Improved Prediction of Regulatory Element Using Hybrid Abelian Complexity Features with DNA Sequences


Abstract
:1. Introduction
2. Results
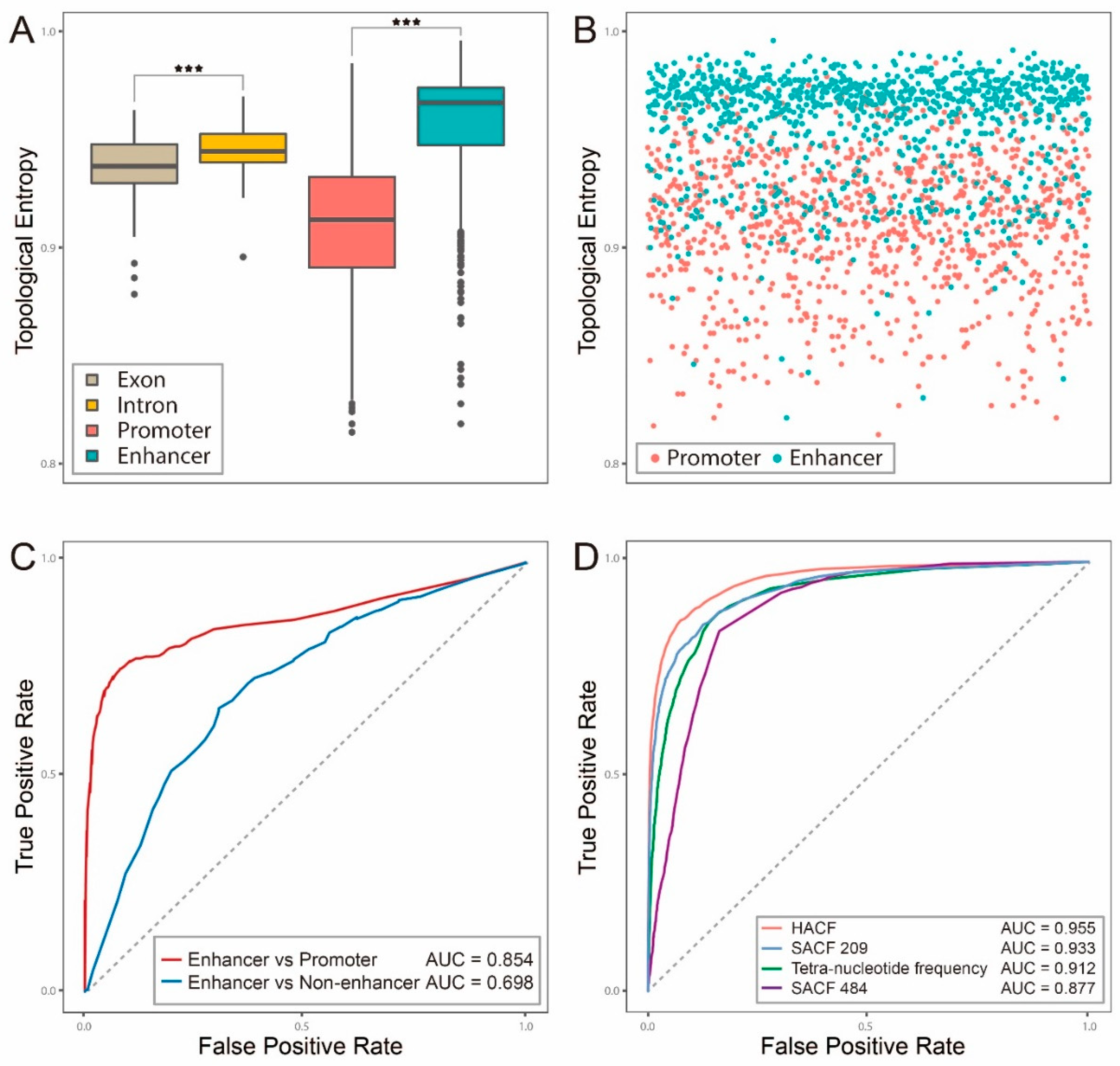
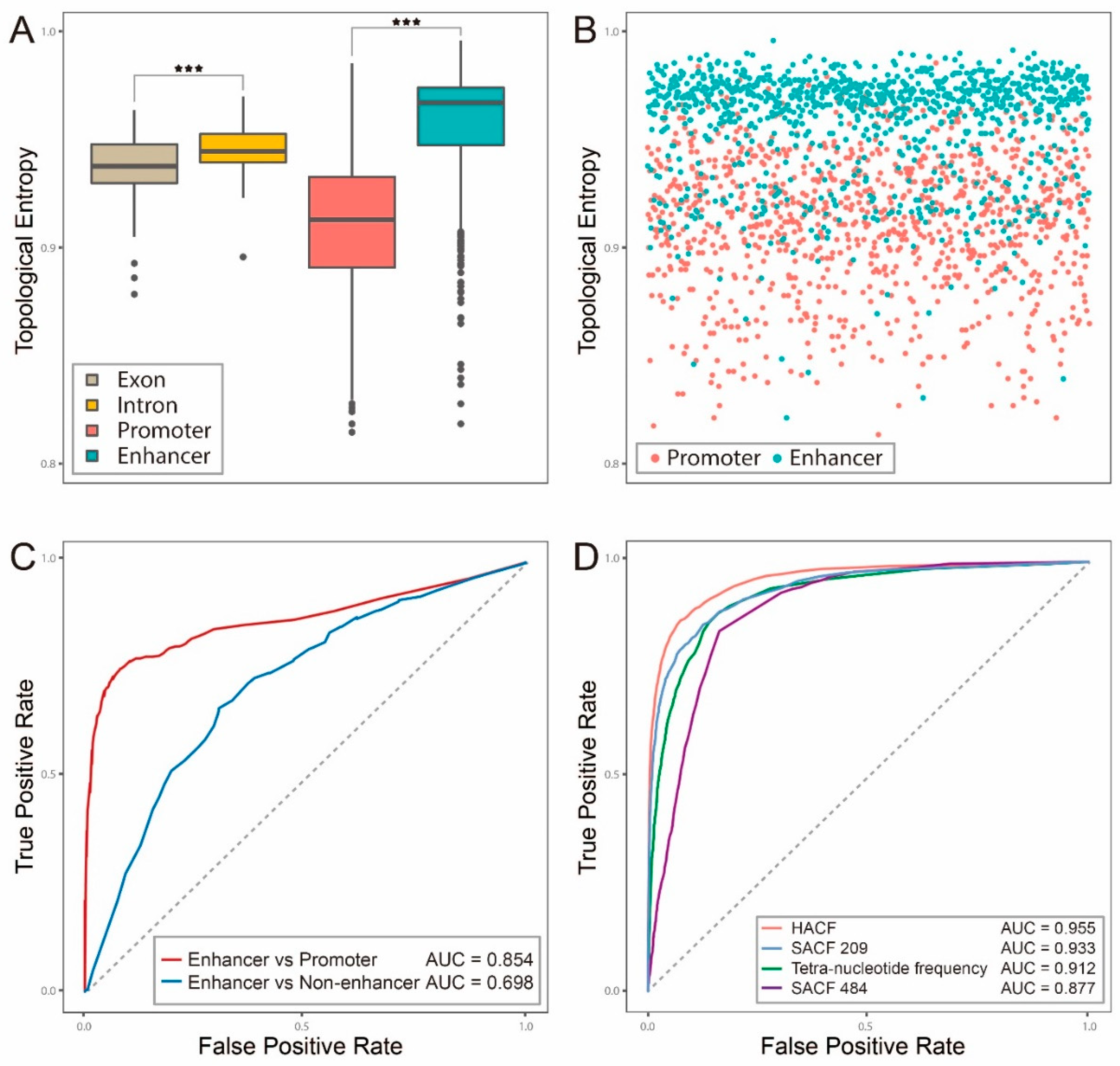
2.1. Topological Entropy of Enhancer is Larger than that of Exon, Intron or Promoter
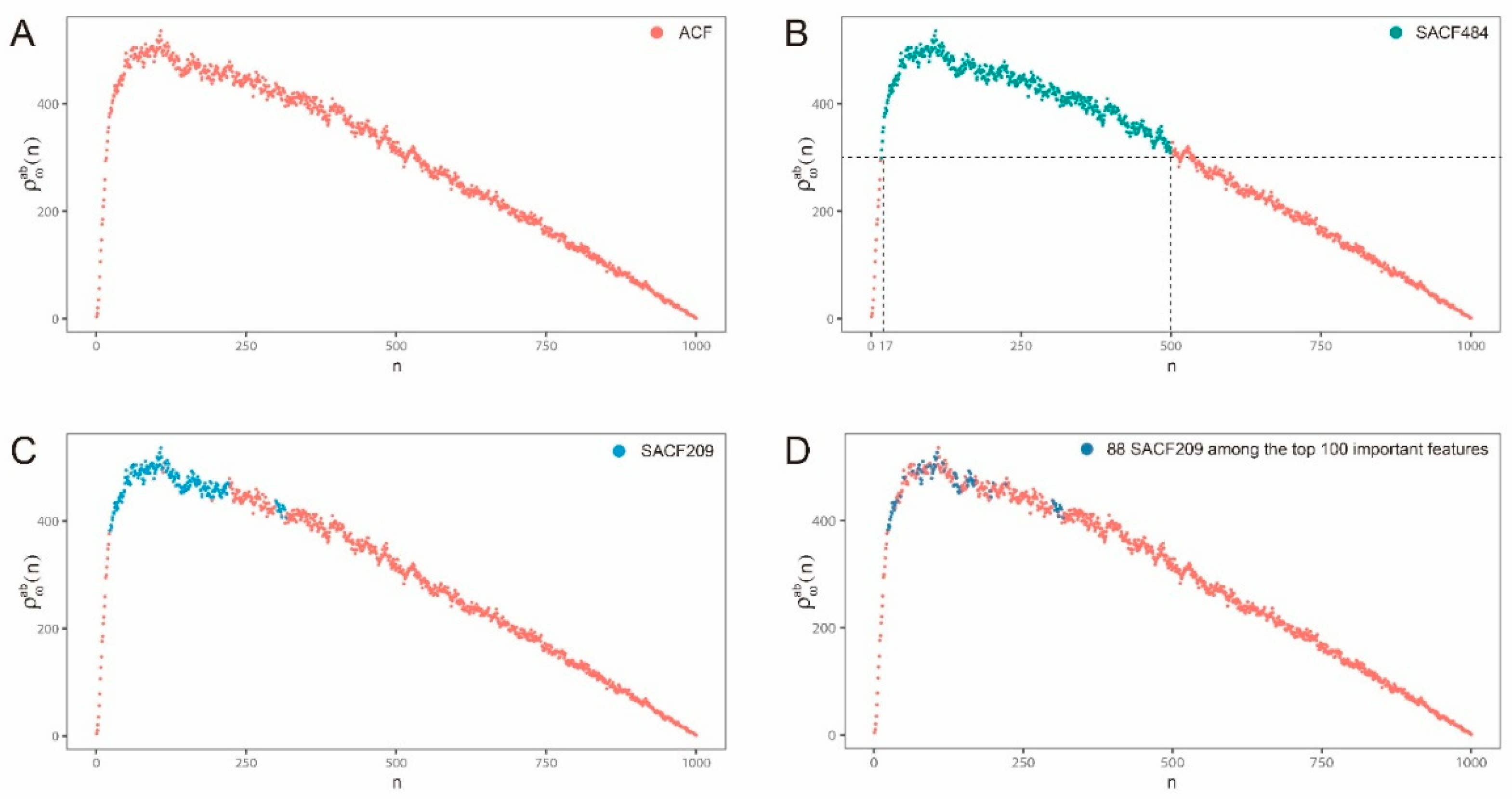
2.2. Hybrid Abelian Complexity Features are Predictive Features for Enhancers
2.3. Comparisons with Other Existing Methods
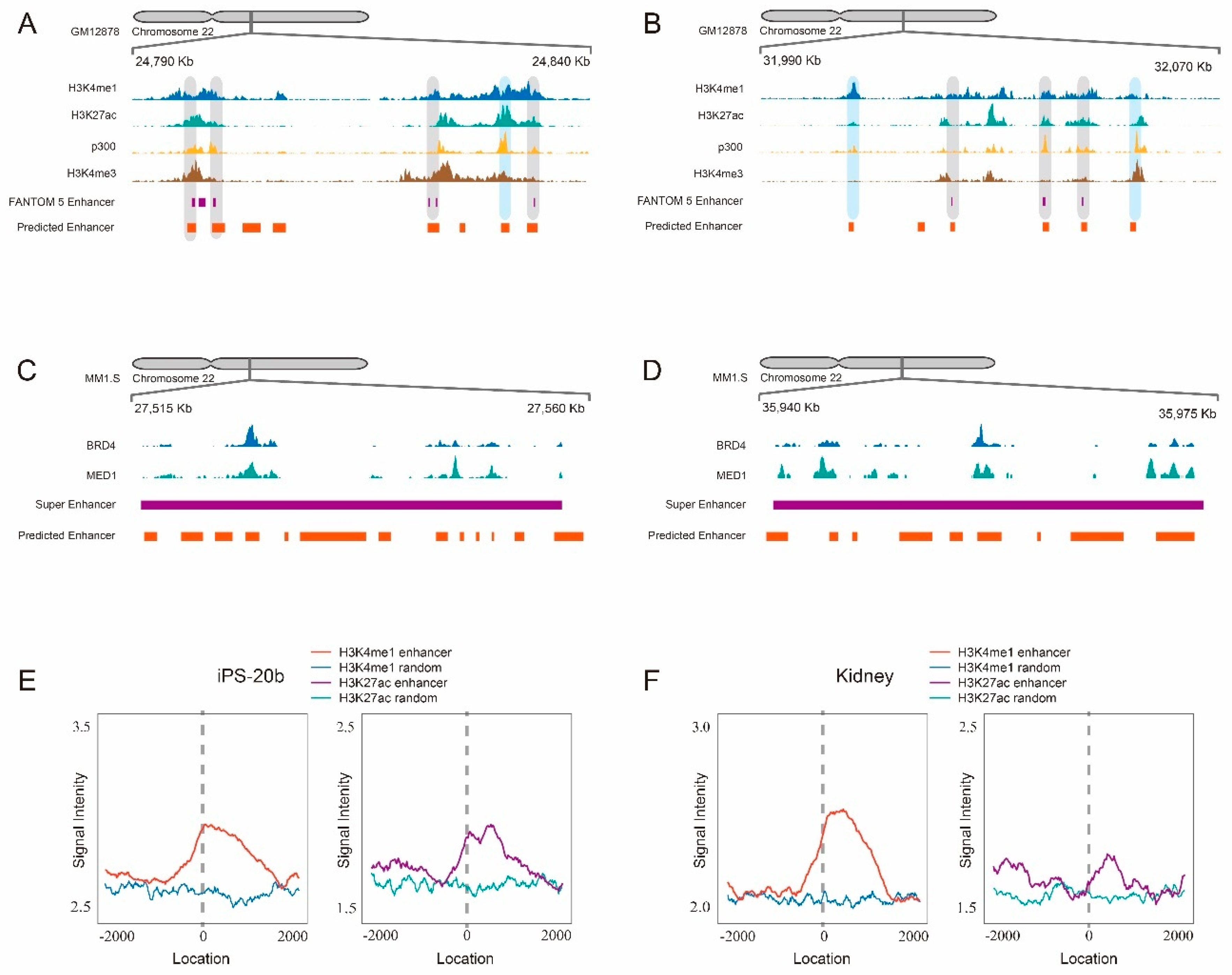
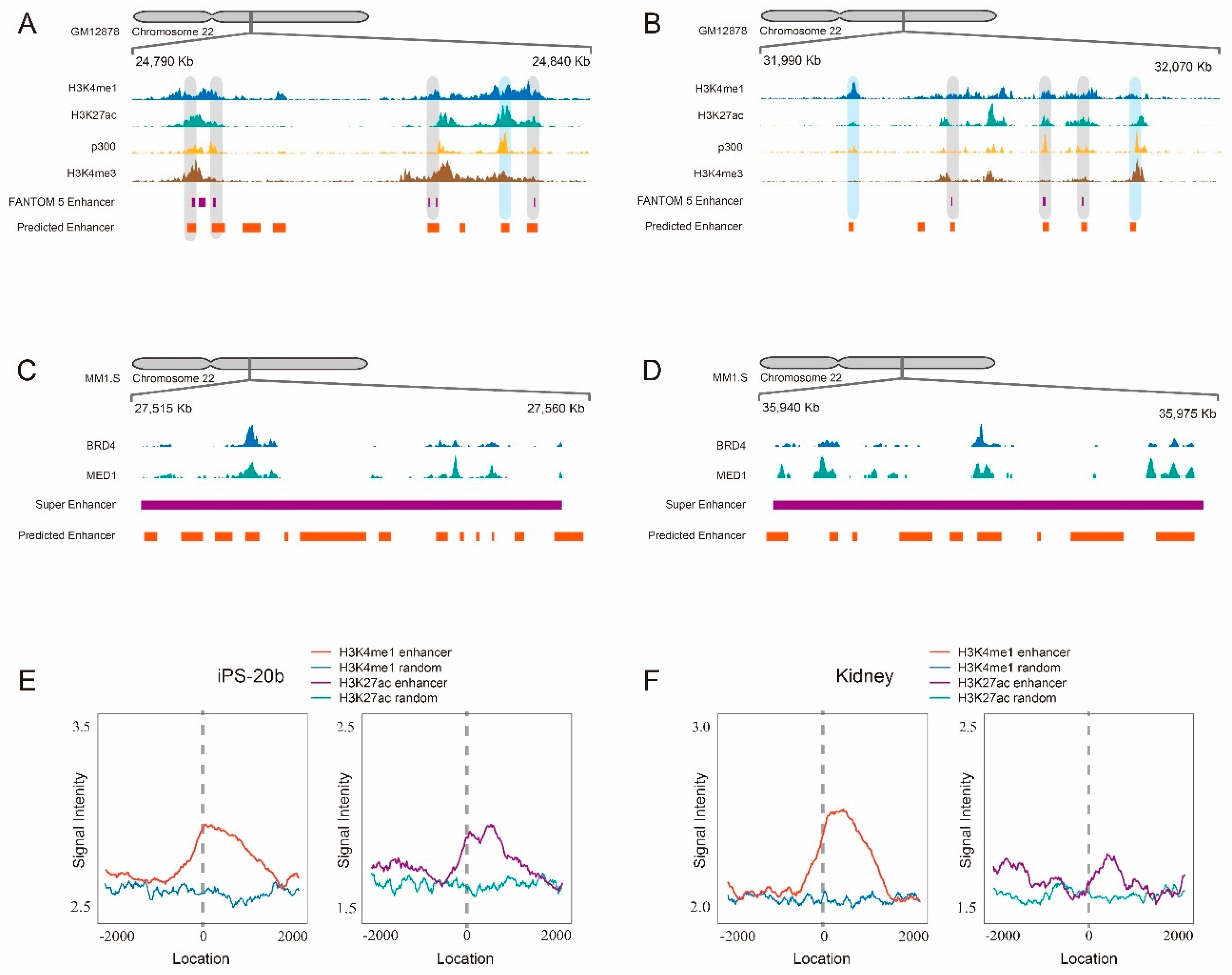
2.4. Scanning Chromosome 22 Identifies Known Disease-Related Super-Enhancers as Well as Novel Candidate Enhancers
3. Methods
3.1. Dataset Preparation
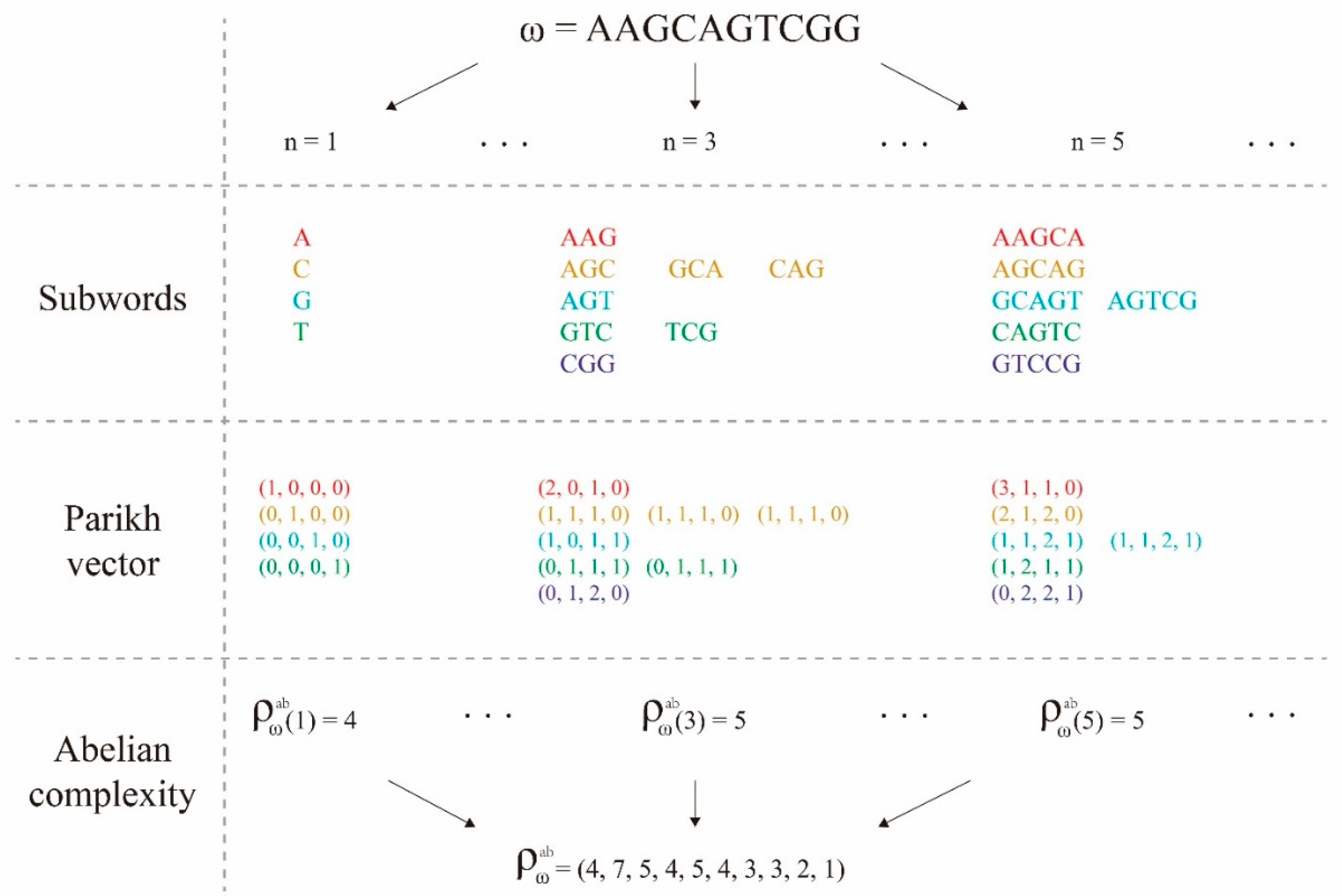
3.2. Subword Complexity Function and Topological Entropy
3.3. Abelian Complexity Function and Its computing Resource Consumption
3.4. Upper Bound Theorem and Feature Selection
3.5. Evaluation of the Prediction Performance
4. Discussion
- (1)
- New contribution to prediction methodology: for the first time, we proposed an innovative method, i.e., selected abelian complexity features together with DNA composition for accurate enhancer identification. In comparison with three state-of-the-art enhancer prediction tools, our method shows greater accuracy and more robust performances on imbalanced datasets.
- (2)
- New contribution to the prediction of novel candidate enhancers: when we applied our trained model to scan human chromosome 22, 7045 genomic regions were identified as enhancers. Some of them are consistent with known FANTOM5 enhancer, and the majority of them are considered as candidate enhancers by the evidences of surrounding histone modification markers including H3K4me1 and H3K27ac. This demonstrates that the predicted enhancers by our scanning approach have the potential to be true enhancers.
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Additional Information
References
- Kleftogiannis, D.; Kalnis, P.; Bajic, V.B. Progress and challenges in bioinformatics approaches for enhancer identification. Brief. Bioinform. 2015, 17, 967–979. [Google Scholar] [CrossRef] [PubMed]
- Shlyueva, D.; Stampfel, G.; Stark, A. Transcriptional enhancers: From properties to genome-wide predictions. Nat. Rev. Genet. 2014, 15, 272. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Notani, D.; Rosenfeld, M.G. Enhancers as non-coding RNA transcription units: Recent insights and future perspectives. Nat. Rev. Genet. 2016, 17, 207–223. [Google Scholar] [CrossRef]
- Bulger, M.; Groudine, M. Functional and mechanistic diversity of distal transcription enhancers. Cell 2011, 144, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Kellis, M. ChromHMM: Automating chromatin-state discovery and characterization. Nat. Methods 2012, 9, 215. [Google Scholar] [CrossRef]
- Hoffman, M.M.; Buske, O.J.; Wang, J.; Weng, Z.; Bilmes, J.A.; Noble, W.S. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat. Methods 2012, 9, 473. [Google Scholar] [CrossRef] [PubMed]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Iny Stein, T.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef]
- Sur, I.K.; Hallikas, O.; Vähärautio, A.; Yan, J.; Turunen, M.; Enge, M.; Taipale, M.; Karhu, A.; Aaltonen, L.A.; Taipale, J. Mice lacking a Myc enhancer that includes human SNP rs6983267 are resistant to intestinal tumors. Science 2012, 338, 1360–1363. [Google Scholar] [CrossRef]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Diao, Y.; Fang, R.; Li, B.; Meng, Z.; Yu, J.; Qiu, Y.; Lin, K.C.; Huang, H.; Liu, T.; Marina, R.J.; et al. A tiling-deletion-based genetic screen for cis-regulatory element identification in mammalian cells. Nat. Methods 2017, 14, 629. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; McKeown, M.R.; Lin, C.Y.; Monti, S.; Roemer, M.G.; Qi, J.; Rahl, P.B.; Sun, H.H.; Yeda, K.T.; Doench, J.G.; et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 2013, 24, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Visel, A.; Minovitsky, S.; Dubchak, I.; Pennacchio, L.A. VISTA Enhancer Browser—A database of tissue-specific human enhancers. Nucleic Acids Res. 2007, 35, D88–D92. [Google Scholar] [CrossRef] [PubMed]
- Visel, A.; Blow, M.J.; Li, Z.; Zhang, T.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; Chen, F.; et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 2009, 457, 854. [Google Scholar] [CrossRef]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Van Calcar, S.; Qu, C.; Ching, K.A.; et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311. [Google Scholar] [CrossRef]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W.; et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108. [Google Scholar] [CrossRef]
- Melnikov, A.; Murugan, A.; Zhang, X.; Tesileanu, T.; Wang, L.; Rogov, P.; Feizi, S.; Gnirke, A.; Callan, C.G., Jr.; Kinney, J.B.; et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat. Biotechnol. 2012, 30, 271–277. [Google Scholar] [CrossRef]
- Kwasnieski, J.C.; Fiore, C.; Chaudhari, H.G.; Cohen, B.A. High-throughput functional testing of ENCODE segmentation predictions. Genome Res. 2014, 24, 1595–1602. [Google Scholar] [CrossRef]
- Shen, S.Q.; Myers, C.A.; Hughes, A.E.; Byrne, L.C.; Flannery, J.G.; Corbo, J.C. Massively parallel cis-regulatory analysis in the mammalian central nervous system. Genome Res. 2015, 26, 238. [Google Scholar] [CrossRef]
- Arnold, C.D.; Gerlach, D.; Stelzer, C.; Boryń, Ł.M.; Rath, M.; Stark, A. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 2013, 339, 1074–1077. [Google Scholar] [CrossRef] [PubMed]
- Andersson, R.; Gebhard, C.; Miguel-Escalada, I.; Hoof, I.; Bornholdt, J.; Boyd, M.; Chen, Y.; Zhao, X.; Schmidl, C.; Suzuki, T.; et al. An atlas of active enhancers across human cell types and tissues. Nature 2014, 507, 455. [Google Scholar] [CrossRef]
- Kim, T.K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S.; et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010, 465, 182. [Google Scholar] [CrossRef] [PubMed]
- Lai, F.; Gardini, A.; Zhang, A.; Shiekhattar, R. Integrator mediates the biogenesis of enhancer RNAs. Nature 2015, 525, 399. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, G.; Lopes, R.; Ugalde, A.P.; Nevedomskaya, E.; Han, R.; Myacheva, K.; Zwart, W.; Elkon, R.; Agami, R. Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 192. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Cuna, J.O.; Arnold, C.D.; Stampfel, G.; Boryń, L.M.; Gerlach, D.; Rath, M.; Stark, A. Dissection of thousands of cell type-specific enhancers identifies dinucleotide repeat motifs as general enhancer features. Genome Res. 2014, 24, 1147–1156. [Google Scholar] [CrossRef]
- Kvon, E.Z.; Stampfel, G.; Yáñez-Cuna, J.O.; Dickson, B.J.; Stark, A. HOT regions function as patterned developmental enhancers and have a distinct cis-regulatory signature. Genes Dev. 2012, 26, 908–913. [Google Scholar] [CrossRef]
- Catarino, R.R.; Stark, A. Assessing sufficiency and necessity of enhancer activities for gene expression and the mechanisms of transcription activation. Genes Dev. 2018, 32, 202–223. [Google Scholar] [CrossRef]
- Yáñez-Cuna, J.O.; Kvon, E.Z.; Stark, A. Deciphering the transcriptional cis-regulatory code. Trends Genet. 2013, 29, 11–22. [Google Scholar] [CrossRef]
- Lee, D.; Karchin, R.; Beer, M.A. Discriminative prediction of mammalian enhancers from DNA sequence. Genome Res. 2011, 21, 2167–2180. [Google Scholar] [CrossRef]
- Kleftogiannis, D.; Kalnis, P.; Bajic, V.B. DEEP: A general computational framework for predicting enhancers. Nucleic Acids Res. 2014, 43, e6. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Fang, L.; Long, R.; Lan, X.; Chou, K.-C. iEnhancer-2L: A two-layer predictor for identifying enhancers and their strength by pseudo k-tuple nucleotide composition. Bioinformatics 2015, 32, 362–369. [Google Scholar] [CrossRef]
- Yang, B.; Liu, F.; Ren, C.; Ouyang, Z.; Xie, Z.; Bo, X.; Shu, W. BiRen: Predicting enhancers with a deep-learning-based model using the DNA sequence alone. Bioinformatics 2017, 33, 1930–1936. [Google Scholar] [CrossRef]
- Beer, M.A. Predicting enhancer activity and variant impact using gkm-SVM. Hum. Mutat. 2017, 38, 1251–1258. [Google Scholar] [CrossRef]
- Lothaire, M. Applied Combinatorics on Words; Cambridge University Press: Cambridge, UK, 2005; Volume 105. [Google Scholar]
- Koslicki, D. Topological entropy of DNA sequences. Bioinformatics 2011, 27, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Tan, R.; Jiang, Q.; Xu, L.; Peng, J.; Wang, Y.; Wang, Y. A generalized topological entropy for analyzing the complexity of DNA sequences. PLoS ONE 2014, 9, e88519. [Google Scholar] [CrossRef] [PubMed]
- FANTOM Consortium and the RIKEN PMI and CLST (DGT); Forrest, A.R.; Kawaji, H.; Rehli, M.; Baillie, J.K.; de Hoon, M.J.; Haberle, V.; Lassmann, T.; Kulakovskiy, I.V.; Lizio, M.; Itoh, M.; et al. A promoter-level mammalian expression atlas. Nature 2014, 507, 462–470. [Google Scholar] [PubMed]
- Erwin, G.D.; Oksenberg, N.; Truty, R.M.; Kostka, D.; Murphy, K.K.; Ahituv, N.; Pollard, K.S.; Capra, J.A. Integrating diverse datasets improves developmental enhancer prediction. PLoS Comput. Biol. 2014, 10, e1003677. [Google Scholar] [CrossRef]
- Richomme, G.; Saari, K.; Zamboni, L.Q. Abelian complexity of minimal subshifts. J. Lond. Math. Soc. 2010, 83, 79–95. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef]
- Rajagopal, N.; Xie, W.; Li, Y.; Wagner, U.; Wang, W.; Stamatoyannopoulos, J.; Ernst, J.; Kellis, M.; Ren, B. RFECS: A random-forest based algorithm for enhancer identification from chromatin state. PLoS Comput. Biol. 2013, 9, e1002968. [Google Scholar] [CrossRef]
- He, Y.; Gorkin, D.U.; Dickel, D.E.; Nery, J.R.; Castanon, R.G.; Lee, A.Y.; Shen, Y.; Visel, A.; Pennacchio, L.A.; Ren, B.; et al. Improved regulatory element prediction based on tissue-specific local epigenomic signatures. Proc. Natl. Acad. Sci. USA 2017, 114, E1633–E1640. [Google Scholar] [CrossRef]
- Wang, M.; Tai, C.; Wei, L. DeFine: Deep convolutional neural networks accurately quantify intensities of transcription factor-DNA binding and facilitate evaluation of functional non-coding variants. Nucleic Acids Res. 2018, 46, e69. [Google Scholar] [CrossRef]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Colosimo, A.; De Luca, A. Special factors in biological strings. J. Theor. Biol. 2000, 204, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Kirillova, O.V. Entropy concepts and DNA investigations. Phys. Lett. A 2000, 274, 247–253. [Google Scholar] [CrossRef]
- Troyanskaya, O.G.; Arbell, O.; Koren, Y.; Landau, G.M.; Bolshoy, A. Sequence complexity profiles of prokaryotic genomic sequences: A fast algorithm for calculating linguistic complexity. Bioinformatics 2002, 18, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Yao, S.; Li, X.; Chen, C.; Hu, X. Genome-Wide Prediction of DNA Methylation Using DNA Composition and Sequence Complexity in Human. Int. J. Mol. Sci. 2017, 18, 420. [Google Scholar] [CrossRef] [PubMed]
- Allouche, J.-P.; Shallit, J. Automatic Sequences: Theory, Applications, Generalizations; Cambridge University Press: Cambridge, UK, 2003. [Google Scholar]
- Zabidi, M.A.; Arnold, C.D.; Schernhuber, K.; Pagani, M.; Rath, M.; Frank, O.; Stark, A. Enhancer-core-promoter specificity separates developmental and housekeeping gene regulation. Nature 2014, 518, 556–559. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Dimension | ACC | AUC | MCC | Sens | Spec | GM |
|---|---|---|---|---|---|---|---|
| SACF484 | 484 | 0.910 | 0.877 | 0.299 | 0.914 | 0.554 | 0.432 |
| SACF209 | 209 | 0.947 | 0.933 | 0.579 | 0.981 | 0.674 | 0.715 |
| TNF | 256 | 0.931 | 0.912 | 0.491 | 0.959 | 0.631 | 0.639 |
| HACF | 465 | 0.956 | 0.955 | 0.627 | 0.989 | 0.701 | 0.755 |
| Comparison Targets | Method | ACC | AUC | Sens | Spec | GM | |
|---|---|---|---|---|---|---|---|
| EnhancerFinder [40] | EnhancerFinder | \ | 0.960 | \ | \ | \ | |
| HACF | 0.915 | 0.962 | 0.904 | 0.928 | 0.916 | ||
| DEEP [29] | Heart | DEEP | 0.822 | \ | 0.802 | 0.824 | 0.812 |
| HACF | 0.941 | 0.919 | 0.964 | 0.690 | 0.734 | ||
| Liver | DEEP | 0.745 | \ | 0.740 | 0.755 | 0.741 | |
| HACF | 0.923 | 0.918 | 0.935 | 0.664 | 0.691 | ||
| Brain | DEEP | 0.853 | \ | 0.832 | 0.855 | 0.843 | |
| HACF | 0.940 | 0.918 | 0.960 | 0.698 | 0.744 | ||
| iEnhancer-2L [32] | Layer I | iEnhancer-2L | 0.769 | 0.850 | 0.781 | 0.759 | \ |
| HACF | 0.837 | 0.907 | 0.846 | 0.828 | 0.838 | ||
| Layer II | iEnhancer-2L | 0.619 | 0.660 | 0.622 | 0.618 | \ | |
| HACF | 0.714 | 0.789 | 0.713 | 0.716 | 0.715 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.; Chen, J.; Liu, Y.; Hu, X. Improved Prediction of Regulatory Element Using Hybrid Abelian Complexity Features with DNA Sequences. Int. J. Mol. Sci. 2019, 20, 1704. https://doi.org/10.3390/ijms20071704
Wu C, Chen J, Liu Y, Hu X. Improved Prediction of Regulatory Element Using Hybrid Abelian Complexity Features with DNA Sequences. International Journal of Molecular Sciences. 2019; 20(7):1704. https://doi.org/10.3390/ijms20071704
Chicago/Turabian StyleWu, Chengchao, Jin Chen, Yunxia Liu, and Xuehai Hu. 2019. "Improved Prediction of Regulatory Element Using Hybrid Abelian Complexity Features with DNA Sequences" International Journal of Molecular Sciences 20, no. 7: 1704. https://doi.org/10.3390/ijms20071704