N-Terminal Domain Mediated Regulation of RORα1 Inhibits Invasive Growth in Prostate Cancer


Abstract
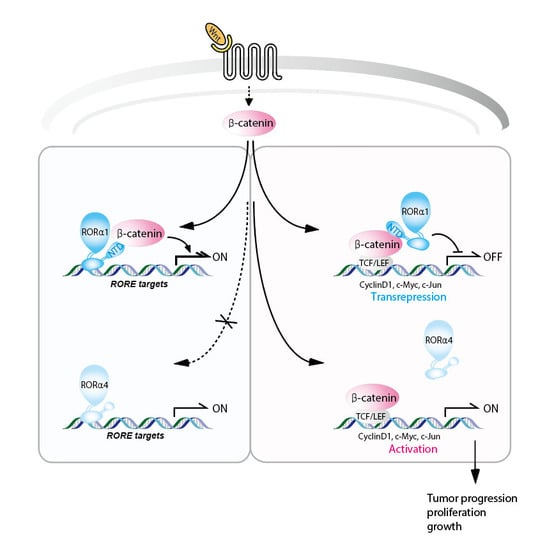
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
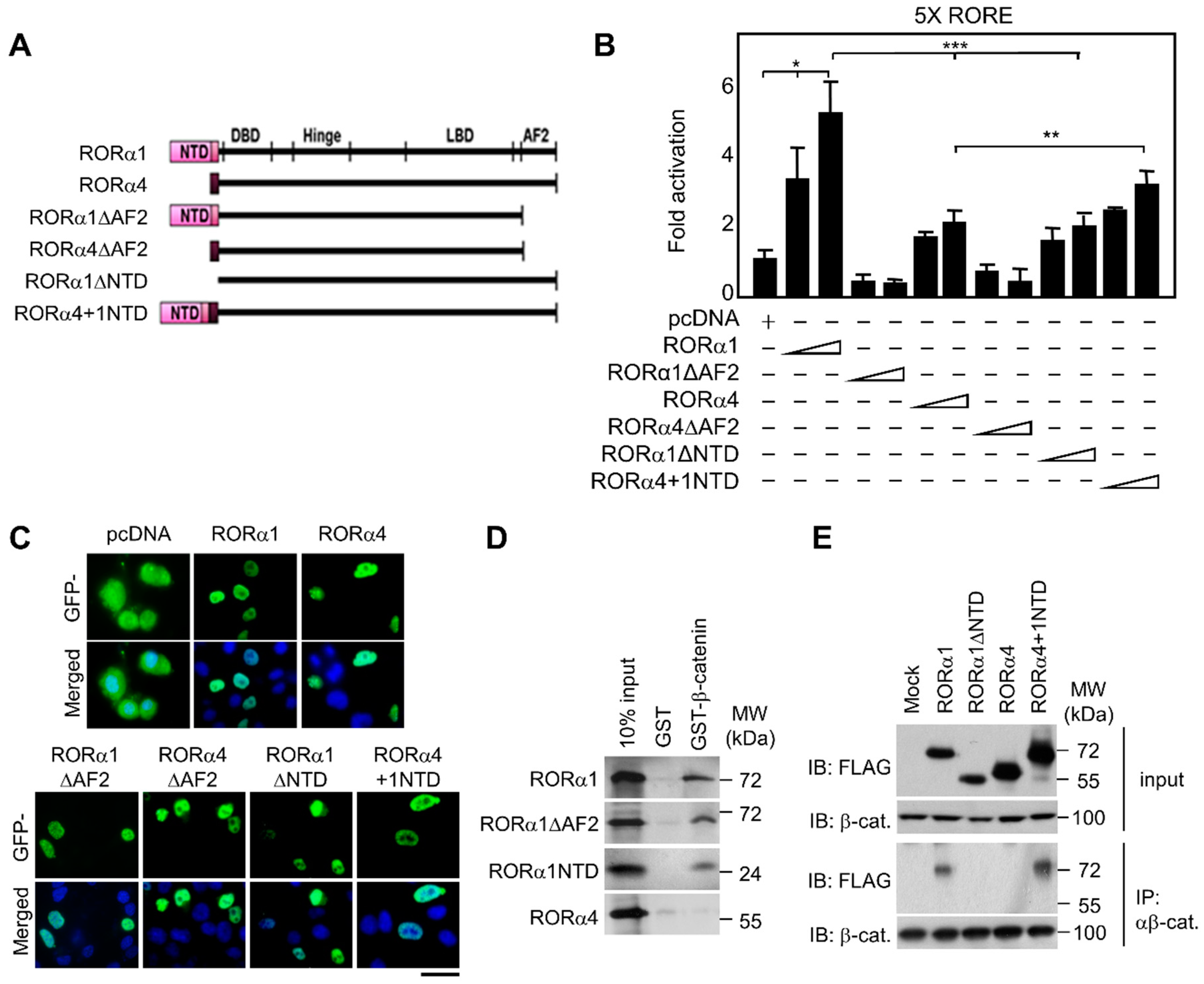
2.1. Selective Linkage of RORα1, not RORα4, in the Downstream Signaling Pathway
2.2. Wnt Signaling Is Hyperactivated in RORα-Null Mouse Embryonic Fibroblasts
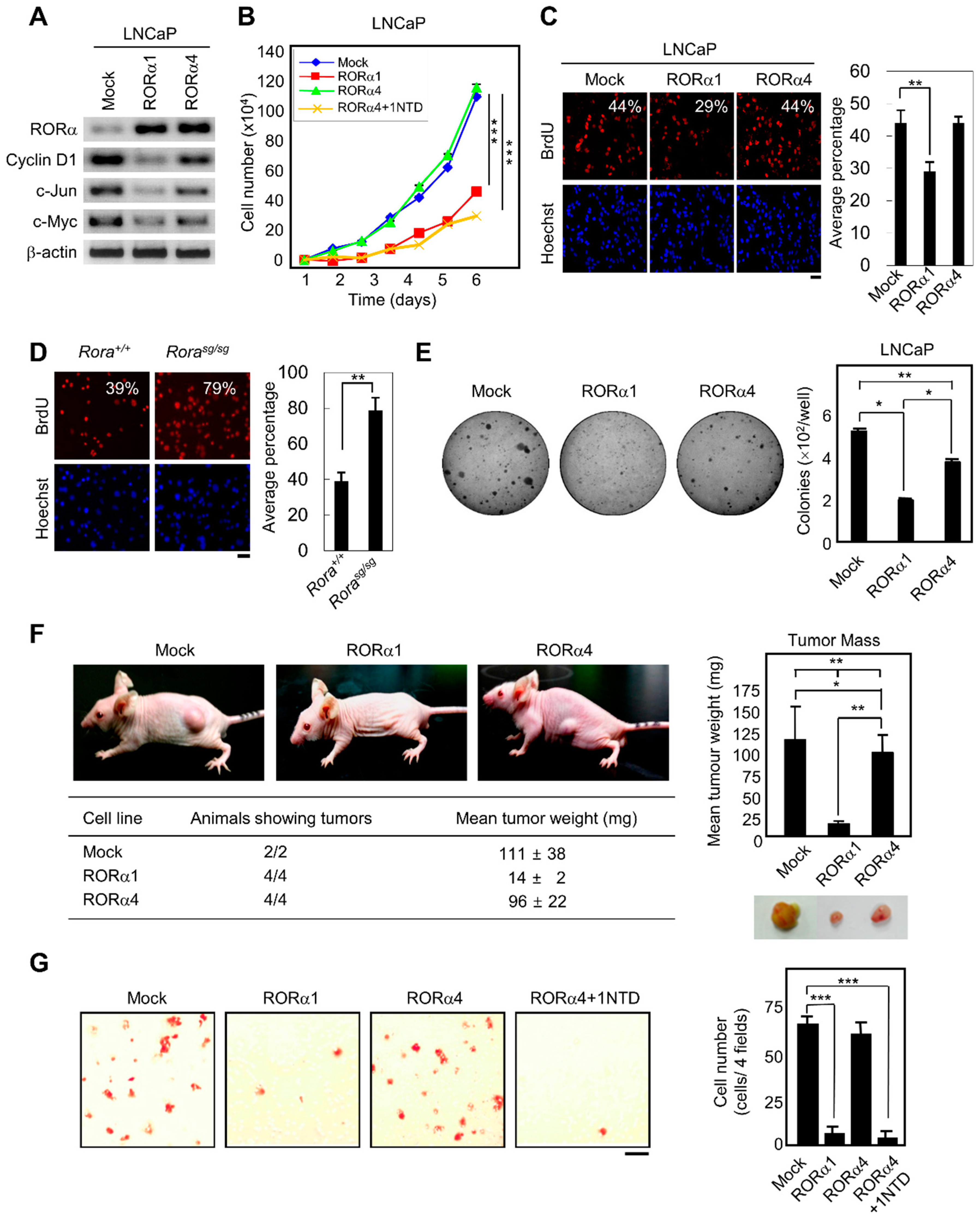
2.3. RORα1 Specifically Inhibits Cell Proliferation via the Downregulation of Wnt Target Genes
2.4. RORα1 Influences in Vivo Tumorigenesis and Metastatic Potential
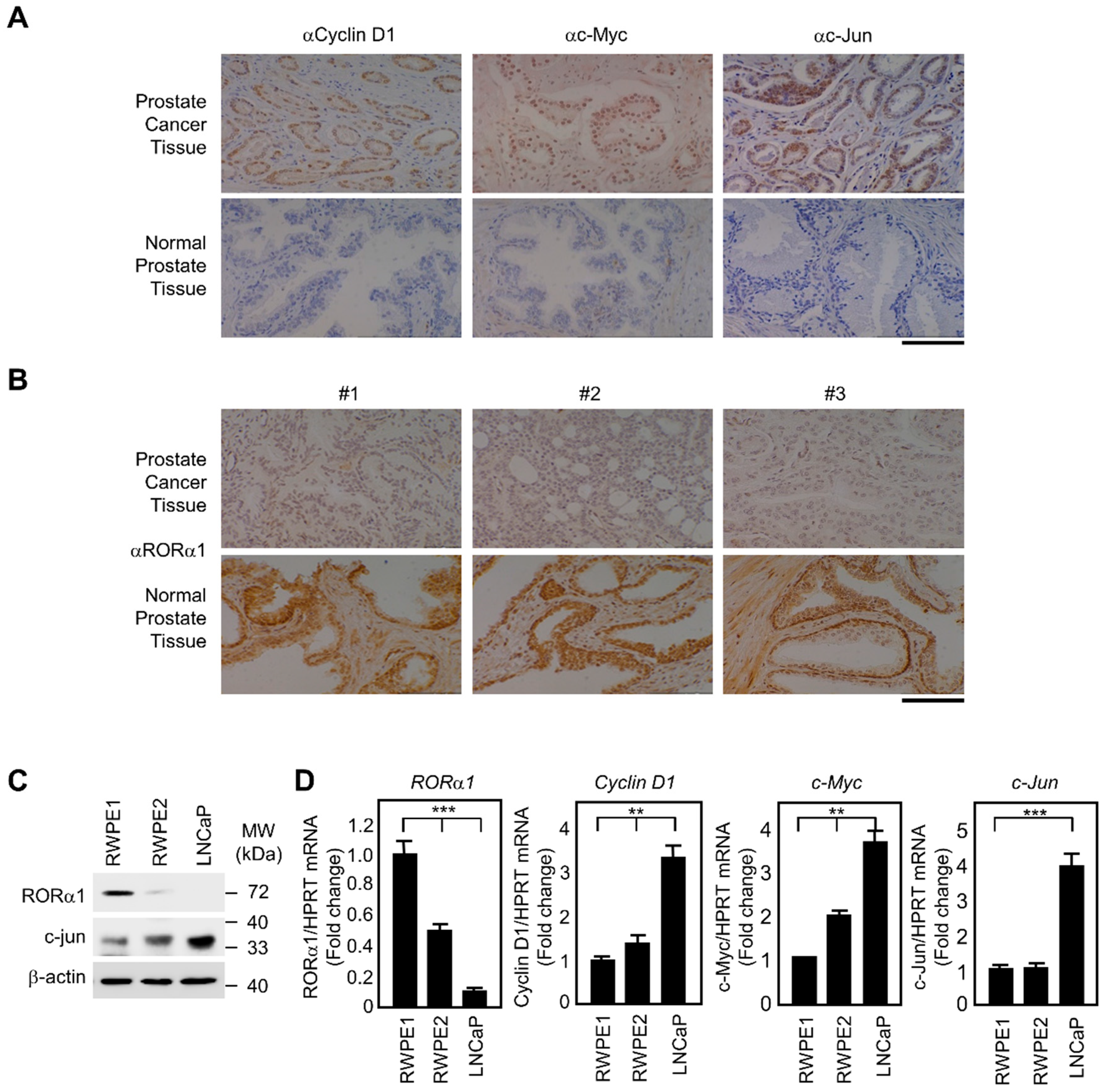
2.5. Inverse Correlation of RORα1 and Wnt Target Genes in Prostate Carcinoma Tissues
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Mice and Preparation of Primary Mouse Embryonic Fibroblasts
4.3. GST Pull-down Assays
4.4. Tissue Array and Immunohistochemistry
4.5. Chromatin Immunoprecipitation (ChIP)
4.6. Real-Time Q-PCR
4.7. Cell Proliferation Assay and BrdU Incorporation Assay
4.8. Matrigel Invasion Assay
4.9. Cell Transformation Assay
4.10. Tumorigenicity Assay
4.11. Quantification and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| RORα | Retinoid acid-related orphan receptor α |
| ONR | Orphan Nuclear Receptor |
| NTD | N-terminal domain |
| DBD | DNA-binding domain |
| LBD | Ligand-binding domain |
| TCF | T cell factor |
| LEF | lymphoid enhancer factor |
| CKI | Casein kinase I |
| GSK-3β | Glycogen synthase kinase-3β |
| Dvl | Dishevelled |
| MEF | Mouse embryonic fibroblast |
| ChIP | Chromatin immunoprecipitation |
References
- Mangelsdorf, D.J.; Evans, R.M. The RXR heterodimers and orphan receptors. Cell 1995, 83, 841–850. [Google Scholar] [CrossRef] [Green Version]
- Giguère, V. Orphan nuclear receptors: From gene to function. Endocr. Rev. 1999, 20, 689–725. [Google Scholar] [CrossRef]
- Danielian, P.S.; White, R.; Lees, J.A.; Parker, M.G. Identification of a conserved region required for hormone dependent transcriptional activation by steroid hormone receptors. EMBO J. 1992, 11, 1025–1033. [Google Scholar] [CrossRef]
- Pissios, P.; Tzameli, I.; Moore, D.D. New insights into receptor ligand binding domains from a novel assembly assay. J. Steroid Biochem. Mol. Biol. 2001, 76, 3–7. [Google Scholar] [PubMed]
- Giguère, V.; Tini, M.; Flock, G.; Ong, E.; Evans, R.M.; Otulakowski, G. Isoform-specific amino-terminal domains dictate DNA-binding properties of ROR alpha, a novel family of orphan hormone nuclear receptors. Genes Dev. 1994, 8, 538–553. [Google Scholar] [PubMed]
- Beckerandré, M.; André, E.; Delamarter, J.F. Identification of nuclear receptor mRNAs by RT-PCR Amplification of conserved zinc-finger motif sequences. Biochem. Biophys. Res. Commun. 1993, 194, 1371–1379. [Google Scholar] [CrossRef]
- Matysiak-Scholze, U.; Nehls, M. The structural integrity of RORα isoforms is mutated in staggerer mice: Cerebellar coexpression of RORα1 and RORα4. Genomics 1997, 43, 78–84. [Google Scholar] [CrossRef]
- Dussault, I.; Giguere, V. Differential regulation of the N-myc proto-oncogene by ROR alpha and RVR, two orphan members of the superfamily of nuclear hormone receptors. Mol. Cell. Biol. 1997, 17, 1860–1867. [Google Scholar] [CrossRef] [PubMed]
- Gold, D.A.; Baek, S.H.; Schork, N.J.; Rose, D.W.; Larsen, D.D.; Sachs, B.D.; Rosenfeld, M.G.; Hamilton, B.A. RORα coordinates reciprocal signaling in cerebellar development through sonic hedgehog and calcium-dependent pathways. Neuron 2003, 40, 1119–1131. [Google Scholar] [CrossRef]
- Hamilton, B.A.; Frankel, W.N.; Kerrebrock, A.W.; Hawkins, T.L.; FitzHugh, W.; Kusumi, K.; Russell, L.B.; Mueller, K.L.; van Berkel, V.; Birren, B.W.; et al. Disruption of the nuclear hormone receptor RORalpha in staggerer mice. Nature 1996, 379, 736–739. [Google Scholar] [CrossRef]
- Lau, P.; Nixon, S.J.; Parton, R.J.; Muscat, G.E. RORα regulates the expression of genes involved in lipid homeostasis in skeletal muscle cells: Caveolin-3 and CPT-1 are direct targets of ROR. J. Biol. Chem. 1994, 279, 36828–36840. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T. Transcriptional regulation of a purkinje cell-specific gene through a functional interaction between ROR alpha and RAR. Genes Cells 1997, 220, 405–410. [Google Scholar]
- Akashi, M.; Takumi, T. The orphan nuclear receptor RORalpha regulates circadian transcription of the mammalian core-clock Bmal1. Nat. Struct. Mol. Biol. 2005, 12, 441–448. [Google Scholar] [CrossRef]
- Kim, H.; Lee, J.M.; Lee, G.; Bhin, J.; Oh, S.K.; Kim, K.; Pyo, K.E.; Lee, J.S.; Yim, H.Y.; Kim, K.I.; et al. DNA damage-induced RORalpha is crucial for p53 stabilization and increased apoptosis. Mol. Cell 2011, 44, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.O.; Pappu, B.P.; Nurieva, R.; Akimzhanov, A.; Kang, H.S.; Chung, Y.; Ma, L.; Shah, B.; Panopoulos, A.D.; Schluns, K.S.; et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity 2008, 28, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Lee, J.S.; Kim, H.; Kim, K.; Park, H.; Kim, J.Y.; Lee, S.H.; Kim, I.S.; Kim, J.; Lee, M.; et al. EZH2 generates a methyl degron that is recognized by the DCAF1/DDB1/CUL4 E3 ubiquitin ligase complex. Mol. Cell 2012, 48, 572–586. [Google Scholar] [CrossRef]
- Kim, K.; Boo, K.; Yu, Y.S.; Oh, S.K.; Kim, H.; Jeon, Y.; Bhin, J.; Hwang, D.; Kim, K.I.; Lee, J.S.; et al. RORalpha controls hepatic lipid homeostasis via negative regulation of PPARgamma transcriptional network. Nat. Commun. 2017, 8, 162. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, D.J.; Dedhar, S.; Coetzee, G.A.; Nelson, C.C. Interaction of nuclear receptors with the Wnt/beta-catenin/Tcf signaling axis: Wnt you like to know? Endocr. Rev. 2005, 26, 898–915. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Kim, I.S.; Kim, H.; Lee, J.S.; Kim, K.; Yim, H.Y.; Jeong, J.; Kim, J.H.; Kim, J.Y.; Lee, H.; et al. RORalpha attenuates Wnt/beta-catenin signaling by PKCalpha-dependent phosphorylation in colon cancer. Mol. Cell 2010, 37, 183–195. [Google Scholar] [CrossRef]
- Korinek, V.; Barker, N.; Morin, J.P.; van Wichen, D.; de Weger, R.; Kinzler, K.W.; Vogelstein, B.; Clevers, H. Constitutivetranscriptional activation by a β-catenin-Tcf complex in APC/colon carcinoma. Science 1997, 275, 1784–1787. [Google Scholar] [CrossRef]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef]
- Peifer, M.; Polakis, P. Wnt signaling in oncogenesis and embryogenesis—A look outside the nucleus. Science 2000, 287, 1606–1609. [Google Scholar] [CrossRef] [PubMed]
- Chesire, D.R.; Isaacs, W.B. β-Catenin signaling in prostate cancer: An early perspective. Endocr. Relat. Cancer 2003, 10, 537–560. [Google Scholar] [CrossRef]
- Moon, R.T.; Bowerman, B.; Boutros, M.; Perrimon, N. The promise and perils of Wnt signaling through beta-catenin. Science 2002, 296, 1644–1646. [Google Scholar] [CrossRef] [PubMed]
- Polakis, P. Wnt signaling and cancer. Genes Dev. 2000, 14, 1837–1851. [Google Scholar] [CrossRef] [PubMed]
- Van Es, J.H.; Barker, N.; Clevers, H. You Wnt some, you lose some: Oncogenes in the Wnt signaling pathway. Curr. Opin. Genet. Dev. 2003, 13, 26–33. [Google Scholar] [CrossRef]
- Behrens, J.; von Kries, J.P.; Kuhl, M.; Bruhn, L.; Wedlich, D.; Grosschedl, R.; Birchmeier, W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature 1996, 382, 638–642. [Google Scholar] [CrossRef]
- Molenaar, M.; van de Wetering, M.; Oosterwegel, M.; Peterson-Maduro, J.; Godsave, S.; Korinek, V.; Roose, J.; Destree, O.; Clevers, H. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell 1996, 86, 391–399. [Google Scholar] [CrossRef]
- Orford, K.; Crockett, C.; Jensen, J.P.; Weissman, A.M.; Byers, S.W. Serine phosphorylation-regulated ubiquitination and degradation of b-catenin. J. Biol. Chem. 1997, 272, 24735–24738. [Google Scholar] [CrossRef] [PubMed]
- Salic, A.; Lee, E.; Mayer, L.; Kirschner, M.W. Control of β-catenin stability: Reconstitution of the cytoplasmic steps of the wnt pathway in Xenopus egg extracts. Mol. Cell 2000, 5, 523–532. [Google Scholar] [CrossRef]
- Van de Wetering, M.; Sancho, E.; Verweij, C.; de Lau, W.; Oving, I.; Hurlstone, A.; van der Horn, K.; Batlle, E.; Coudreuse, D.; Haramis, A.P.; et al. The β-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 2002, 111, 241–250. [Google Scholar] [CrossRef]
- Byers, S.; Pishvaian, M.; Crockett, C.; Peer, C.; Tozeren, A.; Sporn, M.; Anzano, M.; Lechleider, R. Retinoids increase cell-cell adhesion strength, beta catenin protein stability, and localization to the cell membrane in a breast cancer cell line. A role for serine kinase activity. Endocrinology 1996, 137, 3265–3273. [Google Scholar] [CrossRef]
- Mulholland, D.J.; Read, J.T.; Rennie, P.S.; Cox, M.E.; Nelson, C.C. Functional localization and competition between the androgen receptor and T-cell factor for nuclear β-catenin: A means for inhibition of the Tcf signaling axis. Oncogene 2003, 22, 5602–5613. [Google Scholar] [CrossRef]
- Vleugel, M.M.; Greijer, A.E.; Bos, R.; van der Wall, E.; van Diest, P.J. c-Jun activation is associated with proliferation and angiogenesis in invasive breast cancer. Hum. Pathol. 2006, 37, 668–674. [Google Scholar] [CrossRef]
- Yang, G.; Timme, T.L.; Frolov, A.; Wheeler, T.M.; Thompson, T.C. Combined c-Myc and caveolin-1 expression in human prostate carcinoma predicts prostate carcinoma progression. Cancer 2005, 103, 1186–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinmayr, M.; André, E.; Conquet, F.; Rondi-Reig, L.; Delhaye-Bouchaud, N.; Auclair, N.; Daniel, H.; Crepel, F.; Mariani, J.; Sotelo, C.; et al. Staggerer phenotype in retinoid-related orphan receptor α-deficient mice. Proc. Natl. Acad. Sci. USA 1998, 95, 3960–3965. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Lee, J.M.; Yu, Y.S.; Kim, H.; Nam, H.J.; Moon, H.G.; Noh, D.Y.; Kim, K.I.; Fang, S.; Baek, S.H. RORα2 requires LSD1 to enhance tumor progression in breast cancer. Sci. Rep. 2017, 7, 11194. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Madar, A.; David, G.; Garabedian, M.J.; Dasgupta, R.; Logan, S.K. Inhibition of androgen receptor and β-catenin activity in prostate cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 15710–15715. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.R.; Hwang, K.S.; Yoo, J.; Cho, W.K.; Kim, J.M.; Kim, W.H.; Im, D.S. E2-EPF UCP targets pVHL for degradation and associates with tumor growth and metastasis. Nat. Med. 2006, 12, 809–816. [Google Scholar] [CrossRef]
- Baek, S.H.; Ohgi, K.A.; Rose, D.W.; Koo, E.H.; Glass, C.K.; Rosenfeld, M.G. Exchange of N-CoR Corepressor and Tip60 coactivator Complexes Links Gene Expression by NF-κB and beta-amyloid precursor protein. Cell 2002, 110, 55–67. [Google Scholar] [CrossRef]
- Kim, J.H.; Choi, H.J.; Kim, B.; Kim, M.H.; Lee, J.M.; Kim, I.S.; Lee, M.H.; Choi, S.J.; Kim, K.I.; Kim, S.I.; et al. Roles of SUMOylation of a reptin chromatin remodeling complex in cancer metastasis. Nat. Cell Biol. 2006, 8, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Botrugno, O.A.; Fayard, E.; Annicotte, J.S.; Haby, C.; Brennan, T.; Wendling, O.; Tanaka, T.; Kodama, T.; Thomas, W.; Auwerx, J.; et al. Synergy between LRH-1 and beta-catenin induces G1 cyclin-mediated cell proliferation. Mol. Cell 2004, 15, 499–509. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.C.; Park, I.-G.; Kim, H.; Lee, J.M. N-Terminal Domain Mediated Regulation of RORα1 Inhibits Invasive Growth in Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 1684. https://doi.org/10.3390/ijms20071684
Park SC, Park I-G, Kim H, Lee JM. N-Terminal Domain Mediated Regulation of RORα1 Inhibits Invasive Growth in Prostate Cancer. International Journal of Molecular Sciences. 2019; 20(7):1684. https://doi.org/10.3390/ijms20071684
Chicago/Turabian StylePark, Su Chan, Il-Geun Park, Hyunkyung Kim, and Ji Min Lee. 2019. "N-Terminal Domain Mediated Regulation of RORα1 Inhibits Invasive Growth in Prostate Cancer" International Journal of Molecular Sciences 20, no. 7: 1684. https://doi.org/10.3390/ijms20071684
APA StylePark, S. C., Park, I.-G., Kim, H., & Lee, J. M. (2019). N-Terminal Domain Mediated Regulation of RORα1 Inhibits Invasive Growth in Prostate Cancer. International Journal of Molecular Sciences, 20(7), 1684. https://doi.org/10.3390/ijms20071684