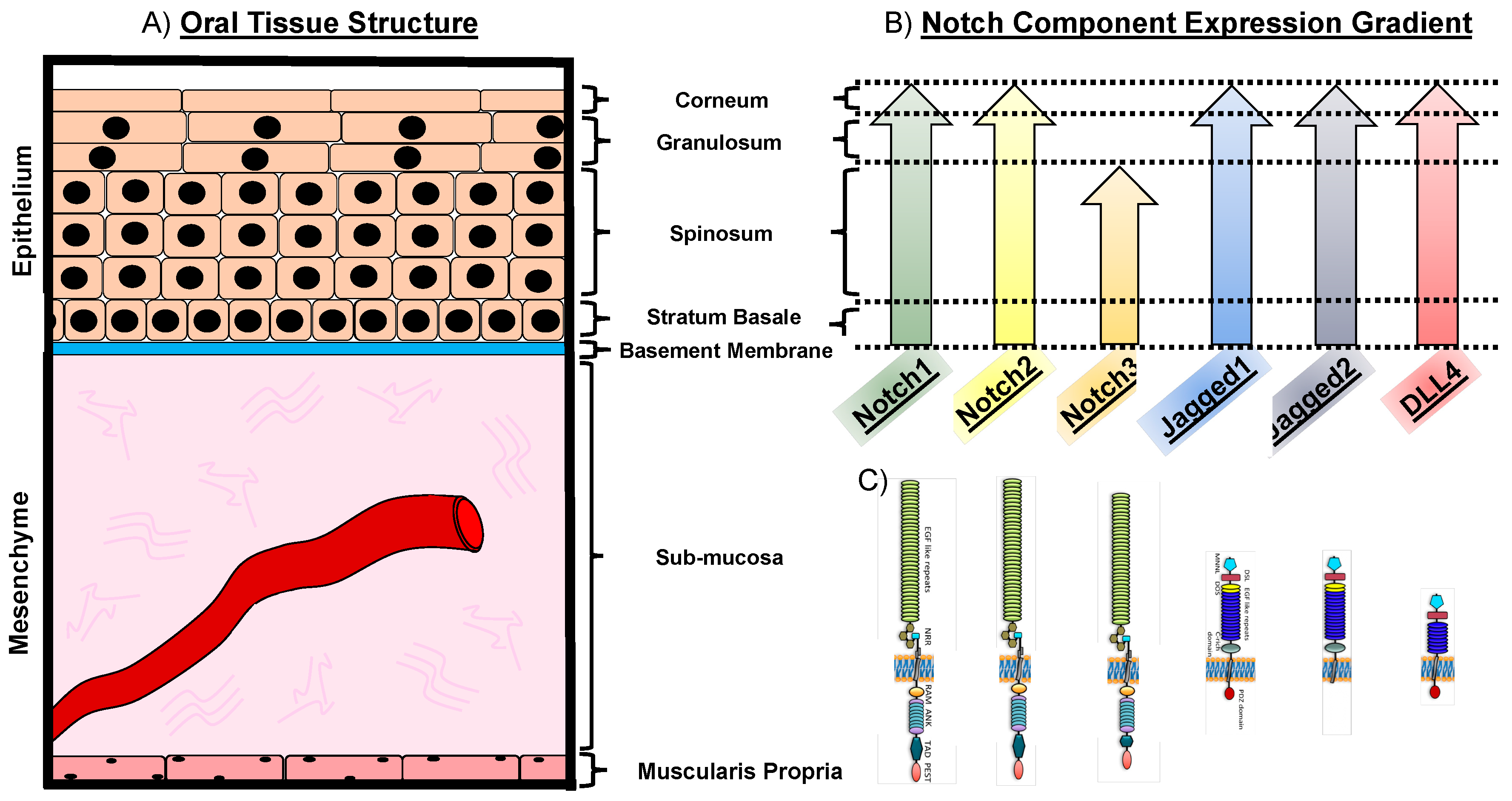
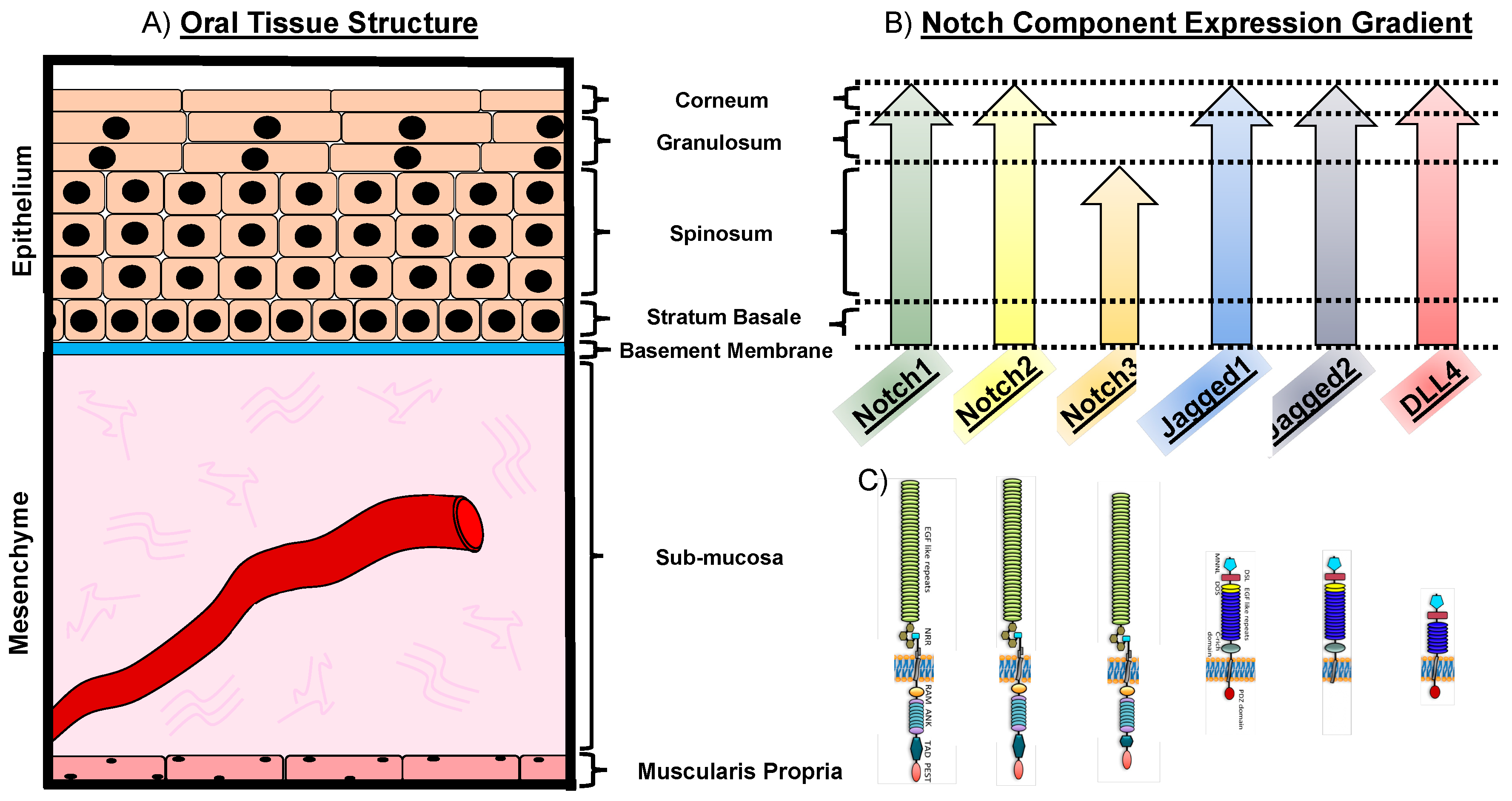
1.2. The Notch Pathway in Oral Physiology
1.3. Notch in Oral Pathological Conditions
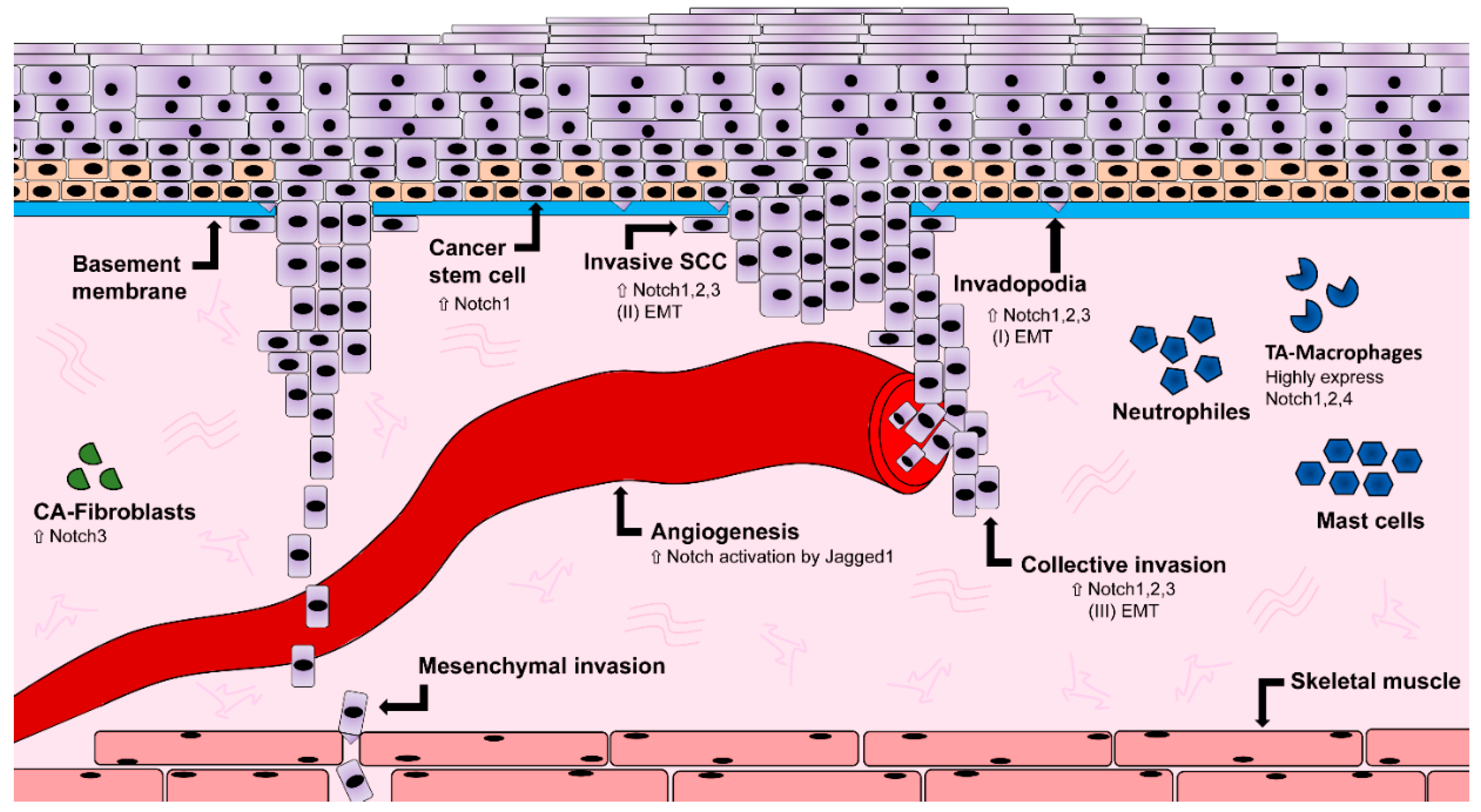
1.4. Notch Expression in Oral Squamous Cell Carcinoma
1.5. Notch and the Influence on Vasculature
1.6. Notch and Epithelial-to-Mesenchymal Transition
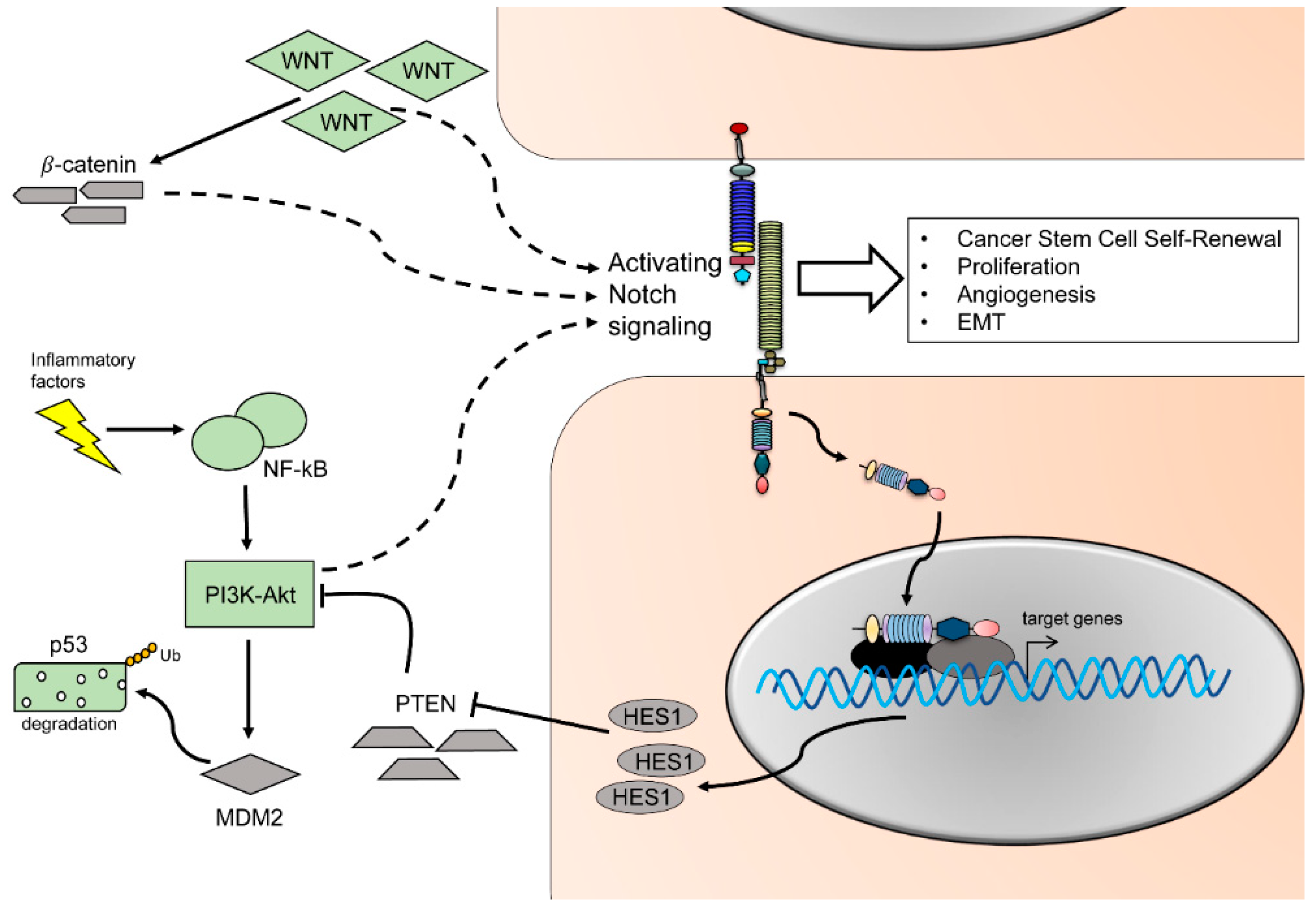
1.7. Notch and Cancer Stem Cells
1.8. The Dual Role of Notch as an Oncogene and a Tumor-Suppressor
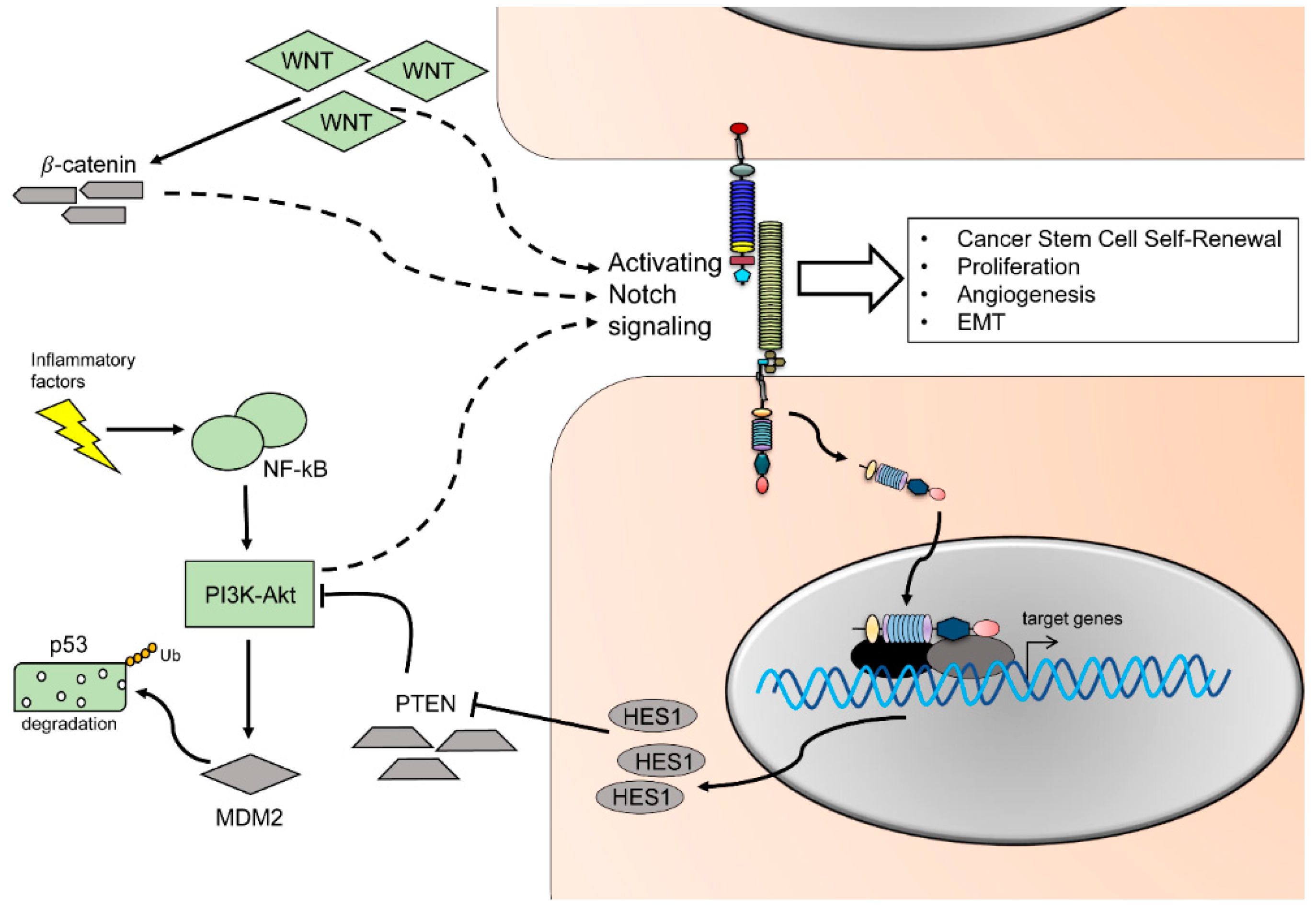
1.9. Crosstalk between Notch and Other Major Pathways
1.10. Current Animal Models for Oral Cancer
1.10.1. Chemically Induced Mouse Models of Oral Cancer
1.10.2. Transgenic Murine Models
1.11. Therapeutic Strategies to Target Notch Signaling
2. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADAM | A Disintegrin And Metalloproteinase |
| APC | Adenomatous polyposis coli |
| CAF | Cancer-associated fibroblast |
| CBF1 | C promoter-binding factor 1 |
| CD44 | Cluster of differentiation 44 |
| cK | Cytokeratin |
| CSC | Cancer stem cell |
| CUP | Carcinoma of unknown primary |
| CXCR4 | CXC chemokine receptor 4 |
| DC | Dendritic cells |
| DLL | Delta-like |
| EGF(R) | Epidermal growth factor (receptor) |
| EMTEphHB3 | Epithelial-to-mesenchymal transitionEphrin type-B receptor 3 |
| ERK | Extracellular regulated kinase |
| GSI DAPT | Gamma-secretase inhibitors N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester |
| Hes | Hairy and enhancer of split |
| Hey | Hairy/enhancer-of-split related with YRPW motif |
| HGF | Hepatocyte growth factor |
| HH | Hedgehog |
| Hif1a | Hypoxia-inducible factor 1 alpha subunit |
| HNSCC | Head and neck squamous cell carcinoma |
| Ikk2 | Inhibitor of nuclear factor kappa-B kinase subunit kinase β |
| IL | Interleukin |
| ILC | Innate lymphoid cell |
| JNK | c-Jun N-terminal kinase |
| KLF5 | Kruppel like factor 5 |
| K-Ras | Kirsten rat sarcoma viral oncogene homolog |
| Lfng | Lunatic Fringe |
| LNR | Cysteine-rich Lin-12/Notch Repeats |
| mAb | Monoclonal antibody |
| MAPK | Mitogen-activated protein kinase |
| MDM2 | Mouse double minute homolog 2 |
| MMP | Matrix metalloproteinase |
| Mnfg | Maniac Fringe |
| Myc | Myc proto-oncogene |
| NCR | Natural cytotoxicity receptor |
| NECD | Notch extracellular domain |
| NEXT | Notch extracellular truncation |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NICD | Notch intracellular domain |
| NK | Natural killer cells |
| NLS | Nuclear localizing sequence |
| Nox1 | Nicotidamide adenine dinucleotide phosphate oxidase 1 |
| OCT4 | Octamer-binding transcription factor 4 |
| OSCC | Oral Squamous Cell Carcinoma |
| PI3K-AKT | Phosphoinositide 3-kinase v-Akt murine thymoma viral oncogene |
| Pparg | Peroxisome proliferator-activated receptor gamma |
| PTEN | Phosphatase and tensin homolog |
| RBPj | Recombining binding protein suppressor of hairless |
| Rfng | Radical Fringe |
| Shh | Sonic hedgehog |
| STAT3 | Signal transducer and activator of transcription 3 |
| SOX2 | Sex determining region Y-box 2 |
| TAM | Tumor associated macrophages |
| TCF | T-cell factor/lymphoid enhancer factor |
| T-ALL | T cell acute lymphoblastic leukemia |
| TCF | T-cell factor |
| TGF-β | Transforming growth factor-β |
| Tgfbr1 | Transforming growth factor-β receptor 1 |
| TNF-α | Tumor necrosis factor alpha |
| VEGF(R) | Vascular endothelial growth factor (receptor) |
| WHO | World Health Organization |
| Wnt | Wingless/Integrated |
| α-SMA | α-smooth muscle actin |
| 4NQO | 4-nitroquinoline 1-oxide |
References
- Sanderson, R.J.; Ironside, J.A.D. Squamous cell carcinomas of the head and neck. BMJ 2002, 325, 822–827. [Google Scholar] [CrossRef]
- Vargas, H.; Pitman, K.T.; Johnson, J.T.; Galati, L.T. More aggressive behavior of squamous cell carcinoma of the anterior tongue in young women. Laryngoscope 2000, 110, 1623–1626. [Google Scholar] [CrossRef]
- Gilroy, J.S.; Morris, C.G.; Amdur, R.J.; Mendenhall, W.M. Impact of young age on prognosis for head and neck cancer: A matched-pair analysis. Head Neck 2005, 27, 269–273. [Google Scholar] [CrossRef]
- Goldenberg, D.; Brooksby, C.; Hollenbeak, C.S. Age as a determinant of outcomes for patients with oral cancer. Oral Oncol. 2009, 45, e57–e61. [Google Scholar] [CrossRef] [PubMed]
- Ascani, G.; Balercia, P.; Messi, M.; Lupi, L.; Goteri, G.; Filosa, A.; Stramazzotti, D.; Pieramici, T.; Rubini, C. Angiogenesis in oral squamous cell carcinoma. Acta Otorhinol. Ital. Organo Uff. Della Soc. Ital. Otorinolaringol. E Chir. Cerv.-Facc. 2005, 25, 13–17. [Google Scholar]
- Warnakulasuriya, S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 2009, 45, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Leethanakul, C.; Patel, V.; Gillespie, J.; Pallente, M.; Ensley, J.F.; Koontongkaew, S.; Liotta, L.A.; Emmert-Buck, M.; Gutkind, J.S. Distinct pattern of expression of differentiation and growth-related genes in squamous cell carcinomas of the head and neck revealed by the use of laser capture microdissection and cDNA arrays. Oncogene 2000, 19, 3220–3224. [Google Scholar] [CrossRef]
- Baik, F.M.; Hansen, S.; Knoblaugh, S.E.; Sahetya, D.; Mitchell, R.M.; Xu, C.; Olson, J.M.; Parrish-Novak, J.; Méndez, E. Fluorescence Identification of Head and Neck Squamous Cell Carcinoma and High-Risk Oral Dysplasia With BLZ-100, a Chlorotoxin-Indocyanine Green Conjugate. JAMA Otolaryngol. Head Neck Surg. 2016, 142, 330–338. [Google Scholar] [CrossRef]
- Gillespie, M.B.; Albergotti, W.G.; Eisele, D.W. Recurrent salivary gland cancer. Curr. Treat. Options Oncol. 2012, 13, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.N.; Hoffman, M.P. Salivary gland development: A template for regeneration. Semin. Cell Dev. Biol. 2014, 25, 52–60. [Google Scholar] [CrossRef]
- Emmerson, E.; Knox, S.M. Salivary gland stem cells: A review of development, regeneration and cancer. Genesis 2018, 56, e23211. [Google Scholar] [CrossRef] [PubMed]
- Konings, A.W.T.; Coppes, R.P.; Vissink, A. On the mechanism of salivary gland radiosensitivity. Int. J. Radiat. Oncol. Biol. Phys. 2005, 62, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Konings, A.W.T.; Faber, H.; Cotteleer, F.; Vissink, A.; Coppes, R.P. Secondary radiation damage as the main cause for unexpected volume effects: A histopathologic study of the parotid gland. Int. J. Radiat. Oncol. Biol. Phys. 2006, 64, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.S.; Fu, K.; Marks, J.; Silverman, S. Late effects of radiation therapy in the head and neck region. Int. J. Radiat. Oncol. Biol. Phys. 1995, 31, 1141–1164. [Google Scholar] [CrossRef]
- Saman, D.M. A review of the epidemiology of oral and pharyngeal carcinoma: Update. Head Neck Oncol. 2012, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Frederick, M.J.; Pickering, C.R.; Bettegowda, C.; Chang, K.; Li, R.J.; Fakhry, C.; Xie, T.X.; Zhang, J.; Wang, J.; Zhang, N.; et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011, 333, 1154–1157. [Google Scholar] [CrossRef]
- Huber, M.A.; Tantiwongkosi, B. Oral and oropharyngeal cancer. Med. Clin. North Am. 2014, 98, 1299–1321. [Google Scholar] [CrossRef]
- Gillison, M.L.; Broutian, T.; Pickard, R.K.; Tong, Z.Y.; Xiao, W.; Kahle, L.; Graubard, B.I.; Chaturvedi, A.K. Prevalence of oral HPV infection in the United States, 2009–2010. JAMA 2012, 307, 693–703. [Google Scholar] [CrossRef]
- Bernier, J.; Cooper, J.S.; Pajak, T.F.; van Glabbeke, M.; Bourhis, J.; Forastiere, A.; Ozsahin, E.M.; Jacobs, J.R.; Jassem, J.; Ang, K.K.; Lefèbvre, J.L. Defining risk levels in locally advanced head and neck cancers: A comparative analysis of concurrent postoperative radiation plus chemotherapy trials of the EORTC (#22931) and RTOG (# 9501). Head Neck 2005, 27, 843–850. [Google Scholar]
- Sommers, L.W.; Steenbakkers, R.J.H.M.; Bijl, H.P.; Vemer-van den Hoek, J.G.M.; Roodenburg, J.L.N.; Oosting, S.F.; Halmos, G.B.; de Rooij, S.E.; Langendijk, J.A. Survival Patterns in Elderly Head and Neck Squamous Cell Carcinoma Patients Treated with Definitive Radiation Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2017, 98, 793–801. [Google Scholar] [CrossRef]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: Cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef]
- Meloty-Kapella, L.; Shergill, B.; Kuon, J.; Botvinick, E.; Weinmaster, G. Notch ligand endocytosis generates mechanical pulling force dependent on dynamin, epsins, and actin. Dev. Cell 2012, 22, 1299–1312. [Google Scholar] [CrossRef] [PubMed]
- Ahimou, F.; Mok, L.-P.; Bardot, B.; Wesley, C. The adhesion force of Notch with Delta and the rate of Notch signaling. J. Cell Biol. 2004, 167, 1217–1229. [Google Scholar] [CrossRef]
- Huenniger, K.; Krämer, A.; Soom, M.; Chang, I.; Köhler, M.; Depping, R.; Kehlenbach, R.H.; Kaether, C. Notch1 signaling is mediated by importins alpha 3, 4, and 7. Cell. Mol. Life Sci. CMLS 2010, 67, 3187–3196. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Teng, L.; Bailey, S.K.; Frost, A.R.; Bland, K.I.; LoBuglio, A.F.; Ruppert, J.M.; Lobo-Ruppert, S.M. Epithelial transformation by KLF4 requires Notch1 but not canonical Notch1 signaling. Cancer Biol. Ther. 2009, 8, 1840–1851. [Google Scholar] [CrossRef]
- Raafat, A.; Lawson, S.; Bargo, S.; Klauzinska, M.; Strizzi, L.; Goldhar, A.S.; Buono, K.; Salomon, D.; Vonderhaar, B.K.; Callahan, R. Rbpj conditional knockout reveals distinct functions of Notch4/Int3 in mammary gland development and tumorigenesis. Oncogene 2009, 28, 219–230. [Google Scholar] [CrossRef]
- Lahmar, M.; Catelain, C.; Poirault, S.; Dorsch, M.; Villeval, J.L.; Vainchenker, W.; Albagli, O.; Lauret, E. Distinct effects of the soluble versus membrane-bound forms of the notch ligand delta-4 on human CD34+CD38low cell expansion and differentiation. Stem Cells Dayt. Ohio 2008, 26, 621–629. [Google Scholar] [CrossRef]
- Liao, W.R.; Hsieh, R.H.; Hsu, K.W.; Wu, M.Z.; Tseng, M.J.; Mai, R.T.; Wu Lee, Y.H.; Yeh, T.S. The CBF1-independent Notch1 signal pathway activates human c-myc expression partially via transcription factor YY1. Carcinogenesis 2007, 28, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Ikawa, T.; Kawamoto, H.; Goldrath, A.W.; Murre, C. E proteins and Notch signaling cooperate to promote T cell lineage specification and commitment. J. Exp. Med. 2006, 203, 1329–1342. [Google Scholar] [CrossRef] [PubMed]
- Stockhausen, M.-T.; Sjölund, J.; Axelson, H. Regulation of the Notch target gene Hes-1 by TGFalpha induced Ras/MAPK signaling in human neuroblastoma cells. Exp. Cell Res. 2005, 310, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Nichols, J.T.; Miyamoto, A.; Weinmaster, G. Notch signaling—Constantly on the move. Traffic 2007, 8, 959–969. [Google Scholar] [CrossRef]
- Panin, V.M.; Papayannopoulos, V.; Wilson, R.; Irvine, K.D. Fringe modulates Notch-ligand interactions. Nature 1997, 387, 908–912. [Google Scholar] [CrossRef]
- Cohen, B.; Bashirullah, A.; Dagnino, L.; Campbell, C.; Fisher, W.W.; Leow, C.C.; Whiting, E.; Ryan, D.; Zinyk, D.; Boulianne, G. Fringe boundaries coincide with Notch-dependent patterning centres in mammals and alter Notch-dependent development in Drosophila. Nat. Genet. 1997, 16, 283–288. [Google Scholar] [CrossRef]
- Xu, K.; Usary, J.; Kousis, P.C.; Prat, A.; Wang, D.Y.; Adams, J.R.; Wang, W.; Loch, A.J.; Deng, T.; Zhao, W.; et al. Lunatic fringe deficiency cooperates with the Met/Caveolin gene amplicon to induce basal-like breast cancer. Cancer Cell 2012, 21, 626–641. [Google Scholar] [CrossRef]
- Collins, L.M.; Dawes, C. The surface area of the adult human mouth and thickness of the salivary film covering the teeth and oral mucosa. J. Dent. Res. 1987, 66, 1300–1302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.H.; Liu, H.C.; Zhu, L.J.; Chu, M.; Liang, Y.J.; Liang, L.Z.; Liao, G.Q. Activation of Notch signaling in human tongue carcinoma. J. Oral Pathol. Med. 2011, 40, 37–45. [Google Scholar] [CrossRef]
- Mandasari, M.; Sawangarun, W.; Katsube, K.; Kayamori, K.; Yamaguchi, A.; Sakamoto, K. A facile one-step strategy for the generation of conditional knockout mice to explore the role of Notch1 in oroesophageal tumorigenesis. Biochem. Biophys. Res. Commun. 2016, 469, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.L.; Zhang, L.; Huang, C.F.; Ma, S.R.; Bu, L.L.; Liu, J.F.; Yu, G.T.; Liu, B.; Gutkind, J.S.; Kulkarni, A.B.; Zhang, W.F.; Sun, Z.J. NOTCH1 inhibition enhances the efficacy of conventional chemotherapeutic agents by targeting head neck cancer stem cell. Sci. Rep. 2016, 6, 24704. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S. Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef]
- Zhang, T.H.; Liu, H.C.; Liang, Y.J.; Liang, L.Z.; Zheng, G.S.; Huang, H.Z.; Wu, J.N.; Liao, G.Q. Suppression of tongue squamous cell carcinoma growth by inhibition of Jagged1 in vitro and in vivo. J. Oral Pathol. Med. 2013, 42, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Dang, H.; Lin, A.L.; Zhang, B.; Zhang, H.-M.; Katz, M.S.; Yeh, C.-K. Role for Notch signaling in salivary acinar cell growth and differentiation. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2009, 238, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Lan, Y.; Chapman, H.D.; Shawber, C.; Norton, C.R.; Serreze, D.V.; Weinmaster, G.; Gridley, T. Defects in limb, craniofacial, and thymic development in Jagged2 mutant mice. Genes Dev. 1998, 12, 1046–1057. [Google Scholar] [CrossRef]
- Casey, L.M.; Lan, Y.; Cho, E.-S.; Maltby, K.M.; Gridley, T.; Jiang, R. Jag2-Notch1 signaling regulates oral epithelial differentiation and palate development. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2006, 235, 1830–1844. [Google Scholar] [CrossRef]
- Kamath, B.M.; Bauer, R.C.; Loomes, K.M.; Chao, G.; Gerfen, J.; Hutchinson, A.; Hardikar, W.; Hirschfield, G.; Jara, P.; Krantz, I.D.; et al. NOTCH2 mutations in Alagille syndrome. J. Med. Genet. 2012, 49, 138–144. [Google Scholar] [CrossRef]
- Mitsiadis, T.A.; Graf, D.; Luder, H.; Gridley, T.; Bluteau, G. BMPs and FGFs target Notch signalling via jagged 2 to regulate tooth morphogenesis and cytodifferentiation. Dev. Camb. Engl. 2010, 137, 3025–3035. [Google Scholar] [CrossRef]
- Mitsiadis, T.A.; Henrique, D.; Thesleff, I.; Lendahl, U. Mouse Serrate-1 (Jagged-1): Expression in the developing tooth is regulated by epithelial-mesenchymal interactions and fibroblast growth factor-4. Dev. Camb. Engl. 1997, 124, 1473–1483. [Google Scholar]
- Mitsiadis, T.A.; de Bari, C.; About, I. Apoptosis in developmental and repair-related human tooth remodeling: A view from the inside. Exp. Cell Res. 2008, 314, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Mitsiadis, T.A.; Graf, D. Cell fate determination during tooth development and regeneration. Birth Defects Res. Part C Embryo Today Rev. 2009, 87, 199–211. [Google Scholar] [CrossRef]
- Mitsiadis, T.A.; Lardelli, M.; Lendahl, U.; Thesleff, I. Expression of Notch 1, 2 and 3 is regulated by epithelial-mesenchymal interactions and retinoic acid in the developing mouse tooth and associated with determination of ameloblast cell fate. J. Cell Biol. 1995, 130, 407–418. [Google Scholar] [CrossRef]
- Hill-Felberg, S.; Wu, H.H.; Toms, S.A.; Dehdashti, A.R. Notch receptor expression in human brain arteriovenous malformations. J. Cell. Mol. Med. 2015, 19, 1986–1993. [Google Scholar] [CrossRef]
- Conboy, I.M.; Rando, T.A. Aging, stem cells and tissue regeneration: Lessons from muscle. Cell Cycle 2005, 4, 407–410. [Google Scholar] [CrossRef]
- Church, J.E.; Trieu, J.; Chee, A.; Naim, T.; Gehrig, S.M.; Lamon, S.; Angelini, C.; Russell, A.P.; Lynch, G.S. Alterations in Notch signalling in skeletal muscles from mdx and dko dystrophic mice and patients with Duchenne muscular dystrophy. Exp. Physiol. 2014, 99, 675–687. [Google Scholar] [CrossRef]
- Van den Engel-Hoek, L.; Erasmus, C.E.; Hendriks, J.C.; Geurts, A.C.; Klein, W.M.; Pillen, S.; Sie, L.T.; de Swart, B.J.; de Groot, I.J. Oral muscles are progressively affected in Duchenne muscular dystrophy: Implications for dysphagia treatment. J. Neurol. 2013, 260, 1295–1303. [Google Scholar] [CrossRef]
- Vieira, N.M.; Elvers, I.; Alexander, M.S.; Moreira, Y.B.; Eran, A.; Gomes, J.P.; Marshall, J.L.; Karlsson, E.K.; Verjovski-Almeida, S.; Lindblad-Toh, K.; et al. Jagged 1 Rescues the Duchenne Muscular Dystrophy Phenotype. Cell 2015, 163, 1204–1213. [Google Scholar] [CrossRef]
- Marelli, F.; Persani, L. Role of Jagged1-Notch pathway in thyroid development. J. Endocrinol. Investig. 2018, 41, 75–81. [Google Scholar] [CrossRef]
- Kumar, L.K.S.; Kurien, N.M.; Jacob, M.M.; Menon, P.V.; Khalam, S.A. Lingual thyroid. Ann. Maxillofac. Surg. 2015, 5, 104–107. [Google Scholar]
- Demehri, S.; Turkoz, A.; Kopan, R. Epidermal Notch1 loss promotes skin tumorigenesis by impacting the stromal microenvironment. Cancer Cell 2009, 16, 55–66. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, C.-X.; Wu, Z.-T.; Ge, L.; Zhou, H.-M. Mining proteins associated with oral squamous cell carcinoma in complex networks. Asian Pac. J. Cancer Prev. APJCP 2013, 14, 4621–4625. [Google Scholar] [CrossRef]
- Sakamoto, K. Notch signaling in oral squamous neoplasia. Pathol. Int. 2016, 66, 609–617. [Google Scholar] [CrossRef]
- Sun, W.; Gaykalova, D.A.; Ochs, M.F.; Mambo, E.; Arnaoutakis, D.; Liu, Y.; Loyo, M.; Agrawal, N.; Howard, J.; Li, R.; et al. Activation of the NOTCH pathway in head and neck cancer. Cancer Res. 2014, 74, 1091–1104. [Google Scholar] [CrossRef] [PubMed]
- Frise, E.; Knoblich, J.A.; Younger-Shepherd, S.; Jan, L.Y.; Jan, Y.N. The Drosophila Numb protein inhibits signaling of the Notch receptor during cell-cell interaction in sensory organ lineage. Proc. Natl. Acad. Sci. USA 1996, 93, 11925–11932. [Google Scholar] [CrossRef]
- Guo, M.; Jan, L.Y.; Jan, Y.N. Control of daughter cell fates during asymmetric division: Interaction of Numb and Notch. Neuron 1996, 17, 27–41. [Google Scholar] [CrossRef]
- Berdnik, D.; Török, T.; González-Gaitán, M.; Knoblich, J.A. The endocytic protein alpha-Adaptin is required for numb-mediated asymmetric cell division in Drosophila. Dev. Cell 2002, 3, 221–231. [Google Scholar] [CrossRef]
- Hong, J.; Liu, Z.; Zhu, H.; Zhang, X.; Liang, Y.; Yao, S.; Wang, F.; Xie, X.; Zhang, B.; Tan, T.; et al. The tumor suppressive role of NUMB isoform 1 in esophageal squamous cell carcinoma. Oncotarget 2014, 5, 5602–5614. [Google Scholar] [CrossRef] [PubMed]
- Hung, P.S.; Liu, C.J.; Chou, C.S.; Kao, S.Y.; Yang, C.C.; Chang, K.W.; Chiu, T.H.; Lin, S.C. miR-146a enhances the oncogenicity of oral carcinoma by concomitant targeting of the IRAK1, TRAF6 and NUMB genes. PLoS ONE 2013, 8, e79926. [Google Scholar] [CrossRef] [PubMed]
- Hijioka, H.; Setoguchi, T.; Miyawaki, A.; Gao, H.; Ishida, T.; Komiya, S.; Nakamura, N. Upregulation of Notch pathway molecules in oral squamous cell carcinoma. Int. J. Oncol. 2010, 36, 817–822. [Google Scholar] [PubMed]
- Simabuco, F.M.; Kawahara, R.; Yokoo, S.; Granato, D.C.; Miguel, L.; Agostini, M.; Aragão, A.Z.; Domingues, R.R.; Flores, I.L.; Macedo, C.C.; et al. ADAM17 mediates OSCC development in an orthotopic murine model. Mol. Cancer 2014, 13, 24. [Google Scholar] [CrossRef]
- Hellström, M.; Phng, L.K.; Hofmann, J.J.; Wallgard, E.; Coultas, L.; Lindblom, P.; Alva, J.; Nilsson, A.K.; Karlsson, L.; Gaiano, N.; et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 2007, 445, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Suchting, S.; Freitas, C.; le Noble, F.; Benedito, R.; Bréant, C.; Duarte, A.; Eichmann, A. The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc. Natl. Acad. Sci. USA 2007, 104, 3225–3230. [Google Scholar] [CrossRef] [PubMed]
- Leslie, J.D.; Ariza-McNaughton, L.; Bermange, A.L.; McAdow, R.; Johnson, S.L.; Lewis, J. Endothelial signalling by the Notch ligand Delta-like 4 restricts angiogenesis. Dev. Camb. Engl. 2007, 134, 839–844. [Google Scholar] [CrossRef]
- Siekmann, A.F.; Lawson, N.D. Notch signalling limits angiogenic cell behaviour in developing zebrafish arteries. Nature 2007, 445, 781–784. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.K.; Li, J.-L.; Murga, M.; Harris, A.L.; Tosato, G. Up-regulation of the Notch ligand Delta-like 4 inhibits VEGF-induced endothelial cell function. Blood 2006, 107, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Benedito, R.; Roca, C.; Sörensen, I.; Adams, S.; Gossler, A.; Fruttiger, M.; Adams, R.H. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell 2009, 137, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- Mazzieri, R.; Pucci, F.; Moi, D.; Zonari, E.; Ranghetti, A.; Berti, A.; Politi, L.S.; Gentner, B.; Brown, J.L.; Naldini, L.; et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell 2011, 19, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Outtz, H.H.; Wu, J.K.; Wang, X.; Kitajewski, J. Notch1 deficiency results in decreased inflammation during wound healing and regulates vascular endothelial growth factor receptor-1 and inflammatory cytokine expression in macrophages. J. Immunol. 2010, 185, 4363–4373. [Google Scholar] [CrossRef]
- Patel, N.S.; Li, J.-L.; Generali, D.; Poulsom, R.; Cranston, D.W.; Harris, A.L. Up-regulation of delta-like 4 ligand in human tumor vasculature and the role of basal expression in endothelial cell function. Cancer Res. 2005, 65, 8690–8697. [Google Scholar] [CrossRef]
- Noguera-Troise, I.; Daly, C.; Papadopoulos, N.J.; Coetzee, S.; Boland, P.; Gale, N.W.; Lin, H.C.; Yancopoulos, G.D.; Thurston, G. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 2006, 444, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- Zaghloul, N.; Hernandez, S.L.; Bae, J.O.; Huang, J.; Fisher, J.C.; Lee, A.; Kadenhe-Chiweshe, A.; Kandel, J.J.; Yamashiro, D.J. Vascular endothelial growth factor blockade rapidly elicits alternative proangiogenic pathways in neuroblastoma. Int. J. Oncol. 2009, 34, 401–407. [Google Scholar]
- Hasina, R.; Lingen, M.W. Angiogenesis in oral cancer. J. Dent. Educ. 2001, 65, 1282–1290. [Google Scholar]
- Zeng, Q.; Li, S.; Chepeha, D.B.; Giordano, T.J.; Li, J.; Zhang, H.; Polverini, P.J.; Nor, J.; Kitajewski, J.; Wang, C.Y. Crosstalk between tumor and endothelial cells promotes tumor angiogenesis by MAPK activation of Notch signaling. Cancer Cell 2005, 8, 13–23. [Google Scholar] [CrossRef]
- Kayamori, K.; Katsube, K.; Sakamoto, K.; Ohyama, Y.; Hirai, H.; Yukimori, A.; Ohata, Y.; Akashi, T.; Saitoh, M.; Harada, K.; et al. NOTCH3 Is Induced in Cancer-Associated Fibroblasts and Promotes Angiogenesis in Oral Squamous Cell Carcinoma. PLoS ONE 2016, 11, e0154112. [Google Scholar] [CrossRef]
- Wang, W.M.; Zhao, Z.L.; Ma, S.R.; Yu, G.T.; Liu, B.; Zhang, L.; Zhang, W.F.; Kulkarni, A.B.; Sun, Z.J.; Zhao, Y.F. Epidermal growth factor receptor inhibition reduces angiogenesis via hypoxia-inducible factor-1α and Notch1 in head neck squamous cell carcinoma. PLoS ONE 2015, 10, e0119723. [Google Scholar] [CrossRef]
- Chan, C.J.; Heisenberg, C.-P.; Hiiragi, T. Coordination of Morphogenesis and Cell-Fate Specification in Development. Curr. Biol. 2017, 27, R1024–R1035. [Google Scholar] [CrossRef]
- Voulgari, A.; Pintzas, A. Epithelial-mesenchymal transition in cancer metastasis: Mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim. Biophys. Acta 2009, 1796, 75–90. [Google Scholar] [CrossRef]
- Fragiadaki, M.; Mason, R.M. Epithelial-mesenchymal transition in renal fibrosis-evidence for and against. Int. J. Exp. Pathol. 2011, 92, 143–150. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Tse, J.C.; Kalluri, R. Mechanisms of metastasis: Epithelial-to-mesenchymal transition and contribution of tumor microenvironment. J. Cell. Biochem. 2007, 101, 816–829. [Google Scholar] [CrossRef]
- Gravdal, K.; Halvorsen, O.J.; Haukaas, S.A.; Akslen, L.A. A switch from E-cadherin to N-cadherin expression indicates epithelial to mesenchymal transition and is of strong and independent importance for the progress of prostate cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 7003–7011. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, X.; Gang, H.; Li, X.; Li, Z.; Wang, T.; Han, J.; Luo, T.; Wen, F.; Wu, X. Up-regulation of gastric cancer cell invasion by Twist is accompanied by N-cadherin and fibronectin expression. Biochem. Biophys. Res. Commun. 2007, 358, 925–930. [Google Scholar] [CrossRef]
- Saad, S.; Stanners, S.R.; Yong, R.; Tang, O.; Pollock, C.A. Notch mediated epithelial to mesenchymal transformation is associated with increased expression of the Snail transcription factor. Int. J. Biochem. Cell Biol. 2010, 42, 1115–1122. [Google Scholar] [CrossRef]
- Stoyianni, A.; Goussia, A.; Pentheroudakis, G.; Siozopoulou, V.; Ioachim, E.; Krikelis, D.; Golfinopoulos, V.; Cervantes, A.; Bobos, M.; Fotsis, T.; et al. Immunohistochemical study of the epithelial-mesenchymal transition phenotype in cancer of unknown primary: Incidence, correlations and prognostic utility. AntiCancer Res. 2012, 32, 1273–1281. [Google Scholar]
- Xie, M.; Zhang, L.; He, C.S.; Xu, F.; Liu, J.L.; Hu, Z.H.; Zhao, L.P.; Tian, Y. Activation of Notch-1 enhances epithelial-mesenchymal transition in gefitinib-acquired resistant lung cancer cells. J. Cell. Biochem. 2012, 113, 1501–1513. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Hijioka, H.; Kume, K.; Miyawaki, A.; Nakamura, N. Notch signaling induces EMT in OSCC cell lines in a hypoxic environment. Oncol. Lett. 2013, 6, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Krisanaprakornkit, S.; Iamaroon, A. Epithelial-mesenchymal transition in oral squamous cell carcinoma. ISRN Oncol. 2012, 2012, 681469. [Google Scholar] [CrossRef] [PubMed]
- Bigas, A.; Porcheri, C. Notch and Stem Cells. Adv. Exp. Med. Biol. 2018, 1066, 235–263. [Google Scholar]
- Pannuti, A.; Foreman, K.; Rizzo, P.; Osipo, C.; Golde, T.; Osborne, B.; Miele, L. Targeting Notch to target cancer stem cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 3141–3152. [Google Scholar] [CrossRef] [PubMed]
- Sansone, P.; Storci, G.; Giovannini, C.; Pandolfi, S.; Pianetti, S.; Taffurelli, M.; Santini, D.; Ceccarelli, C.; Chieco, P.; Bonafé, M. p66Shc/Notch-3 interplay controls self-renewal and hypoxia survival in human stem/progenitor cells of the mammary gland expanded in vitro as mammospheres. Stem Cells Dayt. Ohio 2007, 25, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Harrison, H.; Farnie, G.; Howell, S.J.; Rock, R.E.; Stylianou, S.; Brennan, K.R.; Bundred, N.J.; Clarke, R.B. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010, 70, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Matsui, W.; Khaki, L.; Stearns, D.; Chun, J.; Li, Y.M.; Eberhart, C.G. Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res. 2006, 66, 7445–7452. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Liu, Y.; Ying, H.; Xue, Y.; Zhang, Z.; Wang, P.; Liu, L.; Zhang, H. Increasing of blood-tumor barrier permeability through paracellular pathway by low-frequency ultrasound irradiation in vitro. J. Mol. Neurosci. 2011, 43, 541–548. [Google Scholar] [CrossRef]
- Yao, Z.; Mishra, L. Cancer stem cells and hepatocellular carcinoma. Cancer Biol. Ther. 2009, 8, 1691–1698. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xue, L.; Cao, Q.; Lin, Y.; Ding, Y.; Yang, P.; Che, L. Expression of Notch1, Jagged1 and beta-catenin and their clinicopathological significance in hepatocellular carcinoma. Neoplasma 2009, 56, 533–541. [Google Scholar] [CrossRef]
- Domingo-Domenech, J.; Vidal, S.J.; Rodriguez-Bravo, V.; Castillo-Martin, M.; Quinn, S.A.; Rodriguez-Barrueco, R.; Bonal, D.M.; Charytonowicz, E.; Gladoun, N.; de la Iglesia-Vicente, J.; et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer Cell 2012, 22, 373–388. [Google Scholar] [CrossRef]
- Crabtree, J.S.; Miele, L. Breast Cancer Stem Cells. Biomedicines 2018, 6. [Google Scholar] [CrossRef]
- Pandya, K.; Meeke, K.; Clementz, A.G.; Rogowski, A.; Roberts, J.; Miele, L.; Albain, K.S.; Osipo, C. Targeting both Notch and ErbB-2 signalling pathways is required for prevention of ErbB-2-positive breast tumour recurrence. Br. J. Cancer 2011, 105, 796–806. [Google Scholar] [CrossRef]
- De Andrade, N.P.; Rodrigues, S.D.; Rodini, C.O.; Nunes, F.D. Cancer stem Cell cytokeratins and epithelial to mesenchymal transition markers expression in oral squamous cell carcinoma derived from ortothopic xenoimplantation of CD44high cells. Pathol. Res. Pract. 2017, 213, 235–244. [Google Scholar] [CrossRef]
- Chen, J.; Zhou, J.; Lu, J.; Xiong, H.; Shi, X.; Gong, L. Significance of CD44 expression in head and neck cancer: A systemic review and meta-analysis. BMC Cancer 2014, 14, 5. [Google Scholar] [CrossRef] [PubMed]
- Zeineddine, D.; Hammoud, A.A.; Mortada, M.; Boeuf, H. The Oct4 protein: More than a magic stemness marker. Am. J. Stem Cells 2014, 3, 74–82. [Google Scholar] [PubMed]
- Huang, C.-F.; Xu, X.-R.; Wu, T.-F.; Sun, Z.-J.; Zhang, W.-F. Correlation of ALDH1, CD44, OCT4 and SOX2 in tongue squamous cell carcinoma and their association with disease progression and prognosis. J. Oral Pathol. Med. 2014, 43, 492–498. [Google Scholar] [CrossRef]
- Baillie, R.; Itinteang, T.; Yu, H.H.; Brasch, H.D.; Davis, P.F.; Tan, S.T. Cancer stem cells in moderately differentiated oral tongue squamous cell carcinoma. J. Clin. Pathol. 2016, 69, 742–744. [Google Scholar] [CrossRef] [PubMed]
- Zimmerer, R.M.; Ludwig, N.; Kampmann, A.; Bittermann, G.; Spalthoff, S.; Jungheim, M.; Gellrich, N.C.; Tavassol, F. CD24+ tumor-initiating cells from oral squamous cell carcinoma induce initial angiogenesis in vivo. Microvasc. Res. 2017, 112, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, G.; Devaraj, H. Aberrant expression of CD133 and musashi-1 in preneoplastic and neoplastic human oral squamous epithelium and their correlation with clinicopathological factors. Head Neck 2012, 34, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Irollo, E.; Pirozzi, G. CD133: To be or not to be, is this the real question? Am. J. Transl. Res. 2013, 5, 563–581. [Google Scholar] [PubMed]
- Lee, S.H.; Hong, H.S.; Liu, Z.X.; Kim, R.H.; Kang, M.K.; Park, N.H.; Shin, K.H. TNFα enhances cancer stem cell-like phenotype via Notch-Hes1 activation in oral squamous cell carcinoma cells. Biochem. Biophys. Res. Commun. 2012, 424, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, P.; Weaver, K.L.; Capobianco, A.J. Notch signalling in solid tumours: A little bit of everything but not all the time. Nat. Rev. Cancer 2011, 11, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Radtke, F.; MacDonald, H.R.; Tacchini-Cottier, F. Regulation of innate and adaptive immunity by Notch. Nat. Rev. Immunol. 2013, 13, 427–437. [Google Scholar] [CrossRef]
- Wilson, A.; MacDonald, H.R.; Radtke, F. Notch 1-deficient common lymphoid precursors adopt a B cell fate in the thymus. J. Exp. Med. 2001, 194, 1003–1012. [Google Scholar] [CrossRef]
- Yashiro-Ohtani, Y.; He, Y.; Ohtani, T.; Jones, M.E.; Shestova, O.; Xu, L.; Fang, T.C.; Chiang, M.Y.; Intlekofer, A.M.; Blacklow, S.C.; et al. Pre-TCR signaling inactivates Notch1 transcription by antagonizing E2A. Genes Dev. 2009, 23, 1665–1676. [Google Scholar] [CrossRef]
- Kyoizumi, S.; Kubo, Y.; Kajimura, J.; Yoshida, K.; Hayashi, T.; Nakachi, K.; Moore, M.A.; van den Brink, M.R.M.; Kusunoki, Y. Fate Decision Between Group 3 Innate Lymphoid and Conventional NK Cell Lineages by Notch Signaling in Human Circulating Hematopoietic Progenitors. J. Immunol. 2017, 199, 2777–2793. [Google Scholar] [CrossRef]
- Schroeder, T.; Kohlhof, H.; Rieber, N.; Just, U. Notch signaling induces multilineage myeloid differentiation and up-regulates PU.1 expression. J. Immunol. 2003, 170, 5538–5548. [Google Scholar] [CrossRef]
- Cheng, P.; Nefedova, Y.; Corzo, C.A.; Gabrilovich, D.I. Regulation of dendritic-cell differentiation by bone marrow stroma via different Notch ligands. Blood 2007, 109, 507–515. [Google Scholar] [CrossRef]
- Jundt, F.; Pröbsting, K.S.; Anagnostopoulos, I.; Muehlinghaus, G.; Chatterjee, M.; Mathas, S.; Bargou, R.C.; Manz, R.; Stein, H.; Dörken, B. Jagged1-induced Notch signaling drives proliferation of multiple myeloma cells. Blood 2004, 103, 3511–3515. [Google Scholar] [CrossRef]
- Maniati, E.; Bossard, M.; Cook, N.; Candido, J.B.; Emami-Shahri, N.; Nedospasov, S.A.; Balkwill, F.R.; Tuveson, D.A.; Hagemann, T. Crosstalk between the canonical NF-κB and Notch signaling pathways inhibits Pparγ expression and promotes pancreatic cancer progression in mice. J. Clin. Investig. 2011, 121, 4685–4699. [Google Scholar] [CrossRef]
- Bano, N.; Yadav, M.; Mohania, D.; Das, B.C. The role of NF-κB and miRNA in oral cancer and cancer stem cells with or without HPV16 infection. PLoS ONE 2018, 13, e0205518. [Google Scholar] [CrossRef]
- Sun, Y.; Zhu, D.; Wang, G.; Wang, D.; Zhou, H.; Liu, X.; Jiang, M.; Liao, L.; Zhou, Z.; Hu, J. Pro-Inflammatory Cytokine IL-1β Up-Regulates CXC Chemokine Receptor 4 via Notch and ERK Signaling Pathways in Tongue Squamous Cell Carcinoma. PLoS ONE 2015, 10, e0132677. [Google Scholar] [CrossRef]
- Erdogan, B.; Webb, D.J. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem. Soc. Trans. 2017, 45, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.S.; Tan, M.J.; Sng, M.K.; Teo, Z.; Phua, T.; Choo, C.C.; Li, L.; Zhu, P.; Tan, N.S. Cancer-associated fibroblasts enact field cancerization by promoting extratumoral oxidative stress. Cell Death Dis. 2017, 8, e2562. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xu, Q.; Wu, Y.; Li, J.; Tang, D.; Han, L.; Fan, Q. A CCL2/ROS autoregulation loop is critical for cancer-associated fibroblasts-enhanced tumor growth of oral squamous cell carcinoma. Carcinogenesis 2014, 35, 1362–1370. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Kong, R.; Ferrari, M.L.; Radtke, F.; Capobianco, A.J.; Liu, Z.-J. Notch1 Pathway Activity Determines the Regulatory Role of Cancer-Associated Fibroblasts in Melanoma Growth and Invasion. PLoS ONE 2015, 10, e0142815. [Google Scholar] [CrossRef]
- Lim, K.P.; Cirillo, N.; Hassona, Y.; Wei, W.; Thurlow, J.K.; Cheong, S.C.; Pitiyage, G.; Parkinson, E.K.; Prime, S.S. Fibroblast gene expression profile reflects the stage of tumour progression in oral squamous cell carcinoma. J. Pathol. 2011, 223, 459–469. [Google Scholar] [CrossRef]
- Vsiansky, V.; Gumulec, J.; Raudenska, M.; Masarik, M. Prognostic role of c-Met in head and neck squamous cell cancer tissues: A meta-analysis. Sci. Rep. 2018, 8, 10370. [Google Scholar] [CrossRef]
- Marsh, D.; Suchak, K.; Moutasim, K.A.; Vallath, S.; Hopper, C.; Jerjes, W.; Upile, T.; Kalavrezos, N.; Violette, S.M.; Weinreb, P.H.; et al. Stromal features are predictive of disease mortality in oral cancer patients. J. Pathol. 2011, 223, 470–481. [Google Scholar] [CrossRef]
- Lim, Y.C.; Han, J.H.; Kang, H.J.; Kim, Y.S.; Lee, B.H.; Choi, E.C.; Kim, C.H. Overexpression of c-Met promotes invasion and metastasis of small oral tongue carcinoma. Oral Oncol. 2012, 48, 1114–1119. [Google Scholar] [CrossRef]
- Dong, G.; Chen, Z.; Li, Z.Y.; Yeh, N.T.; Bancroft, C.C.; van Waes, C. Hepatocyte growth factor/scatter factor-induced activation of MEK and PI3K signal pathways contributes to expression of proangiogenic cytokines interleukin-8 and vascular endothelial growth factor in head and neck squamous cell carcinoma. Cancer Res. 2001, 61, 5911–5918. [Google Scholar]
- Knowles, L.M.; Stabile, L.P.; Egloff, A.M.; Rothstein, M.E.; Thomas, S.M.; Gubish, C.T.; Lerner, E.C.; Seethala, R.R.; Suzuki, S.; Quesnelle, K.M.; et al. HGF and c-Met participate in paracrine tumorigenic pathways in head and neck squamous cell cancer. Clin. Cancer Res. 2009, 15, 3740–3750. [Google Scholar] [CrossRef]
- Dajee, M.; Lazarov, M.; Zhang, J.Y.; Cai, T.; Green, C.L.; Russell, A.J.; Marinkovich, M.P.; Tao, S.; Lin, Q.; Kubo, Y.; et al. NF-kappaB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature 2003, 421, 639–643. [Google Scholar] [CrossRef]
- Nicolas, M.; Wolfer, A.; Raj, K.; Kummer, J.A.; Mill, P.; van Noort, M.; Hui, C.C.; Clevers, H.; Dotto, G.P.; Radtke, F. Notch1 functions as a tumor suppressor in mouse skin. Nat. Genet. 2003, 33, 416–421. [Google Scholar] [CrossRef]
- Thélu, J.; Rossio, P.; Favier, B. Notch signalling is linked to epidermal cell differentiation level in basal cell carcinoma, psoriasis and wound healing. BMC Dermatol. 2002, 2, 7. [Google Scholar] [CrossRef]
- Yap, L.F.; Lee, D.; Khairuddin, A.; Pairan, M.F.; Puspita, B.; Siar, C.H.; Paterson, I.C. The opposing roles of NOTCH signalling in head and neck cancer: A mini review. Oral Dis. 2015, 21, 850–857. [Google Scholar] [CrossRef]
- Pickering, C.R.; Zhang, J.; Yoo, S.Y.; Bengtsson, L.; Moorthy, S.; Neskey, D.M.; Zhao, M.; Ortega Alves, M.V.; Chang, K.; Drummond, J.; et al. Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discov. 2013, 3, 770–781. [Google Scholar] [CrossRef]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef]
- Beverly, L.J.; Felsher, D.W.; Capobianco, A.J. Suppression of p53 by Notch in lymphomagenesis: Implications for initiation and regression. Cancer Res. 2005, 65, 7159–7168. [Google Scholar] [CrossRef]
- Mungamuri, S.K.; Yang, X.; Thor, A.D.; Somasundaram, K. Survival signaling by Notch1: Mammalian target of rapamycin (mTOR)-dependent inhibition of p53. Cancer Res. 2006, 66, 4715–4724. [Google Scholar] [CrossRef]
- Nair, P.; Somasundaram, K.; Krishna, S. Activated Notch1 inhibits p53-induced apoptosis and sustains transformation by human papillomavirus type 16 E6 and E7 oncogenes through a PI3K-PKB/Akt-dependent pathway. J. Virol. 2003, 77, 7106–7112. [Google Scholar] [CrossRef]
- Kim, S.B.; Chae, G.W.; Lee, J.; Park, J.; Tak, H.; Chung, J.H.; Park, T.G.; Ahn, J.K.; Joe, C.O. Activated Notch1 interacts with p53 to inhibit its phosphorylation and transactivation. Cell Death Differ. 2007, 14, 982–991. [Google Scholar] [CrossRef]
- Huang, Q.; Raya, A.; DeJesus, P.; Chao, S.H.; Quon, K.C.; Caldwell, J.S.; Chanda, S.K.; Izpisua-Belmonte, J.C.; Schultz, P.G. Identification of p53 regulators by genome-wide functional analysis. Proc. Natl. Acad. Sci. USA 2004, 101, 3456–3461. [Google Scholar] [CrossRef]
- Qi, R.; An, H.; Yu, Y.; Zhang, M.; Liu, S.; Xu, H.; Guo, Z.; Cheng, T.; Cao, X. Notch1 signaling inhibits growth of human hepatocellular carcinoma through induction of cell cycle arrest and apoptosis. Cancer Res. 2003, 63, 8323–8329. [Google Scholar]
- Henning, K.; Heering, J.; Schwanbeck, R.; Schroeder, T.; Helmbold, H.; Schäfer, H.; Deppert, W.; Kim, E.; Just, U. Notch1 activation reduces proliferation in the multipotent hematopoietic progenitor cell line FDCP-mix through a p53-dependent pathway but Notch1 effects on myeloid and erythroid differentiation are independent of p53. Cell Death Differ. 2008, 15, 398–407. [Google Scholar] [CrossRef]
- Duan, L.; Yao, J.; Wu, X.; Fan, M. Growth suppression induced by Notch1 activation involves Wnt-beta-catenin down-regulation in human tongue carcinoma cells. Biol. Cell 2006, 98, 479–490. [Google Scholar] [CrossRef]
- Palomero, T.; Dominguez, M.; Ferrando, A.A. The role of the PTEN/AKT pathway in NOTCH1-induced leukemia. Cell Cycle 2008, 7, 965–970. [Google Scholar] [CrossRef]
- Chang, K.-Y.; Tsai, S.Y.; Chen, S.H.; Tsou, H.H.; Yen, C.J.; Liu, K.J.; Fang, H.L.; Wu, H.C.; Chuang, B.F.; Chou, S.W.; et al. Dissecting the EGFR-PI3K-AKT pathway in oral cancer highlights the role of the EGFR variant III and its clinical relevance. J. Biomed. Sci. 2013, 20, 43. [Google Scholar] [CrossRef]
- Cai, Y.; Dodhia, S.; Su, G.H. Dysregulations in the PI3K pathway and targeted therapies for head and neck squamous cell carcinoma. Oncotarget 2017, 8, 22203–22217. [Google Scholar] [CrossRef]
- Yang, J.; Ren, X.; Zhang, L.; Li, Y.; Cheng, B.; Xia, J. Oridonin inhibits oral cancer growth and PI3K/Akt signaling pathway. Biomed. Pharmacother. Biomed. Pharmacother. 2018, 100, 226–232. [Google Scholar] [CrossRef]
- Wong, G.W.; Knowles, G.C.; Mak, T.W.; Ferrando, A.A.; Zúñiga-Pflücker, J.C. HES1 opposes a PTEN-dependent check on survival, differentiation, and proliferation of TCRβ-selected mouse thymocytes. Blood 2012, 120, 1439–1448. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Wang, Z.; Ding, X.; Zhang, W.; Li, G.; Liu, L.; Wu, H.; Gu, W.; Wu, Y.; Song, X. A novel Notch1 missense mutation (C1133Y) in the Abruptex domain exhibits enhanced proliferation and invasion in oral squamous cell carcinoma. Cancer Cell Int. 2018, 18, 6. [Google Scholar] [CrossRef]
- Rodilla, V.; Villanueva, A.; Obrador-Hevia, A.; Robert-Moreno, A.; Fernández-Majada, V.; Grilli, A.; López-Bigas, N.; Bellora, N.; Albà, M.M.; Torres, F.; et al. Jagged1 is the pathological link between Wnt and Notch pathways in colorectal cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 6315–6320. [Google Scholar] [CrossRef]
- Gekas, C.; D’Altri, C.G.T.; Aligué, R.; González, J.; Espinosa, L.; Bigas, A. β-Catenin is required for T-cell leukemia initiation and MYC transcription downstream of Notch1. Leukemia 2016, 30, 2002–2010. [Google Scholar] [CrossRef]
- Pannone, G.; Bufo, P.; Santoro, A.; Franco, R.; Aquino, G.; Longo, F.; Botti, G.; Serpico, R.; Cafarelli, B.; Abbruzzese, A.; et al. WNT pathway in oral cancer: Epigenetic inactivation of WNT-inhibitors. Oncol. Rep. 2010, 24, 1035–1041. [Google Scholar]
- Buim, M.E.C.; Gurgel, C.A.S.; Ramos, E.A.G.; Lourenço, S.V.; Soares, F.A. Activation of sonic hedgehog signaling in oral squamous cell carcinomas: A preliminary study. Hum. Pathol. 2011, 42, 1484–1490. [Google Scholar] [CrossRef]
- Wang, Y.-F.; Chang, C.J.; Lin, C.P.; Chang, S.Y.; Chu, P.Y.; Tai, S.K.; Li, W.Y.; Chao, K.S.; Chen, Y.J. Expression of hedgehog signaling molecules as a prognostic indicator of oral squamous cell carcinoma. Head Neck 2012, 34, 1556–1561. [Google Scholar] [CrossRef]
- Schneider, S.; Thurnher, D.; Kloimstein, P.; Leitner, V.; Petzelbauer, P.; Pammer, J.; Brunner, M.; Erovic, B.M. Expression of the Sonic hedgehog pathway in squamous cell carcinoma of the skin and the mucosa of the head and neck. Head Neck 2011, 33, 244–250. [Google Scholar] [CrossRef]
- Srinath, S.; Iyengar, A.R.; Mysorekar, V. Sonic hedgehog in oral squamous cell carcinoma: An immunohistochemical study. J. Oral Maxillofac. Pathol. JOMFP 2016, 20, 377–383. [Google Scholar] [CrossRef]
- Stepan, V.; Ramamoorthy, S.; Nitsche, H.; Zavros, Y.; Merchant, J.L.; Todisco, A. Regulation and function of the sonic hedgehog signal transduction pathway in isolated gastric parietal cells. J. Biol. Chem. 2005, 280, 15700–15708. [Google Scholar] [CrossRef]
- Tanaka, T.; Atsumi, N.; Nakamura, N.; Yanai, H.; Komai, Y.; Omachi, T.; Tanaka, K.; Ishigaki, K.; Saiga, K.; Ohsugi, H.; et al. Bmi1-positive cells in the lingual epithelium could serve as cancer stem cells in tongue cancer. Sci. Rep. 2016, 6, 39386. [Google Scholar] [CrossRef]
- Rich, A.M.; Reade, P.C. Epithelial-mesenchymal interactions in experimental oral mucosal carcinogenesis. J. Oral Pathol. Med. 2001, 30, 389–397. [Google Scholar] [CrossRef]
- Chang, N.W.; Tsai, M.H.; Lin, C.; Hsu, H.T.; Chu, P.Y.; Yeh, C.M.; Chiu, C.F.; Yeh, K.T. Fenofibrate exhibits a high potential to suppress the formation of squamous cell carcinoma in an oral-specific 4-nitroquinoline 1-oxide/arecoline mouse model. Biochim. Biophys. Acta 2011, 1812, 558–564. [Google Scholar] [CrossRef]
- Zhou, G.; Hasina, R.; Wroblewski, K.; Mankame, T.P.; Doçi, C.L.; Lingen, M.W. Dual inhibition of vascular endothelial growth factor receptor and epidermal growth factor receptor is an effective chemopreventive strategy in the mouse 4-NQO model of oral carcinogenesis. Cancer Prev. Res. 2010, 3, 1493–1502. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef]
- Nakagawa, H.; Wang, T.C.; Zukerberg, L.; Odze, R.; Togawa, K.; May, G.H.; Wilson, J.; Rustgi, A.K. The targeting of the cyclin D1 oncogene by an Epstein-Barr virus promoter in transgenic mice causes dysplasia in the tongue, esophagus and forestomach. Oncogene 1997, 14, 1185–1190. [Google Scholar] [CrossRef]
- Song, A.J.; Palmiter, R.D. Detecting and Avoiding Problems When Using the Cre-lox System. Trends Genet. 2018, 34, 333–340. [Google Scholar] [CrossRef]
- Bian, Y.; Hall, B.; Sun, Z.J.; Molinolo, A.; Chen, W.; Gutkind, J.S.; Waes, C.V.; Kulkarni, A.B. Loss of TGF-β signaling and PTEN promotes head and neck squamous cell carcinoma through cellular senescence evasion and cancer-related inflammation. Oncogene 2012, 31, 3322–3332. [Google Scholar] [CrossRef]
- O’Hagan, R.C.; Heyer, J. KRAS Mouse Models: Modeling Cancer Harboring KRAS Mutations. Genes Cancer 2011, 2, 335–343. [Google Scholar] [CrossRef]
- Caulin, C.; Nguyen, T.; Longley, M.A.; Zhou, Z.; Wang, X.-J.; Roop, D.R. Inducible activation of oncogenic K-ras results in tumor formation in the oral cavity. Cancer Res. 2004, 64, 5054–5058. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, P.; Osipo, C.; Foreman, K.; Golde, T.; Osborne, B.; Miele, L. Rational targeting of Notch signaling in cancer. Oncogene 2008, 27, 5124–5131. [Google Scholar] [CrossRef] [PubMed]
- Dorai, T.; Aggarwal, B.B. Role of chemopreventive agents in cancer therapy. Cancer Lett. 2004, 215, 129–140. [Google Scholar] [CrossRef]
- Koprowski, S.; Sokolowski, K.; Kunnimalaiyaan, S.; Gamblin, T.C.; Kunnimalaiyaan, M. Curcumin-mediated regulation of Notch1/hairy and enhancer of split-1/survivin: Molecular targeting in cholangiocarcinoma. J. Surg. Res. 2015, 198, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Cecchinato, V.; Chiaramonte, R.; Nizzardo, M.; Cristofaro, B.; Basile, A.; Sherbet, G.V.; Comi, P. Resveratrol-induced apoptosis in human T-cell acute lymphoblastic leukaemia MOLT-4 cells. Biochem. Pharmacol. 2007, 74, 1568–1574. [Google Scholar] [CrossRef]
- Sun, Z.; Zhou, C.; Liu, F.; Zhang, W.; Chen, J.; Pan, Y.; Ma, L.; Liu, Q.; Du, Y.; Yang, J.; Wang, Q. Inhibition of breast cancer cell survival by Xanthohumol via modulation of the Notch signaling pathway in vivo and in vitro. Oncol. Lett. 2018, 15, 908–916. [Google Scholar] [CrossRef]
- Zhang, Q.; Yuan, Y.; Cui, J.; Xiao, T.; Jiang, D. Paeoniflorin inhibits proliferation and invasion of breast cancer cells through suppressing Notch-1 signaling pathway. Biomed. Pharmacother. Biomed. Pharmacother. 2016, 78, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Kiesel, V.A.; Stan, S.D. Diallyl trisulfide, a chemopreventive agent from Allium vegetables, inhibits alpha-secretases in breast cancer cells. Biochem. Biophys. Res. Commun. 2017, 484, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Chen, H.; Sun, X. Baicalein suppresses non small cell lung cancer cell proliferation, invasion and Notch signaling pathway. Cancer Biomark. Sect. Dis. Mark. 2018, 22, 13–18. [Google Scholar] [CrossRef]
- Amin, A.R.M.R.; Karpowicz, P.A.; Carey, T.E.; Arbiser, J.; Nahta, R.; Chen, Z.G.; Dong, J.T.; Kucuk, O.; Khan, G.N.; Huang, G.S.; Mi, S.; Lee, H.Y.; Reichrath, J.; Honoki, K.; Georgakilas, A.G.; et al. Evasion of anti-growth signaling: A key step in tumorigenesis and potential target for treatment and prophylaxis by natural compounds. Semin. Cancer Biol. 2015, 35, S55–S77. [Google Scholar] [CrossRef] [PubMed]
- Koduru, S.; Kumar, R.; Srinivasan, S.; Evers, M.B.; Damodaran, C. Notch-1 inhibition by Withaferin-A: A therapeutic target against colon carcinogenesis. Mol. Cancer Ther. 2010, 9, 202–210. [Google Scholar] [CrossRef]
- Sagiv, E.; Rozovski, U.; Kazanov, D.; Liberman, E.; Arber, N. Gene expression analysis proposes alternative pathways for the mechanism by which celecoxib selectively inhibits the growth of transformed but not normal enterocytes. Clin. Cancer Res. 2007, 13 Pt 1, 6807–6815. [Google Scholar] [CrossRef]
- Shehzad, A.; Lee, Y.S. Molecular mechanisms of curcumin action: Signal transduction. BioFactors Oxf. Engl. 2013, 39, 27–36. [Google Scholar] [CrossRef]
- Singh, A.K.; Sharma, N.; Ghosh, M.; Park, Y.H.; Jeong, D.K. Emerging importance of dietary phytochemicals in fight against cancer: Role in targeting cancer stem cells. Crit. Rev. Food Sci. Nutr. 2017, 57, 3449–3463. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porcheri, C.; Meisel, C.T.; Mitsiadis, T. Multifactorial Contribution of Notch Signaling in Head and Neck Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 1520. https://doi.org/10.3390/ijms20061520
Porcheri C, Meisel CT, Mitsiadis T. Multifactorial Contribution of Notch Signaling in Head and Neck Squamous Cell Carcinoma. International Journal of Molecular Sciences. 2019; 20(6):1520. https://doi.org/10.3390/ijms20061520
Chicago/Turabian StylePorcheri, Cristina, Christian Thomas Meisel, and Thimios Mitsiadis. 2019. "Multifactorial Contribution of Notch Signaling in Head and Neck Squamous Cell Carcinoma" International Journal of Molecular Sciences 20, no. 6: 1520. https://doi.org/10.3390/ijms20061520
APA StylePorcheri, C., Meisel, C. T., & Mitsiadis, T. (2019). Multifactorial Contribution of Notch Signaling in Head and Neck Squamous Cell Carcinoma. International Journal of Molecular Sciences, 20(6), 1520. https://doi.org/10.3390/ijms20061520