3.1. Variant Data Collection and Mapping to Protein Data Bank (PDB)
SRVs were collected from the release of March 2018 of the Humsavar. In order to perform structural and functional analyses, we firstly mapped SRVs from protein sequences on positions in corresponding PDB structures. For this task, we used the PDBSWS website (last updated on July 2018) [
24] and SIFTS mapping [
25]. PDB coverage was computed dividing the number of residues covered in the PDB by the length of the UniprotKB sequence deprived of signal or transit peptides, when present.
In the HVAR3D dataset, we collected all disease-related and neutral variations occurring in protein chains covering more than 70% of the corresponding UniprotKB sequence and endowed with at least one disease-related variation (i.e., we retained only chains having both disease-related and neutral or only disease-related variations).
3.2. Retrieving Protein Annotations
Enzyme commission (EC) numbers and Gene Ontology Molecular Function (GO-MF) terms were retrieved for each protein using cross references directly available from the UniprotKB entry. In computing statistics for variations, all available EC numbers (at any level of annotation) were collapsed into their first-digit classification.
For each GO-MF term annotated, we reconstructed the full path to the ontology root node using the GO-TermFinder Perl software package (
https://metacpan.org/release/GO-TermFinder) [
26] and using the latest version of GO obo ontology file available at the Gene Ontology Consortium website (
http://geneontology.org/page/download-ontology). Complete DAGs for all GO-MF annotated on each protein were then merged together into a single functional annotation. Each protein chain (and corresponding variants therein) were then classified into one (or more) terms from the first-level GO-MF annotation (overall comprising 19 terms).
Positions of ACTIVE and BINDING sites for each protein chain were retrieved directly from features annotated on the UniprotKB entry.
Reactome pathway annotation was retrieved for each protein using the all-level pathway hierarchy mapping file (available at
https://reactome.org/download-data). This file assigns, to each UniprotKB accession, the full Reactome annotation at all levels of the hierarchy. From these, we extracted the first-level pathways used to perform statistics.
CATH classification was performed by downloading, from the CATH website (
http://www.cathdb.info/), annotation file mapping CATH domains on UniprotKB/PDB entries.
3.4. Prediction of the Impact of SRVs on Protein Stability
The effect of different SRVs on protein stability has been computationally evaluated with INPS-MD [
17], a method based on Support Vector Regression (SVR) for estimating the difference folding Gibbs free energy between the wild-type and the mutated forms of the proteins (ΔΔG). INPS-MD analyses two sets of descriptors extracted from protein sequence and structure, respectively. The former includes BLOSUM62 scores, hydrophobicity, Dayhoff mutability index and evolutionary information derived from multiple sequence alignments, while the latter consists of the residue relative solvent exposure and the difference between the native and the variant structure in terms of pairwise contact potential computed in a local structural environment (for further details, refer to [
22,
23]). When assessed on a dataset comprising 2648 variations whose ΔΔG has been experimentally determined, INPS-MD performs with a Pearson’s correlation index of 0.58 and a root mean error of 1.2 kcal/mol.
Each variation can be classified as perturbing or non-perturbing the protein stability according to the value of predicted ΔΔG. In particular, we define perturbing variations as those having |ΔΔG| ≥ 1, non-perturbing otherwise (i.e., |ΔΔG| < 1).
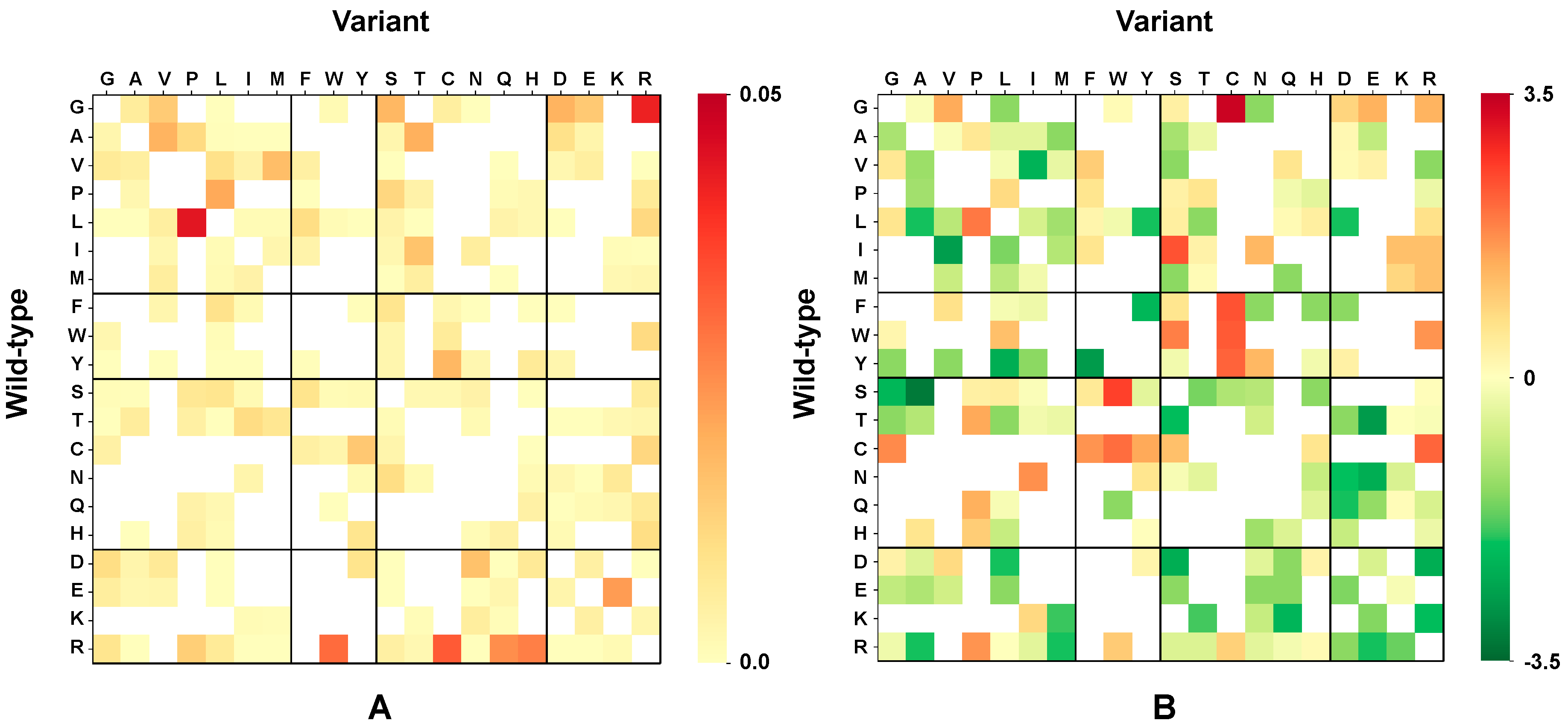
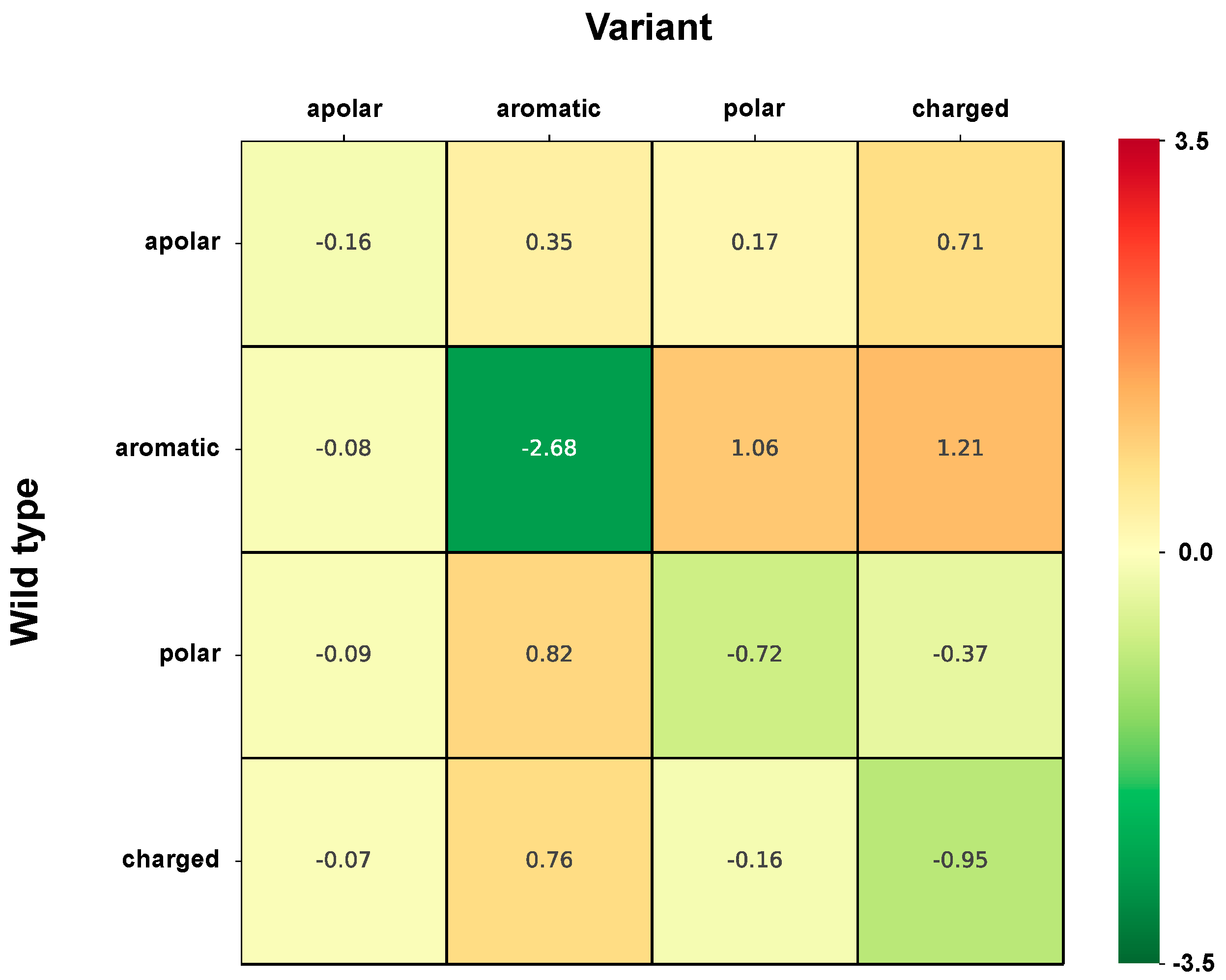
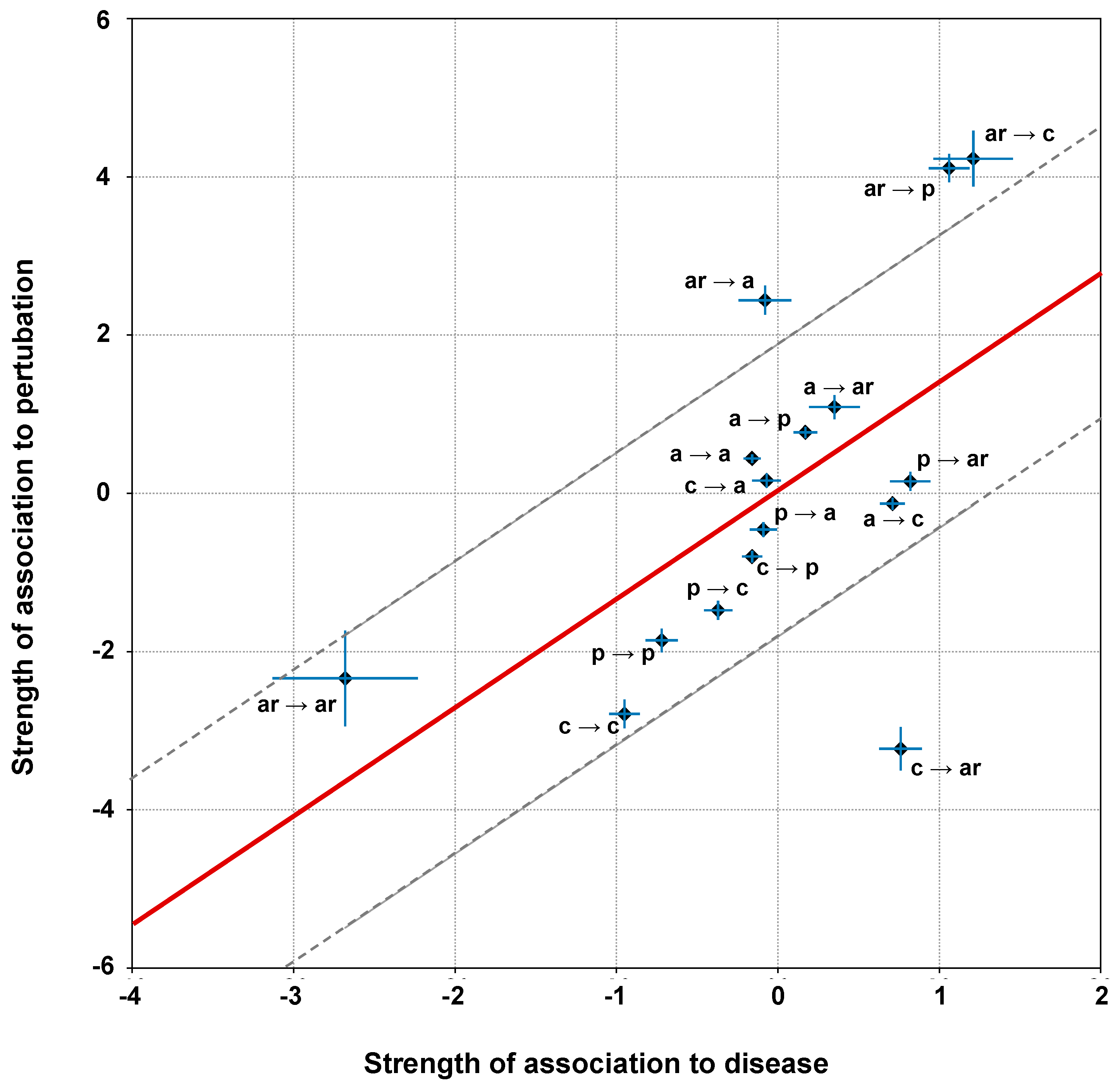
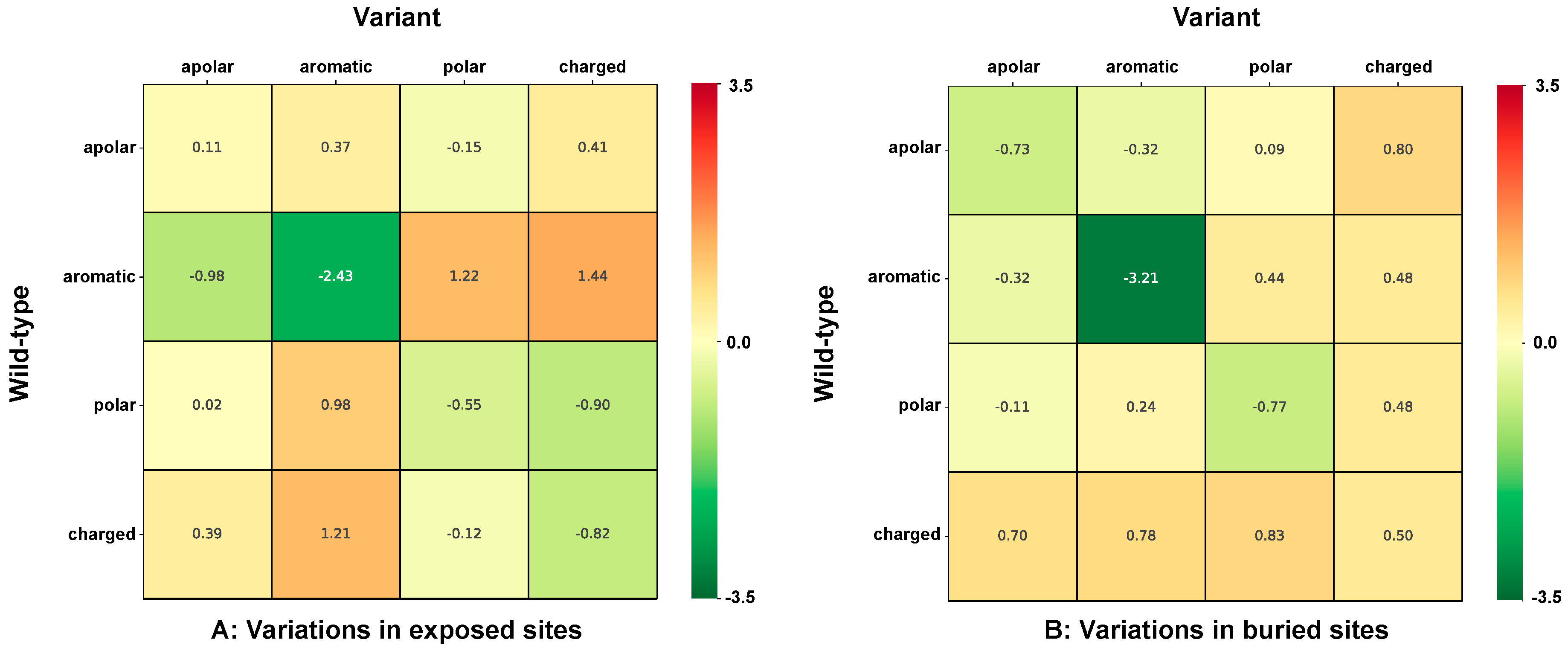
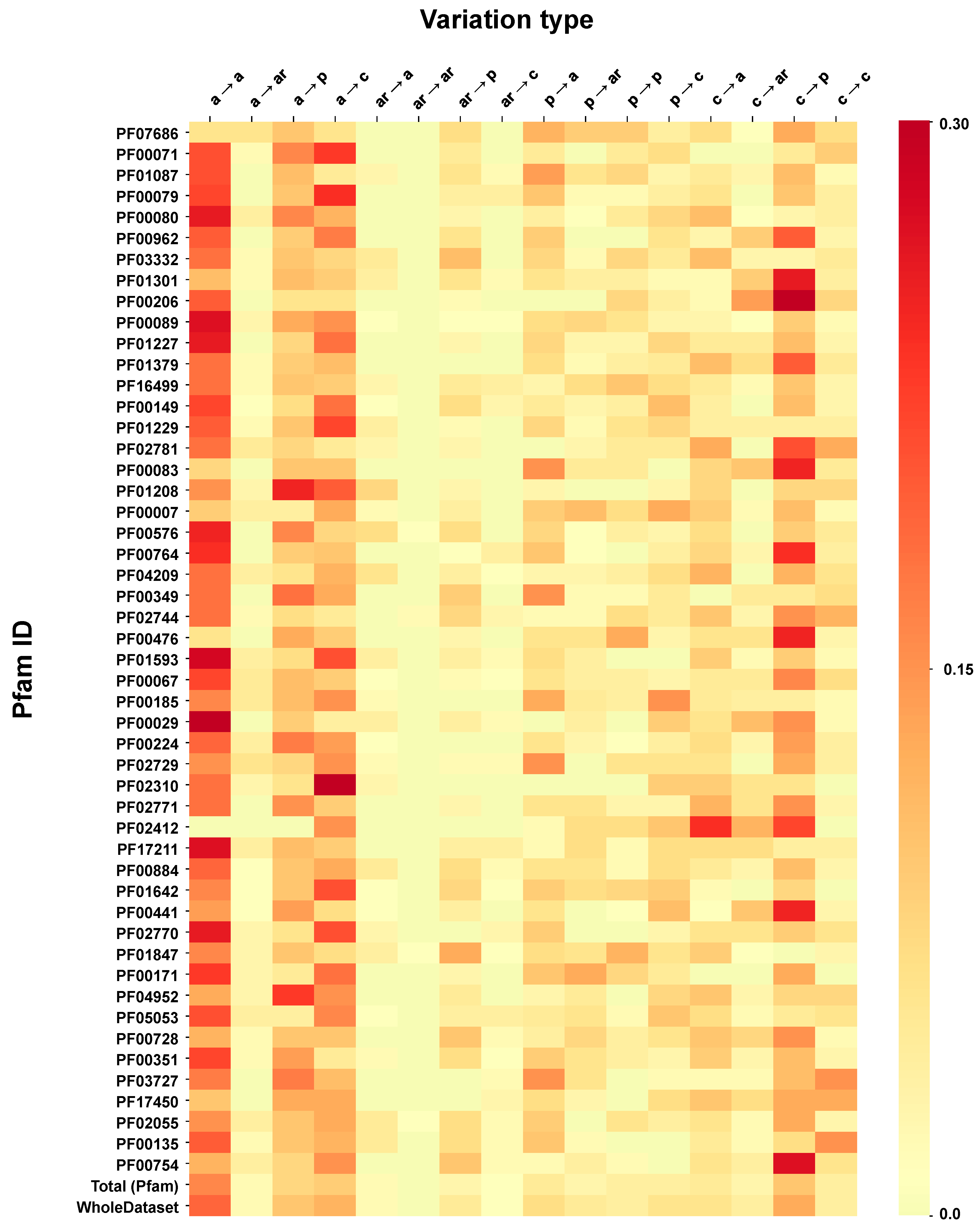
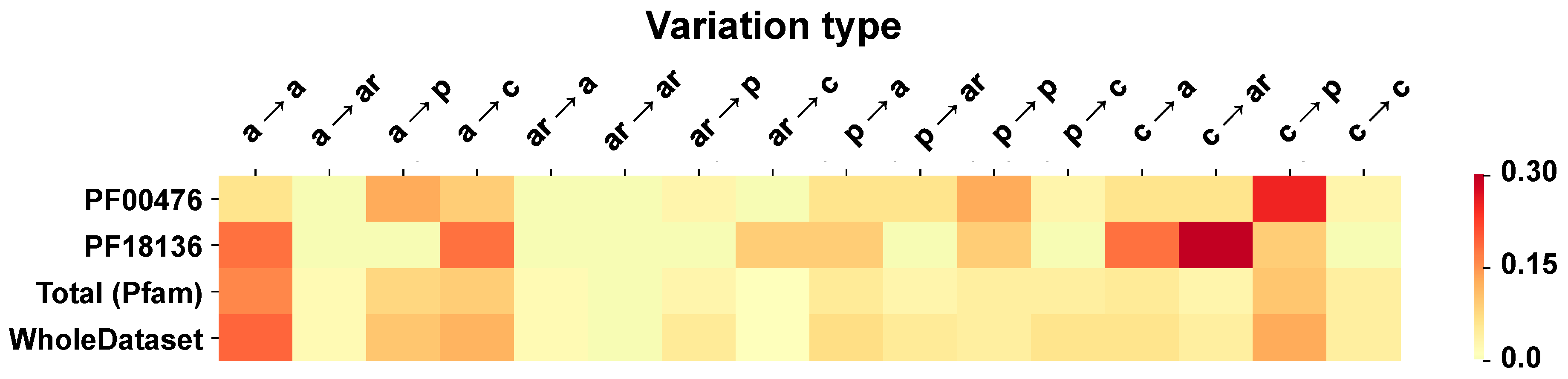
3.5. Computing the Association Strength of Each Variation Type to Disease and Pertubation
The association strength of each variation type to disease has been evaluated from the HVAR3D dataset. In order to strengthen our statistical analysis, HVAR3D was complemented using an additional dataset comprising 2667 neutral variations occurring on protein chains endowed with only neutral variations and covering more than 70% of the corresponding UniprotKB sequence (this set of neutral variations was not considered in all other analyses). Moreover, from disease-related variations in HVAR3D, we excluded those falling on ACTIVE or BINDING sites as well as those localized on known protein-protein interfaces.
After this procedure, we obtained a dataset of 8701 variations comprising 4990 and 3626 disease-related and neutral variations, respectively. We used this extended dataset to perform association analysis.
Let
be the resulting set of variations comprising
and
disease-related and neutral variations, respectively. For each variation type
from residue type
to
, we can compute the frequencies of occurrence
and
in disease-related and neutral subsets, respectively, as follows:
where
and
are the number of occurrences of the variation type
in the disease-related and neutral subsets, respectively.
The association strength
(Log-odd ratio) of variation type
to disease is then computed as:
An estimate of the standard error on the log odd ratios in Equation (3) can be computed as follows:
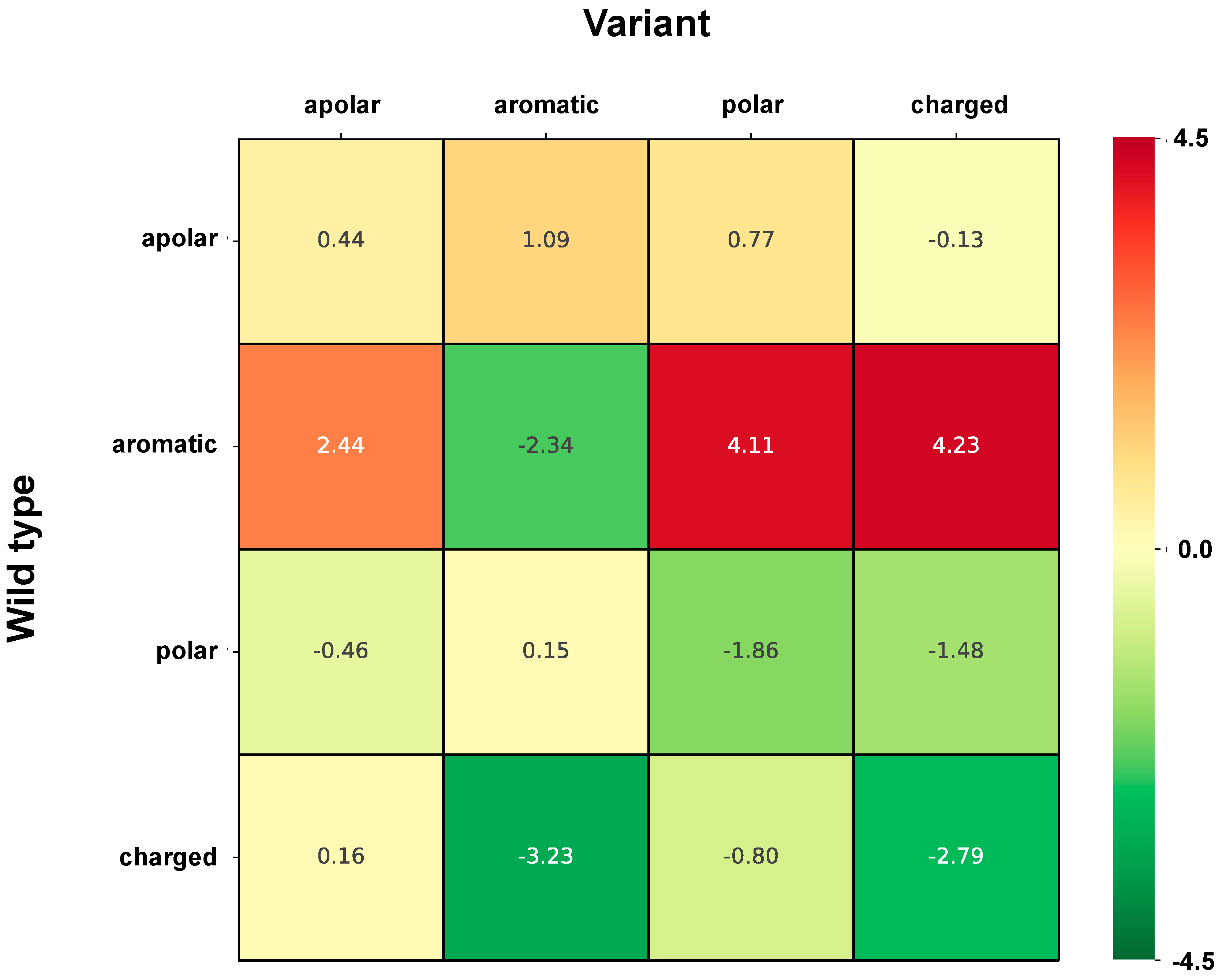
Analogously, associations of variation types to perturbation of protein stability has been evaluated on the same set
of variations as above (i.e., HVAR3D + complement of neutral variations). First, variations in
were classified into perturbing and non-perturbing using INPS-3D as explained in the
Section 3.4. Then, we computed frequencies of occurrence
and
of variation type
on the perturbing and non-perturbing subsets, respectively:
where
and
are the number of occurrences of the variation type
in the perturbing and non-perturbing subsets, respectively, and
and
are the total number of perturbing and non-perturbing variations, respectively.
The association strength
(Log-odd ratio) of variation type
to perturbation is then computed as:
Standard error is computed for as done for (Equation (4)).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}