Structure-Based Virtual Screening and In Vitro Evaluation of New Trypanosoma cruzi Cruzain Inhibitors

 , , , , ,
, , , , , 
Abstract
:
1. Introduction
2. Results and Discussion
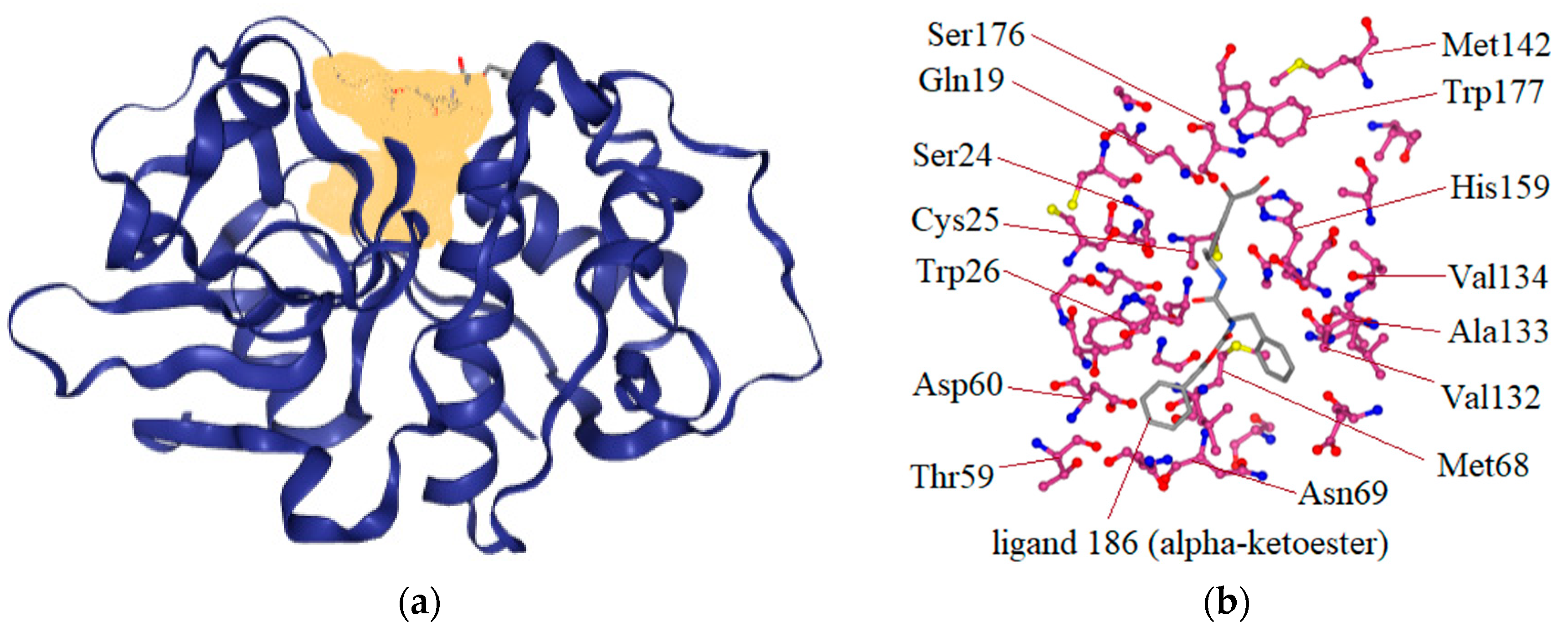
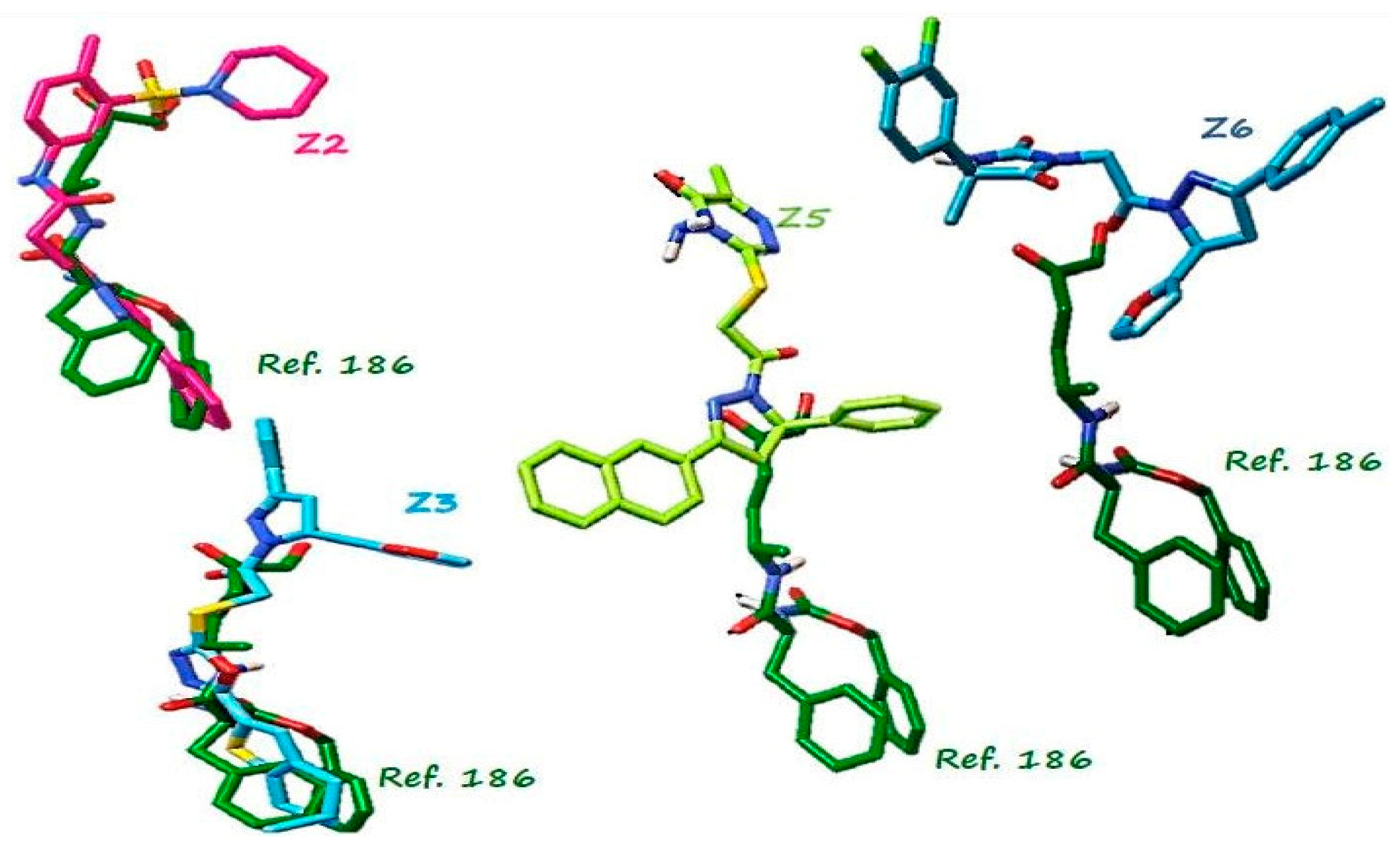
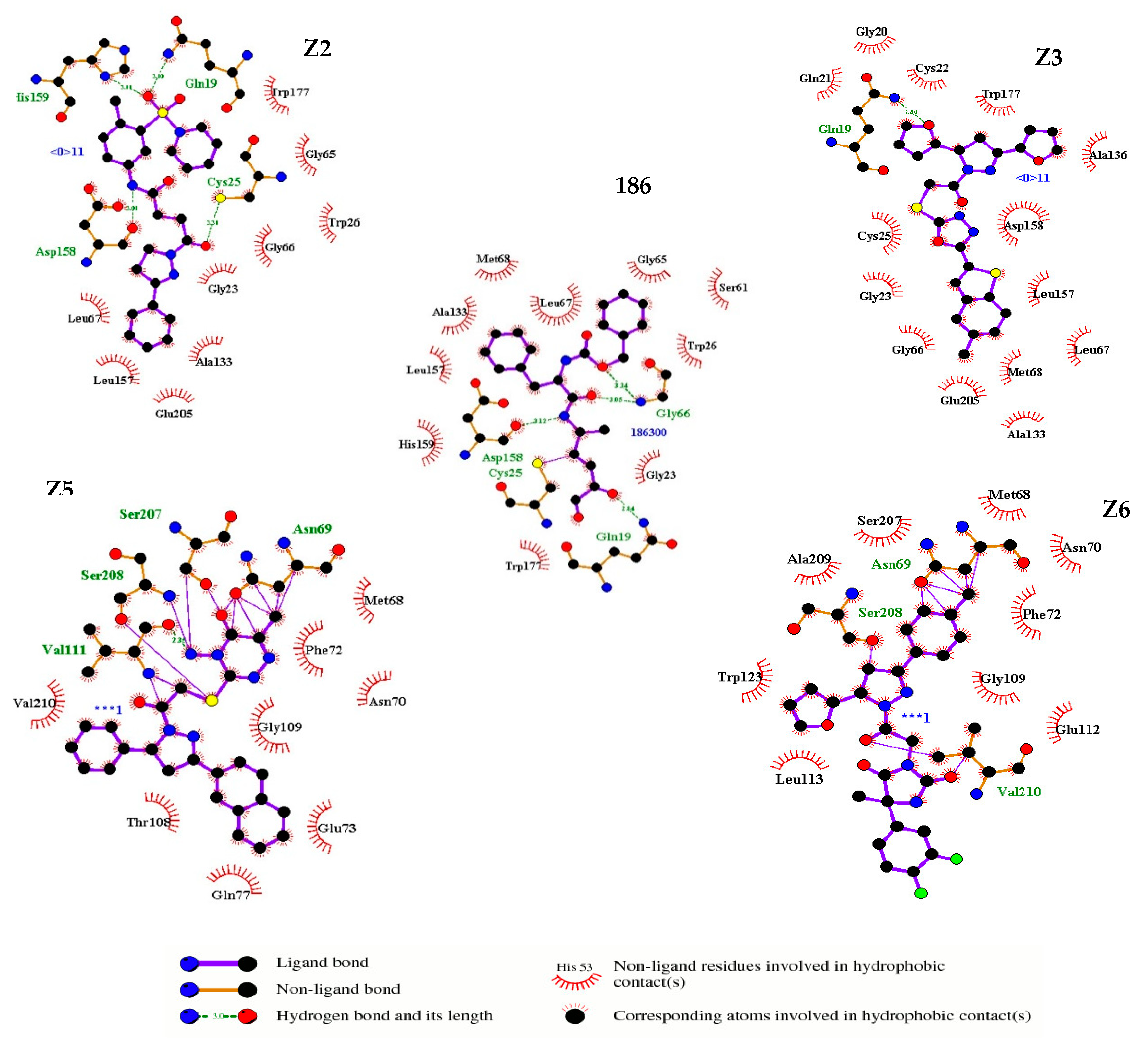
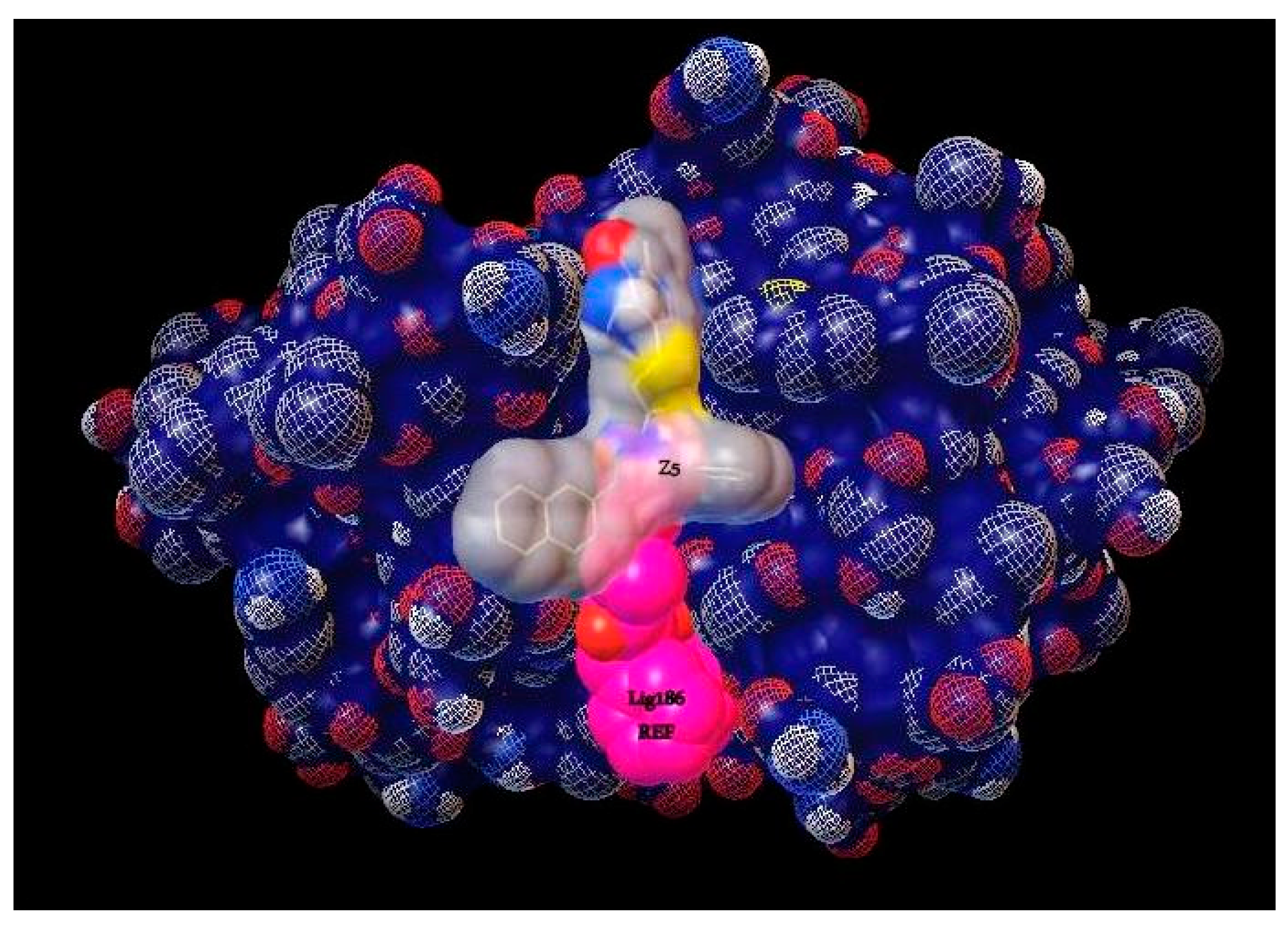
2.1. Virtual Screening
2.2. In Vitro Evaluation
2.3. Enzyme Inhibition
3. Materials and Methods
3.1. Structure-Based Virtual Screening
3.2. In Vitro Evaluation
3.3. Enzyme Inhibition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Nfx | Nifurtimox |
| Bzn | Benznidazole |
| Cz | Cruzain |
| CD | Chagas disease |
| IC50 | Inhibitory concentration |
| LC50 | Lytic concentration |
References
- Bern, C.; Kjos, S.; Yabsley, M.J.; Montgomery, S.P. Trypanosoma cruzi and Chagas’ Disease in the United States. Clin. Microbiol Rev. 2011, 24, 655–681. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). The Global Burden of Disease: 2004 Update; World Health Organization; TDR: Geneva, Switzerland, 2008; p. 160. [Google Scholar]
- World Health Organization (WHO). Chagas Disease (American Trypanosomiasis). 2016. Available online: https://www.who.int/chagas/en/ (accessed on 11 August 2018).
- Zaldini, H.M.; Montenegro, R.M.; Lima, L.A.C.; Oliveira, C.M.V.; Magalhaes, M.D.R.; Brondani, D.J.; Simone, C.A.; Campos, R.L.; Assis, S.M.; Alves, P.V.R.; et al. Studies toward the structural optimization of novel thiazolylhydrazone-based potent antitrypanosomal agents. Bioorg. Med. Chem. 2010, 18, 7826–7835. [Google Scholar] [CrossRef]
- Nichi, S.A.R.; Kimoto, K.Y.; Steinde, M.; Grisard, E.C.; Gomes, M.L. Limit of detection of PCR/RFLP analysis of cytochrome oxidase II for the identification of genetic groups of Trypanosoma cruzi and Trypanosoma rangeli in biological material from vertebrate hosts. Parasitol. Res. 2018, 117, 2403–2410. [Google Scholar] [CrossRef]
- Maguire, J.H. Trypanosoma. In Infectious Diseases, 2nd ed.; Gorbach, S., Bartlett, J., Blacklow, N., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2004; pp. 2327–2334. [Google Scholar]
- Villar, J.C.; Marin, N.J.A.; Ebrahim, S.; Yusuf, S. Trypanocidal drugs for chronic asymptomatic Trypanosoma cruzi infection. Cochrane Database Syst. Rev. 2002, 1, 1–22. [Google Scholar] [CrossRef]
- Siles, R.; Chen, E.C.; Zhou, M.; Pinney, K.G.; Trawick, M.L. Design, synthesis, and biochemical evaluation of novel cruzain inhibitors with potential application in the treatment of Chagas’ disease. Bioorg Med. Chem. Lett. 2006, 16, 4405–4409. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, C.; Zulantay, I.; Apt, W.; Ortiz, S.; Schijman, A.G.; Bisio, M.; Ferrada, V.; Herrera, C.; Martínez, G.; Solarib, A. Evaluation of nifurtimox treatment of chronic Chagas disease by means of several parasitological methods. Antimicrob. Agents Chemother. 2013, 57, 4518–4523. [Google Scholar] [CrossRef]
- Sajid, M.; McKerrow, J.H. Cysteine proteases of parasitic organisms. Mol. Biochem. Parasitol. 2002, 120, 1–21. [Google Scholar] [CrossRef]
- Choe, Y.; Brinen, L.S.; Price, M.S.; Engel, J.C.; Lange, M.; Grisostomi, C.; Weston, S.G.; Pallai, P.V.; Cheng, H.; Hardy, L.W.; et al. Development of a-keto-based inhibitors of cruzain, a cysteine protease implicated in Chagas disease. Bioorg Med. Chem. 2005, 13, 2141–2156. [Google Scholar] [CrossRef]
- Delcroix, M.; Sajid, M.; Caffrey, C.R.; Lim, K.C.; Dvorak, J.; Hsieh, I.; Bahgat, M.; Dissous, C.; McKerrow, J.H. A multienzyme network functions in intestinal protein digestion by a platyhelminth parasite. J. Biol. Chem. 2006, 281, 39316–39329. [Google Scholar] [CrossRef]
- McKerrow, J.H.; Rosenthal, P.J.; Swenerton, R.; Doyle, P. Development of protease inhibitors for protozoan infections. Curr. Opin. Infect. Dis. 2008, 21, 668–672. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Hansell, E.; Engel, J.C.; Caffrey, C.R.; Cohen, F.E.; McKerrow, J.H. Aryl ureas represent a new class of anti-trypanosomal agents. Chem. Biol. 2000, 7, 733–742. [Google Scholar] [CrossRef] [Green Version]
- Roush, W.R.; Alvarez, H.A.; McKerrow, J.H.; Selzer, P.M.; Hansell, E.; Engel, J.C. Design, Synthesis and Evaluation of d-Homophenylalanyl Epoxysuccinate Inhibitors of the Trypanosomal Cysteine Protease Cruzain. Tetrahedron 2000, 56, 9747–9762. [Google Scholar] [CrossRef]
- Zanatta, N.; Madruga, C.C.; Marisco, P.C.; Da Rosa, L.S.; Fernandes, L.D.S.; Flores, D.C.; Martins, M.A.P. Synthesis and structural study of a new series of 2-methylsulfanyl-tetrahydropyrimidines from β-alkoxyvinyl trihalomethyl ketones. J. Heterocyclic Chem. 2008, 45, 221–227. [Google Scholar] [CrossRef]
- Borchhardt, D.M.; Mascarello, A.; Chiaradia, L.D.; Nunes, R.J.; Oliva, G.; Yunes, R.A.; Andricopulo, A.D. Biochemical evaluation of a series of synthetic chalcone and hydrazide derivatives as novel inhibitors of cruzain from Trypanosoma cruzi. J. Braz. Chem. Soc. 2010, 21, 142–150. [Google Scholar] [CrossRef] [Green Version]
- Beaulieu, C.; Isabel, E.; Fortier, A.; Massé, F.; Mellon, C.; Méthot, N.; Black, W.C. Identification of potent and reversible cruzipain inhibitors for the treatment of Chagas disease. Bioorg Med. Chem. Lett. 2010, 20, 7444–7449. [Google Scholar] [CrossRef]
- Massarico, S.R.A.; Gonçalves, J.E.; Pereira, S.F.; de Melo, L.A.P.; Storpirtis, S.; Krogh, R.; Defini, A.A.; Dias, L.C.; Igne, F.E. Design, synthesis and biological evaluation of hybrid bioisoster derivatives of N-acylhydrazone and furoxan groups with potential and selective anti-Trypanosoma cruzi activity. Eur. J. Med. Chem. 2014, 82, 418–425. [Google Scholar] [CrossRef]
- Romeiro, N.C.; Aguirre, G.; Hernández, P.; González, M.; Cerecetto, H.; Aldana, I.; Pérez, S.S.; Monge, A.; Barreiro, E.J.; Lima, L.M. Synthesis, trypanocidal activity and docking studies of novel quinoxaline-N-acylhydrazones, designed as cruzain inhibitors candidates. Bioorg Med. Chem. 2009, 17, 641–652. [Google Scholar] [CrossRef]
- Moreira, M.C.; Wang, Y.; Heringer, W.S.; Wessel, N.; Walther, T. Prognostic value of natriuretic peptides in Chagas’ disease: A head-to-head comparison of the 3 natriuretic peptides. Congest Heart Fail. 2009, 15, 75–81. [Google Scholar] [CrossRef]
- Oprea, T.I.; Davis, A.M.; Teague, S.J.; Leeson, P.D. Is there a difference between leads and drugs? A historical perspective. J. Chem. Inf. Comput. Sci. 2001, 41, 1308–1315. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Ferreira, R.S.; Simeonov, A.; Jadhav, A.; Eidam, O.; Mott, B.T.; Keiser, M.J.; McKerrow, J.H.; Maloney, D.J.; Irwin, J.J.; Shoichet, B.K. Complementarity between a docking and a high-throughput screen in discovering new cruzain inhibitors. J. Med. Chem. 2010, 53, 4891–4905. [Google Scholar] [CrossRef]
- Fereidoonnezhad, M.; Mostoufi, A.; Zali, S.; Eskandari, M.; Afshar, D.; Aliyan, F. Molecular Docking and PLIF Studies of Novel Tacrine-Naphtoquinone Hybrids Based on Multi-Target-Directed Ligand Approach for Alzheimer’s Disease. Jundishapur. J. Nat. Pharm. Prod. 2018, 13, 1–10. [Google Scholar] [CrossRef]
- Lara-Ramírez, E.E.; López-Cedillo, J.C.; Nogueda-Torres, B.; Kashif, M.; Garcia-Perez, C.; Bocanegra-García, V.; Agusti, R.; Uhrig, M.L.; Rivera, G. An in vitro and in vivo evaluation of new potential trans-sialidase inhibitors of Trypanosoma cruzi predicted by a computational drug repositioning method. Eur. J. Med. Chem. 2017, 26, 249–261. [Google Scholar] [CrossRef]
- Mahler, G.; Moreira, L.L. Cisteína endopeptidasa de T. cruzi: Cruzipaína. In Enfermedad de Chagas: Estrategias En La Búsqueda de Nuevos Medicamentos, Una Visión Iberoamericana, 1st ed.; Cerecetto, M.H., González, M., Eds.; Laboratorio Silanes: Mexico City, México, 2012; pp. 45–63. [Google Scholar]
- Villarreal, D.; Barnabé, C.; Sereno, D.; Tibayrenc, M. Lack of correlation between in vitro susceptibility to Benznidazole and phylogenetic diversity of Trypanosoma cruzi, the agent of Chagas disease. Exp. Parasitol. 2004, 108, 24–31. [Google Scholar] [CrossRef]
- Toledo, M.J.O.; Bahia, M.T.; Veloso, V.M.; Carneiro, C.M.; Machado-Coelho, G.L.L.; Alves, M.H.R.; Cruz, R.E.; Tafuri, W.L.; Lana, M. Effects of specific treatment on parasitological and histopathological parameters in mice infected with different Trypanosoma cruzi clonal genotypes. J. Antimicrob. Chemother. 2004, 53, 1045–1053. [Google Scholar] [CrossRef]
- Moreno, M.; D’ávila, D.A.; Silva, M.N.; Galvão, L.M.; Macedo, A.M.; Chiari, E.; Gontijo, E.D.; Zingales, B. Trypanosoma cruzi benznidazole susceptibility in vitro does not predict the therapeutic outcome of human Chagas disease. Mem. Inst. Oswaldo. Cruz. 2010, 105, 918–924. [Google Scholar] [CrossRef]
- Muñoz-Calderón, A.; Santaniello, A.; Pereira, A.; Yannuzzi, J.; Díaz-Bello, Z.; Alarcón de Noya, B. Susceptibilidad in vitro a Nifurtimox y Benznidazol de aislados de Trypanosoma cruzi obtenidos de pacientes venezolanos con enfermedad de Chagas infectados por mecanismos de transmisión oral y vectorial. Rev. Ibero-Latinoam. Parasitol. 2012, 71, 14–22. [Google Scholar]
- Boyle, N.O.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 1–14. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Volkamer, A.; Kuhn, D.; Grombacher, T.; Rippmann, F.; Rarey, M. Combining global and local measures for structure-based druggability predictions. J. Chem. Inf. Model. 2012, 52, 360–372. [Google Scholar] [CrossRef]
- Mantsyzov, A.B.; Bouvier, G.; Evrard-Todeschi, N.; Bertho, G. Contact-based ligand-clustering approach for the identification of active compounds in virtual screening. Adv. Appl. Bioinform. Chem. 2012, 5, 61–79. [Google Scholar] [CrossRef] [Green Version]
- Stöver, B.C.; Müller, K.F. TreeGraph 2: Combining and visualizing evidence from different phylogenetic analyses. BMC Bioinform. 2010, 11, 1–9. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand-Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Paveto, C.; Güida, M.C.; Esteva, M.I.; Martino, V.; Coussio, J.; Flawiá, M.M.; Torres, H.N. Anti-Trypanosoma cruzi Activity of Green Tea (Camellia sinensis) Catechins. Antimicrob. Agents Chemother. 2004, 48, 69–74. [Google Scholar] [CrossRef]
- Brener, Z. Biology of Trypanosoma cruzi. Annu. Rev. Microbiol. 1973, 27, 347–382. [Google Scholar] [CrossRef]
- Pizzi, T. Inmunología de la enfermedad de Chagas. Estado actual del problema. Boletín de la Oficina Sanitaria Panamericana 1961, 51, 450–464. [Google Scholar]
- Santos, C.C.; Sant’Anna, C.; Terres, A.; Cunha-e-Silva, N.L.; Scharfstein, J.; Lima, A.P.C.D.A. Chagasin, the endogenous cysteine-protease inhibitor of Trypanosoma cruzi, modulates parasite differentiation and invasion of mammalian cells. J. Cell Sci. 2005, 118, 901–915. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
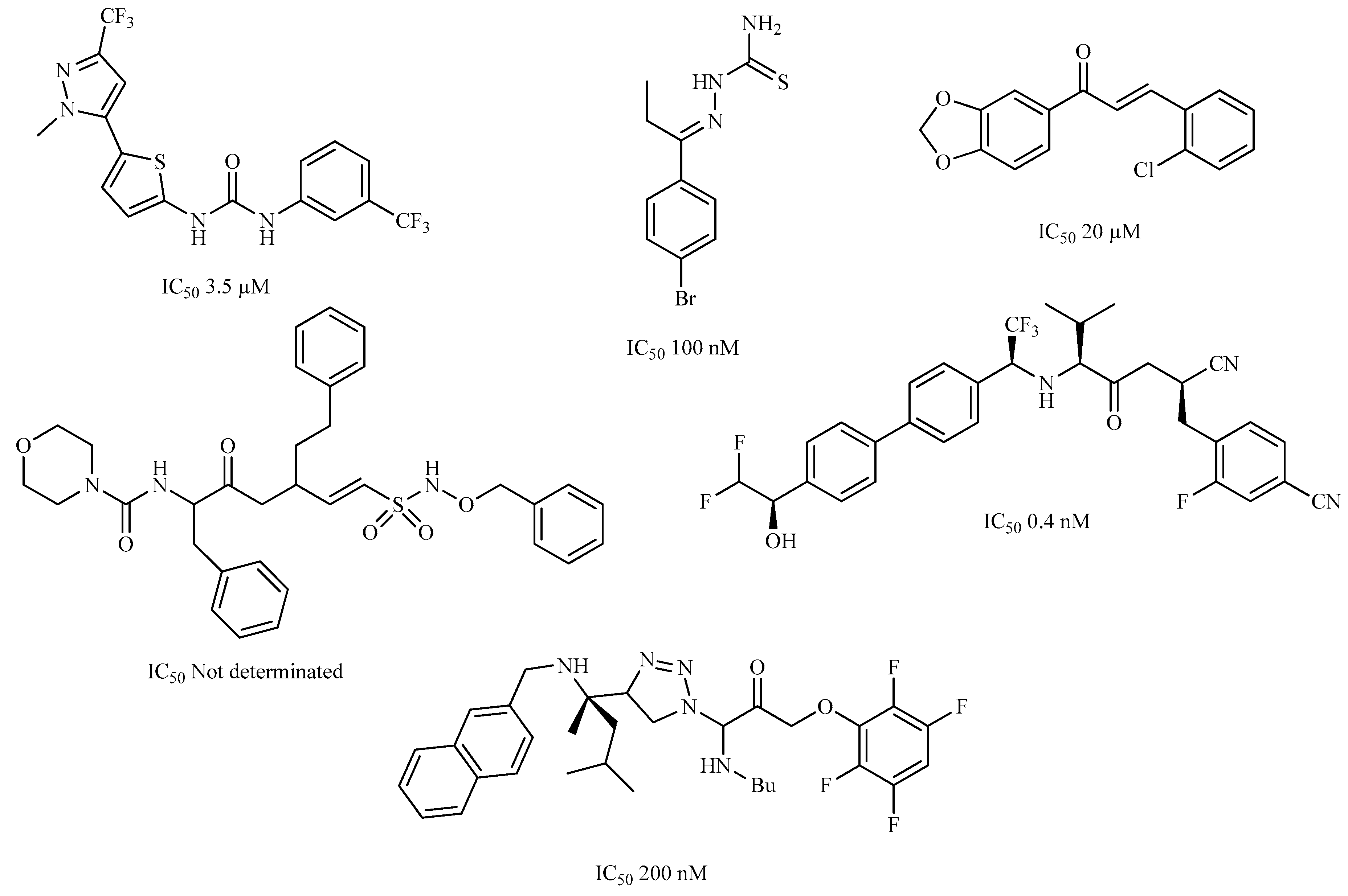
| Compound | Structure | Binding Energy (kcal/mol) | Activity Reported |
|---|---|---|---|
| Z1 ZINC129684040 |  | −8.6 | None |
| Z2 ZINC9873043 | 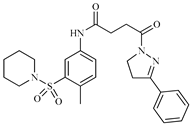 | −8.3 | Inhibitor of cannabinoid receptor type 2 |
| Z3 ZINC9870651 |  | −8.2 | None |
| Z4 ZINC9835465 | 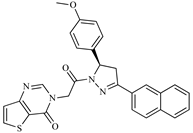 | −8.2 | Amine oxidase inhibitor |
| Z5 ZINC9715287 | 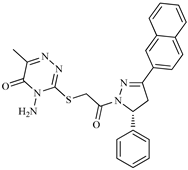 | −8.1 | Amine oxidase inhibitor |
| Z6 ZINC9861447 | 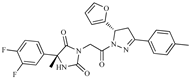 | −8.1 | Amine oxidase inhibitor |
| Z7 ZINC9873693 | 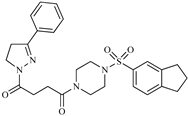 | −8.1 | Inhibitor of aldo keto reductases |
| Z8 ZINC14741665 |  | −8.0 | Inhibitor of cathepsin S |
| Z9 ZINC60293658 | 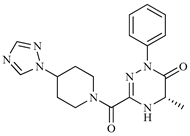 | −8.0 | None |
| Z10 ZINC9867137 | 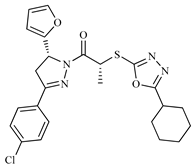 | −8.0 | Amine oxidase inhibitor |
| 186 | 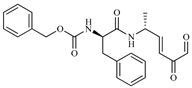 | −6.6 | Cz inhibitor |
| Compound | Epimastigotes | Bloodstream Trypomastigotes | |
|---|---|---|---|
| LC50 (µM) T. cruzi INC-5 | LC50 (µM) | ||
| T. cruzi NINOA | T. cruzi INC-5 | ||
| Z2 | 239.40 ± 9.3 | >250 | >250 |
| Z3 | >250 | >250 | >250 |
| Z5 | 36.26 ± 9.9 | 166.21 ± 14.5 | 185.1 ± 8.5 |
| Z6 | >250 | >250 | >250 |
| S1 | >250 | >250 | >250 |
| Nfx | 38.36 ± 5.2 | 99.41± 11.1 | 117.16 ± 16.36 |
| Bnz | 9.64 ± 4.2 | 183.1 ± 16.2 | 225.40 ± 26.5 |
| Compound | IC50 (µM) |
|---|---|
| Z2 | 1410.05 |
| Z3 | 84.37 |
| Z5 | 56.23 |
| Z6 | 50.35 |
| SI | >200 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrera-Mayorga, V.; Lara-Ramírez, E.E.; Chacón-Vargas, K.F.; Aguirre-Alvarado, C.; Rodríguez-Páez, L.; Alcántara-Farfán, V.; Cordero-Martínez, J.; Nogueda-Torres, B.; Reyes-Espinosa, F.; Bocanegra-García, V.; et al. Structure-Based Virtual Screening and In Vitro Evaluation of New Trypanosoma cruzi Cruzain Inhibitors. Int. J. Mol. Sci. 2019, 20, 1742. https://doi.org/10.3390/ijms20071742
Herrera-Mayorga V, Lara-Ramírez EE, Chacón-Vargas KF, Aguirre-Alvarado C, Rodríguez-Páez L, Alcántara-Farfán V, Cordero-Martínez J, Nogueda-Torres B, Reyes-Espinosa F, Bocanegra-García V, et al. Structure-Based Virtual Screening and In Vitro Evaluation of New Trypanosoma cruzi Cruzain Inhibitors. International Journal of Molecular Sciences. 2019; 20(7):1742. https://doi.org/10.3390/ijms20071742
Chicago/Turabian StyleHerrera-Mayorga, Verónica, Edgar E. Lara-Ramírez, Karla F. Chacón-Vargas, Charmina Aguirre-Alvarado, Lorena Rodríguez-Páez, Verónica Alcántara-Farfán, Joaquín Cordero-Martínez, Benjamín Nogueda-Torres, Francisco Reyes-Espinosa, Virgilio Bocanegra-García, and et al. 2019. "Structure-Based Virtual Screening and In Vitro Evaluation of New Trypanosoma cruzi Cruzain Inhibitors" International Journal of Molecular Sciences 20, no. 7: 1742. https://doi.org/10.3390/ijms20071742