Abstract
Reproductive isolation is an important component of species differentiation. The plastid accD gene coding for the acetyl-CoA carboxylase subunit and the nuclear bccp gene coding for the biotin carboxyl carrier protein were identified as candidate genes governing nuclear-cytoplasmic incompatibility in peas. We examined the allelic diversity in a set of 195 geographically diverse samples of both cultivated (Pisum sativum, P. abyssinicum) and wild (P. fulvum and P. elatius) peas. Based on deduced protein sequences, we identified 34 accD and 31 bccp alleles that are partially geographically and genetically structured. The accD is highly variable due to insertions of tandem repeats. P. fulvum and P. abyssinicum have unique alleles and combinations of both genes. On the other hand, partial overlap was observed between P. sativum and P. elatius. Mapping of protein sequence polymorphisms to 3D structures revealed that most of the repeat and indel polymorphisms map to sequence regions that could not be modeled, consistent with this part of the protein being less constrained by requirements for precise folding than the enzymatically active domains. The results of this study are important not only from an evolutionary point of view but are also relevant for pea breeding when using more distant wild relatives.
1. Introduction
Reproductive isolation is an important component of species differentiation. Mechanisms that create reproductive barriers between once-conspecific organisms have long been a focus of evolutionary biology [1]. Although geographical separation plays a vital role in speciation [2], ecological factors also contribute [3]. Ecological selection favoring a particular cytoplasm has been described from various taxa [4,5]. Hybrid incompatibility due to the genetic divergence between the hybridizing parents has been theorized already by Bateson [6], Dobzhansky [7], and Muller [8]. Hybrid incompatibilities are proposed to be among the first genetic barriers to arise during speciation [9]. Although interspecific hybridization seems to be relatively frequent in plants, comparatively less is known about the reproductive barriers within species [10]. The most classical definition of the species relies on reproductive isolation, namely the inability to produce a viable offspring from inter-species hybridization [11,12]. Reproductive barriers might be broadly classified into prezygotic (pre-pollination) and postzygotic (post-pollination) ones [13]. Pre-pollination isolation mechanisms, such as habitat divergence, temporal isolation, pollinator isolation, and mating system divergence, are usually more effective than post-pollination isolation [2].
Interactions among nuclear-encoded genes can lead to diverse forms of hybrid incompatibility via multiple gametophytic and sporophytic mechanisms [9]. The identification of so-called ‘speciation genes’ is of interest because their knowledge would offer clues to the ecological settings, evolutionary forces, and molecular mechanisms that drive the divergence of populations and species [12,14]. A speciation gene can be strictly defined as a gene that contributes to the splitting of two lineages by reducing the amount of gene flow between them [12].
Until recently, characterization of genetic incompatibility has largely focused on the differences between species and on nuclear incompatibilities [2,12,15,16,17]. As a result, the importance of cytonuclear incompatibility (i.e., incompatibility between the nuclear and organelle genomes) in driving the early stages of speciation received less attention [10]. There has been long co-evolution between the nuclei and organelles. Molecular data indicate a large degree of interdependence between the cellular sub-genomes [18]. The subdivided eukaryotic genome has resulted from a massive restructuring and intermixing of the genomes of the initially free-living symbiotic partner cells with loss, intracellular transfer, and gain of genetic information, with resulting high interdependence and mutual “fine tuning” of both genomes that can easily become disrupted upon intraspecific hybridization. Cytonuclear incompatibilities are predisposed to be substantial contributors to reproductive isolation and speciation [19,20]. Empirical studies have shown that intrinsic postzygotic barriers to reproduction—hybrid inviability and hybrid sterility—evolve through mechanisms consistent with the classic Bateson–Dobzhansky–Muller model [9]. As adaptive or nearly neutral substitutions accumulate in diverging lineages, these may in a particular lineage become fixed in a state incompatible with that in the other lineage. As a result, the hybrid dysfunction occurs when such incompatible alleles are brought together. The genetic basis for hybrid sterility has been studied in several plants, such as Solanum [21], Oryza [22], Mimulus [23], Oenothera [24], Arabidopsis lyrata [25], and A. thaliana [26,27]. There are two classes of cytonuclear hybrid incompatibility: cytoplasmic male sterility (CMS), due to mitochondrial-nuclear mismatch, and cytonuclear chlorosis, caused by plastome–nuclear incompatibilities. Organelle genomes have a reduced population size and lack sexual recombination [28]. These characteristics both increase genetic drift, and lead to potential accumulation of deleterious mutations and selection for compensatory evolution in interacting nuclear genes. Due to these factors, cytonuclear incompatibilities have been proposed to be among the first genetic incompatibilities to arise, influencing the earliest stages of speciation [10,19,20,24,29].
Most known plant sterility loci have been found in the mitochondrial genome, causing CMS characterized by the absence of viable pollen. The genetics of hybrid CMS are remarkably conserved across flowering plants. Molecular genetic studies indicate that CMS typically results from rearrangements in the mitochondrial genome [30,31]. As mitochondria are usually maternally inherited, CMS is typically transmitted through the ovules. In contrast, nuclear genes are transmitted through both ovules and pollen. This difference in inheritance patterns creates a genetic conflict between nuclear and cytoplasmic genes. Hybrid nucleo-organelle dysfunction can result in post-zygotic hybridization barriers that usually manifest as differences in the offspring of reciprocal crosses owing to non-Mendelian inheritance of organelles. Asymmetry in reproductive isolation appears to be common and taxonomically widespread among plant species. Plastids can also contribute to nucleo-cytoplasmic incompatibility. Although cytonuclear chlorosis or albinism of hybrids is not as common as CMS, these have been widely observed, and their implications for speciation were recognized early on [32,33,34,35]. The role of plastids in speciation processes is known from species with a biparental mode of plastid inheritance, e.g., Geranium, Pelargonium and Medicago [36], and mainly from genus Oenothera, which became one of the models for studying plant evolution [24]. Various incompatible phenotypes have also been reported from Rhododendron, Hypericum, Trifolium, Zantedeschia, and Pisum [24]. Cyto-nuclear co-adaptation has been described in Arabidopsis thaliana [18] and demonstrated to affect its adaptive traits [37]. Interestingly, crop domestication may also increase the likelihood that genes causing incompatibility become fixed in the population through genetic hitchhiking [38].
The plastid accD gene coding for the acetyl-CoA carboxylase beta subunit and the nuclear gene bccp coding for the biotin carboxyl carrier protein of acetyl-CoA carboxylase were nominated as candidate genes responsible for nuclear-cytoplasmic incompatibility in peas based on data from crosses between wild and domesticated pea forms [39]. Incompatible hybrids exhibit chlorophyll deficiency, reduction of leaf size low pollen fertility, low seed set, and poorly developed roots [40]. The acetyl-CoA carboxylase (ACCase) complex is involved in the biosynthesis of fatty acids, which takes place in the plastids [40]. ACCase belongs to a group of biotin dependent carboxylases, catalyzing acetyl-coenzyme A carboxylation to malonyl coenzyme A and providing the only entry point for all carbon atoms in the fatty acid synthesis pathway [41]. Uniquely in Eukaryota, plants have two distinct ACCases: one eukaryotic-like homomeric multidomain ACCase in the cytosol and a bacterial-like heteromeric ACCase within the plastids [41]. The heteromeric form of ACCase is found in prokaryotes and the plastids of Viridiplantae. Presumably, all genes encoding ACCase subunits initially resided in the plastid genome after the original endosymbiotic event in algae and underwent sequential transfer to the nuclear genome [42]. Plastid ACCase participates in fatty acid synthesis, whereas the cytosolic enzyme is engaged in the synthesis of very long chain fatty acids, phytoalexins, flavonoids, and anthocyanins. Plastid-localized ACCD enzyme is responsible for catalyzing the initial tightly-regulated and rate-limiting step in fatty acid biosynthesis. Nuclear encoded Biotin Carboxyl Carrier Protein (BCCP) is a part of the enzyme Acetyl-CoA carboxylase complex and serves as a carrier protein for biotin and carboxybiotin throughout the ATP-dependent carboxylation of acetyl-CoA to form malonyl-CoA. The resulting Acetyl-CoA carboxylase is a heterohexamer composed of the biotin carboxyl carrier protein, biotin carboxylase, and two subunits each of the ACCase subunit alpha and the ACCase plastid-coded subunit beta [40].
The plastid ACCase of legumes (Papilionoideae) consists of four subunits, each coded by a separate gene: biotin carboxylase (accC), biotin carboxyl carrier protein (accB=bccp), alpha-carboxyltransferase (accA), and beta-carboxyltransferase (accD). The genes coding accC, accB, and accA are localized in the nuclear genome, whereas the accD gene is localized in the plastid genome [42]. Multiple independent lineages have experienced accelerated rates of substitution in similar subsets of non-photosynthetic genes, including accD (in legumes [43,44,45] and in Oleaceae [46]). In Silene (Caryophyllaceae) species with accelerated plastid genome evolution, the nuclear-encoded subunits of the ACCase complexes are also evolving rapidly, indicating a strong positive selection [47]. Such patterns of molecular evolution in these plastid–nuclear complexes are unusual for ancient conserved enzymes but resemble cases of antagonistic coevolution between pathogens and host immune genes. Genetic characterization of hybrid necrosis in crosses between tomato species [48] and between Arabidopsis ecotypes [49,50] has revealed that incompatibilities among complementary disease resistance genes might play such a role in the evolution of hybrid inviability [51].
In this work, we explored the allelic diversity of accD/bccp in the geographically diverse set of wild pea (Pisum sp.). The accD/bccp are recently identified genes underlying nuclear-cytoplasmic incompatibility in Pisum sp. [39]. We sought to map the allelic combinations of accD/bccp occurring in nature to determine geographic patterns in their distribution, and to identify possible relationships to pea genetic diversity.
2. Results
2.1. Structure and Variation of accD Gene
The accD gene is located between positions 70,882 and 72,654 in the P. sativum cv. Feltham First (HM029370) reference chloroplast genome, resulting in a 1772 bp DNA encoding a protein of 432 amino acid residues. The primers used in our study were designed to match the most conserved region and were located close to the ends of the accD coding sequence. Consequently, we did not capture the very 5′ and 3′ end of the coding sequence due to quality trimming. The beginning and end of the accD sequence, comprising 48 nt from the start codon and 58 nt from the stop codon, consequently missing the first 16 and last 19 codons, were thus excluded from the subsequent analysis.
The length of the accD gene within our studied material ranged from 1403 bp to 1859 bp at DNA level and from 467 to 619 amino acid residues, respectively (GenBank accession numbers MK619486—MK619678). In the studied set of 195 accessions, there was extraordinary variation in the gene length, due to the occurrence of 13 indels whose length varied between 3 and 167 nucleotides. This variation is due to insertions consisting of tandem repeats of 10-150 bp units present in 1 to 37 nearly identical copies, all in the same (i.e., direct) orientation relative to each other (Figure 1). The repetitive sequences can be divided into 6 categories. In the shortest 1403 bp allele (JI1010, P. fulvum) there are four, three, and one repeats of 9 to 12 bp long. These expand in the longest 1859 bp allele (JI267, P. elatius), which has 37 repeats of 10 to 33 bp, 1 repeat of 57 bp, 1 repeat of 102 bp, and 1 repeat of 149 bp. We identified the main five longest tandem repeats blocks, which consist of two or three individual blocks of different lengths and degrees of identity. These blocks are not identical and contain many nucleotide changes and triplet duplications. Such repeats were identified by the presence of small, almost identical blocks, that are part of larger tandem repeats. The first tandem repeat block is the most complex and most degenerate, consisting of three sequential blocks (highlighted in yellow in Figure 1, Figure S1). These blocks are of different lengths and are degenerate to varying degrees from each other. The most similar are first two blocks (89%), which differ by 3 amino acids and by the insertion L-I-L-I for a total of 64 amino acid residues. Characteristic for this tandem repeat is the presence of multiple duplications of three amino acids D-T-N alone or together with D-I-S. The complex, degenerate, and mixed tandem repeat is also the penultimate (3 and 4 grey blocks). This tandem repeat has multiple duplications of five amino acid stretch of S-E-E-E-K. The remaining repeats consist of two blocks separated from each other by 7 or 9 amino acids (Figure 1).
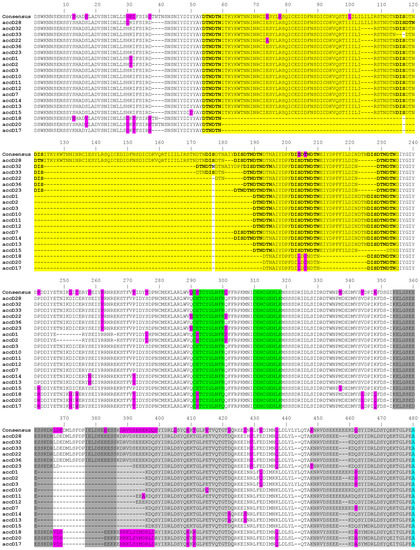
Figure 1.
The alignment of amino acid sequences of all identified accD alleles. The figure only shows the region from 1 to 480 amino acid residues. The colored regions show the 5 translated repeats, polymorphic amino acid exchanges (in magenta), Zn-finger (boxed), acetyl-CoA binding (light blue), coA carboxylation catalytic (dark blue), and carboxybiotin binding (in green) sites. Residues in purple are point mutations in at least one haplotype. There are no indels after position 480 (for full see Figure S1).
2.2. Variation in Nuclear bccp Gene
The predicted ORF of the bccp gene encoding the biotin carboxyl carrier protein of P. sativum cv. Cameor from the pea RNA atlas is 873 bp long and encodes a protein of 290 amino acids. In the pea RNA atlas, this is represented by the ubiquitously expressed PsCam051640 transcript, which corresponds to Tayeh et al. (2015) map PsCam051640 at LGIII. The genomic DNA extracted from the shotgun genome sequence is 5906 bp, with 9 exons interspersed by 8 introns (Exon 1 is 234 bp, exon 2 is 206 bp, exon 3 is 76 bp, exon 4 is 54 bp, exon 5 is 262 bp, exon 6 is 62 bp, exon 7 is 69 bp, exon 8 is 46, and exon 9 is 265 bp). The respective introns are 1170, 541, 263, 874, 111, 856, 84, and 733 bp. The following analysis was conducted on cDNA, avoiding introns. The detected polymorphism, thus, only concerns the coding sequence, and is correspondingly lower than that expected for the complete locus. Notably, to obtain sufficient PCR product we had to perform out two consecutive nested PCR amplifications. This likely reflects the relatively low expression level of the gene in young leaf tissue. There were altogether 39 variable positions and no indels in a total of 195 studied accessions (NCBI accession numbers MK644626—MK644819). These identified 31 protein bccp variants (Table S1). Sixteen analyzed P. fulvum accessions had three bccp alleles (bccp1/2/3) separated by 4 to 10 amino acid changes from the nearest P. elatius alleles. From domesticated P. sativum landraces (60 acc.), 16 had the bccp_22, and six had the bccp_18 allele. From the independently domesticated Ethiopian pea P. abyssinicum (24 acc.), 19 had the specific bccp_26 allele, shared with two P. elatius accessions (PI343978, PI343979 from Turkey), four had the bccp_20 allele, separated by one or two amino acid exchanges from nearest P. elatius. Ninety-five analyzed P. elatius accessions had the largest diversity (all together 28 distinct bccp alleles, Table S1).
2.3. Network and Maximum Parsimony Analyses
Various approaches in the visualization of the data through networks and maximum parsimony (MP) analysis produced a very similar view, with only minimal differences. For further interpretation of clustering of identified alleles into larger groups, the consensus maximum parsimony tree method was used. This produced a very similar clustering of alleles as inspected networks (Median network, NeighborNet, SplitDecomposition networks; not shown). The MP analysis found 18 equally parsimonious trees for the accD gene (length 73 steps) and 19 for the bccp (42 steps) (Figure 2, Figure 3). The resulting trees contained several polytomies. This is because of a large part of the total sequence variability being due to indels in the case of accD, and this information was not included into the MP analysis. In addition, a number of homoplasious mutations were also excluded, with the resulting trees contained several polytomies. However, as we were not interested in the assessment of the gene phylogeny, we did not try to interpret these polytomies. Produced clades (with a rather high bootstrap support) were very similar to groups inferred from the network analyses. Based on the similarity in the grouping of alleles between inspected networks and the MP analysis, the groups of alleles were inferred from the consensus MP tree for both investigated genes. For the accD gene 10 groups (A–J) were inferred; 15 groups were inferred for the bccp gene (A–O) were inferred (Figure 2, Figure 3, Table S1). The accD gene group D (comprising alleles accD_13 and accD_14) was specific for P. abyssinicum, except for one sample of P. sativum from Montenegro (accession n° PI357292), which also possessed the accD_14 allele. Group F (comprising alleles accD_17/18/19/20/21) was specific for P. fulvum. Accessions of P. elatius and landraces of P. sativum were represented by multiple alleles belonging to different groups.
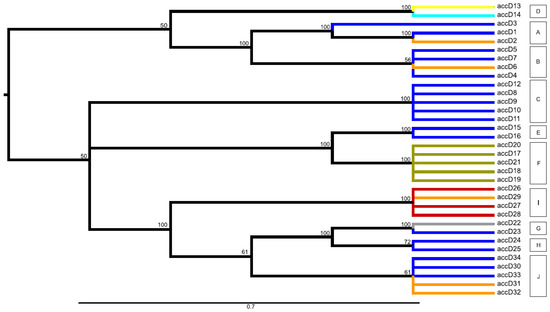
Figure 2.
Midpoint-rooted consensus tree for the accD gene presenting the most parsimonious relationships among the identified 34 alleles within the studied world-wide pea collection. The consensus tree is build up from the 18 equally parsimonious trees (length 73, consistency index 0.900; retention index 0.972; composite index 0.892). Branch coloring follows the species presence of particular alleles: olive green = alleles observed only within P. fulvum; grey = alleles shared among P. fulvum and P. elatius; orange = alleles shared among P. sativum and P. elatius; red = alleles observed only for P. sativum; turquoise = alleles shared among P. abyssinicum and P. sativum; yellow = alleles observed only within P. abyssinicum; blue = alleles observed only within P. elatius. Bootstrap support ≥ 50 is shown above branches.
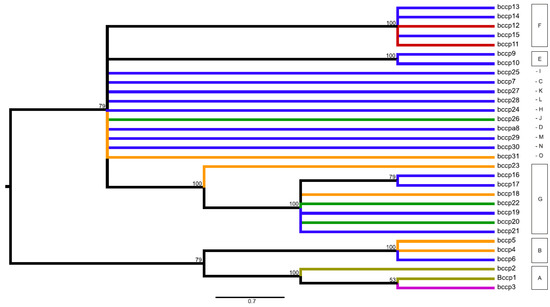
Figure 3.
Midpoint-rooted consensus tree for the bccp gene presenting parsimonious relationships among the identified 31 alleles within the studied world-wide pea collection. The consensus tree built from the 19 equally parsimonious trees (length 42, consistency index 0.762; retention index 0.900; composite index 0.793). Branch coloring follows species presence of particular alleles: olive green = alleles observed only within P. fulvum; magenta = alleles shared among P. fulvum and P. sativum; orange = alleles shared among P. sativum and P. elatius; red = alleles observed only for P. sativum; green = alleles shared among P. abyssinicum, P. sativum and P. elatius; blue = alleles observed only within P. elatius. Bootstrap support ≥ 50 is shown above branches.
In the case of the bccp gene, P. abyssinicum was represented by groups J (allele bccp_26) and G (alleles bccp_20/22). However, in contrast to the accD gene, inferred alleles were not specific for P. abyssinicum, but were also found within P. elatius and samples of P. sativum (Figure 3, Table S1). The three identified alleles observed for P. fulvum (bccp_1/2/3) clustered together and represented group A. Two of these alleles were specific (bccp_1/2) for P. fulvum and one (bccp_3) was shared with two samples of P. sativum from Greece (JI1525 and JI2573). The identified alleles for the investigated accessions of P. elatius fall within 12 groups and for P. sativum within six groups, which were shared between these two species (Table S1).
2.4. Frequency of Amino Acid Substitutions and Their Distribution
Analysis of the nuclear encoded bccp gene in a panel of 179 samples of 809 sites resulting in 269 analyzed codons revealed 196 synonymous sites (Pi(s): 0,00616 Pi(s), Jukes & Cantor: 0,00620) and 610 non-synonymous sites (Pi(a): 0,00553, Pi(a), Jukes & Cantor: 0,00556). This resulted in a Ka/Ks ratio of 0.895. Despite the presence of frequent insertions and deletions, the accD sequence could be translated into protein. The analysis covered 1306 sites (e.g., 425 codons). Nucleotide diversity analysis of accD showed 278 synonymous sites (Pi(s): 0,00390, Pi(s), Jukes & Cantor: 0,00391) and 997 non-synonymous sites (Pi(a): 0,01048, Pi(a), Jukes & Cantor: 0,01064). This resulted in a high Ka/Ks ratio of 2.726, which indicates positive selections and accelerated evolutions.
Analysis of protein sequence revealed that the ACCD protein has a ClpP protease/crotonase domain (IPRO 29045; region of 251 to 296 and 384 to 584 amino acids), coiled coil domain (region of 380 to 407 amino acids), an acetyl-CoA-carboxyltransferase N terminal domain (IPRO 11762; in region of 226 to 590 amino acids), and a zinc finger (230–252 amino acids) domain (Figure 1). The BCCP protein has a biotin/lipoyl attachment (IPRO 000089) domain (region of 207 to 280 amino acids) and a carboxytransferase (CT) interaction site (239G-284F-249G-250A-257D), where 249G is a conserved biotinylation site.
We next attempted to investigate the location of the individual amino acid substitutions, and the conspicuous indels found in accD. This was performed with respect to the 3D folding of both ACCD and BCCP proteins, to the extent that we were able to predict their spatial structure by threading on experimentally characterized related templates. We could produce only partial models for both proteins (File S1, S2 For ACCD, the model covered approximately 43% of the sequence, corresponding to the C-terminal portion of the protein). The N-terminal region and an additional loop within the modelled segment were disordered in the prediction. For the BCCP protein, approximately 45% of the sequence was covered by the best templates but only two short separate fragments from this domain could be reliably modeled; the rest of the molecule was disordered in the prediction (Figure 4, Table 1).

Figure 4.
Parts of the ACCD (A) and BCCP (B) protein sequences covered by the molecular model are marked in bold. Residues on gray background were not covered by the population sequence alignment. Residues exhibiting one, two, or more allelic variants are shown on a colored background. Residues shown in red are deleted only in some alleles. Black arrows indicate the location of insertions in some alleles.

Table 1.
Distribution of protein sequence polymorphisms in structurally modelled versus non-modelled parts of the ACCD and BCCP protein sequences.
Remarkably, mapping of the identified protein sequence polymorphisms revealed that most of the above-described repeat and indel polymorphisms in the ACCD sequence map to sequence regions could not be modelled due to the lack of suitable templates and intrinsic disorder. This is consistent with this part of the protein being less constrained by requirements for precise folding than the enzymatically active domain. Point mutations were also somewhat enriched in the part of the ACCD protein that was not modeled. However, no such bias was detected for BCCP (Figure 4, Table 1).
2.5. Allelic accD/bccp Combinations
We found 34 accD and 31 bccp alleles yielding altogether 1054 possible combinations. Within the wild pea (P. elatius) we detected 61 combinations (Table S2). Most of these combinations (45) were found only once. Cultivated P. sativum landraces had 20 combinations; the most frequent were accD_29/bccp_22 (30), followed by accD_29/bccp_18 (8). P. abyssinicum accessions had 4 distinct combinations, with accD_14/bccp_26 being predominant (17). P. fulvum had 9 combinations, accD_21/bccp_1 (4), accD_20/bccp_1 (3), and accD_17/bccp_3 (2). The only exception in our P. fulvum set was JI2539 from Israel, which had accD_22 (accD_G lineage) shared with P. elatius. There were two bccp alleles (bccp_22 and bccp_31) that formed the highest number of combinations with 18 and 10 accD alleles, respectively. Conversely, two accD alleles, accD_29, accD_25, and bccp_22, bccp_31 formed 8, 9, and 19, 10 combinations, respectively. Notably, the most frequent combination found in P. sativum landraces accD_29/bccp_22 was found in these high occurrence alleles (Figure 5).
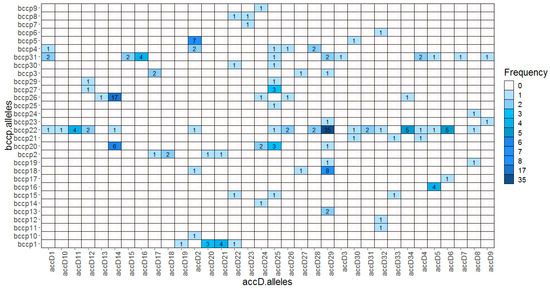
Figure 5.
Heatmap of identified pairwise ACCD/BCCP allelic combinations.
2.6. Relationship to Pisum Genetic Diversity
Having previously analyzed genetic diversity based on genome-wide sampled polymorphism [52,53], we examined the distribution of both accD and bccp alleles within respective genetic groups. Cultivated Pisum sativum accessions can be divided into two (nr. 3 and 6) equally abundant (24 and, 27 accessions, respectively) groups. The independently domesticated Ethiopian pea (P. abyssinicum) forms a separate (nr. 7) group (Table S1). With respect to accD/bccp alleles, accD_29 and bccp_22 alleles predominate in 60 analyzed P. sativum accessions (41, 38 accessions respectively) (Supplementary Table S1), while all 24 P. abyssinicum accessions had single unique accD_14 and bccp_26 (17 acc.), bccp_20 (5 acc.) and bccp_22 (JI1974) alleles corresponding to its separate domestication history and associated bottleneck. P. fulvum as a separate species forms a separate genetic group (nr. 2) and has also distinct and the most distant accD (accD_17-21) and bccp (bccp_1-3) alleles, separated by 39 to 40, and 7 to 8 amino acids, respectively, from the closest P. elatius alleles. On the contrary, wild P. elatius is genetically the most diverse and has seven genetic groups (Trněný et al. 2018), one of which (nr. 3) overlaps with P. sativum. This diversity is also reflected with 22 different accD and 25 bccp alleles, respectively. The most abundant are accD_25 (13 acc.), accD_29 (9 acc.), accD_2 (11 acc.), and bccp_22 (28 acc.), and bccp_31 (16 acc.) (Table S1). There is only a partial relationship between the genome wide DARTseq and accD/bccp based diversity. Genetic group nr. 10 of P. elatius accessions from the Caucasus region has the most distinct accD_30, 31, 34, but not bccp alleles. Similarly, genetic groups nr. 4 and 5 have a high proportion of accD_2 (8 acc.) and accD_15/16 (5 acc.) alleles in samples from Israel or eastern Turkey and Georgia, respectively. No clear genetic group assignment was found for bccp alleles within P. elatius accessions.
2.7. Geographic Distribution of accD/bccp Alleles
Pisum fulvum (16 acc.) is geographically restricted to Israel (7 acc.), Syria (7 acc.), Jordan (1 acc.), and southeastern Turkey (1 acc.), and displays distinct accD/bccp alleles. Genetically and geographically the most diverse set is from P. elatius (96 acc.). Of these, there were 34 accessions from Turkey, which had the highest genetic diversity (Figure 6, Table S1). These accessions have various accD/bccp alleles, although the combination accD_25 and bccp_20 is the most frequent (10). The next large group is P. elatius from Israel, which had 25 accessions that belong to various genetic groups. These also have different accD_2 and bccp_5 (22 alleles occurring in 13 accessions). European samples cover a large region of Western (Spain, Portugal, France), Central (Italy), and Eastern (Greece, Hungary, Serbia) Europe (Table S1). The later samples are distinct by both by genome wide analyses and by accD/bccp alleles analysis. Finally, the most separate group of P. elatius is from Armenia, with unique accD_34 and bccp_21/22 alleles (Figure 6).
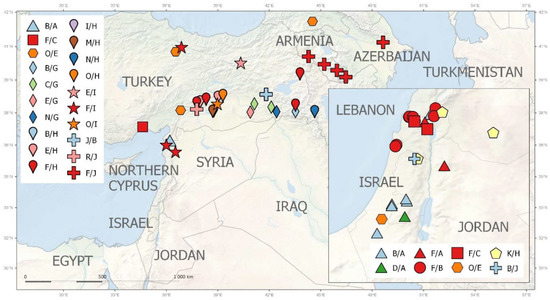
Figure 6.
Geographic distribution of ACCD/BCCP allelic combinations assigned to large groups (for details see Table S1) within the Middle East.
The cultivated pea is geographically less precisely localized, except for P. abyssinicum, which is found only in Ethiopia and Yemen. All P. abyssinicum accessions have accD_14/bccp_20/26 alleles. Landraces of P. sativum originate from 24 countries and span a large geographical area from the Western Mediterranean to Central and Southern Asia. They predominantly have accD_29 (41 acc.) and bccp_22 (36 acc.) alleles typical for cultivated pea. There are few distinct accessions that have different alleles. Two were from Algeria (accD_32/bccp_11/12), and two accessions were from Greece (specific bccp_3 allele). Two accessions from China (ATC6925, ATC6937) have a accD_6 allele shared with P. elatius, while PI560969 from Nepal has distinct accD_2/bccp_5 alleles (Table S1).
3. Discussion
Here we report the allelic composition and geographical distribution of two genes involved in postzygotic reproductive isolation in the pea [39]. Taking advantage of the available germplasm resources [52,53], we analyzed the allelic composition of chloroplast localized accD and nuclear encoded bccp genes. Our results extend the experimental data of Bogdanova et al. [39]. We analyzed the allelic composition of accessions collected from the wild (including all recognized Pisum species) and domesticated peas of various geographical origins.
Postzygotic reproductive isolation, expressed as hybrid sterility or inviability, hybrid weakness or necrosis, and hybrid breakdown, is considered one of the two major fundamental processes leading to speciation [2,9]. The plastome–genome dysfunctions concern various kinds of albinism. Generally, incompatible hybrid materials suffer from reduced pigment content, lower rates of photosynthesis, and an impaired thylakoid structure. We detected the occurrence of albinotic plants in crosses of wild Pisum fulvum or P. elatius with the cultivated pea P. sativum, which upon identification of the respective genes [39] prompted this study.
3.1. Hypervariability of the Chloroplast accD Gene
The region of the chloroplast genome around the accD gene has been found to be prone to accumulation of repeats, resulting in high interspecific variability in numerous species (Pisum and Lathyrus [45], Capsicum [54], Glycine [43], Silene [47], Oenothera [55,56], Cupressophytes [57]) but much less variability at the intraspecific level (Medicago truncatula [44], tea, Camellia sinensis [58], and pea, Pisum sp. [39,40]). Our present study substantially expands the previous reports [39,40] by analyzing 195 pea samples covering the entire geographical and species range [52,59]. Our results on the ratios of nonsynonymous to synonymous substitutions (Ka/Ks) in the pea accD gene agree with data from Oenothera, Silene, and Cupressophytes [47,55,57]. This indicates positive selection, since Ka/Ks values significantly above 1 are unlikely to occur without at least some of the mutations being advantageous. The large variation in plastid-encoded accD gene sequences, both between and within the Pisum species, is consistent with findings in Silene, where positive selection in the phylogenetic context has been detected [47]. In many cases of plastid genome evolution, mutations have disproportionately affected nonsynonymous sites, resulting in elevated ratios of nonsynonymous to synonymous substitution rates. Notably, plastid genome comparison between Lathyrus sativus and Pisum sativum resulted in identification of a region spanning the accD gene with increased mutation rate [45]. Analysis of publicly available accD sequences for Lathyrus and Vicia species supported these findings (unpublished).
Variation detected in the Pisum sp. accD sequence is mainly caused by the insertion of multiple tandem repeated sequences, as found in Cupressophytes [57] and Medicago [44]. In particular, the later study corresponds well to our pea accD data, since each of the 24 studied Medicago truncatula genotypes appears to have a different accD sequence, yet with maintained reading frames despite the high variability. Mapping of the insertion sites onto the predicted protein structure indicated their clustering within the N-terminal part of the ACCD protein that could not be reliably modelled due to intrinsic disorder. Such disordered protein regions are known to be extremely flexible and dynamic, alleviating some structural constraints [60], and were reported to be prone to insertions and deletions [61]. It has been suggested that regions surrounding tandem repeats evolve faster than other non-repeat-containing regions, which results in increased frequency of substitutions near the flanking sequences [62]. As shown in tobacco, a functional accD is essential for development [63]. Interestingly, the relationship to biparental inheritance of plastids was proposed to be related to the plastid competition [56]. Since about 20% of all angiosperms contain plastid DNA in the sperm cell, it is likely that this mechanism of cytonuclear conflict is also present in other systems [64,65,66,67].
3.2. Allelic accD/bccp Combinations Found in Wild and Domesticated Peas
One of our major aims was to detect allelic combinations of both genes occurring in wild peas, as well as in cultivated pea crop. Altogether we found 36 accD and 35 bccp alleles in the set of 195 accessions. Within the wild pea (P. elatius) these occurred in 60 out of 671 possible combinations, indicating a high diversity, while both domesticated P. sativum and P. abyssinicum had only a reduced subset. There was no overlap between P. fulvum and P. elatius, except for one P. fulvum JI2539 accession from Israel, which had accD_22 (G lineage) allele shared with three P. elatius samples from Turkey. Notably, in our previous study [52], we have found in this accession a typical P. elatius trnSG_E6 allele, suggesting some past hybridization event between P. fulvum and P. elatius. Interestingly, in another two P. fulvum accessions (JI2510, JI2521) that also have the trnSG_E6 allele [52], the accD allele was canonical to P. fulvum (accD_20, 21, e.g., F lineage). P. abyssinicum had accD alleles and combinations distinct from P. sativum, supporting its independent domestication [53]. The accD_14 allele of P. abyssinicum was not found in any of P. elatius or P. sativum samples. Notably, two of the most frequent alleles of each gene, accD_29 and bccp_22, contributed to the most frequent combination of accD_30/bccp_25 found in domesticated P. sativum.
It remains to be experimentally tested by crosses if the allelic combinations detected in the natural conditions create barriers against gene flow in natural pea populations. Some experimental crosses between cultivated pea and selected P. fulvum and P. elatius accessions were conducted by Bogdanova et al. [68]. These crosses revealed hybrid sterility, ultimately leading to identification of the respective genes [39]. In our work, we made reciprocal crosses between P. elatius L100 (accD_2/bccp_5) and P. sativum cv. Cameor (accD_29/bccp_22), which resulted in the appearance of albinotic plants (Smýkal, unpublished), while a cross between P. elatius JI64 (accD_30/bccp_5) and P. sativum JI92 (accD_29/bccp_22) was fully viable and fertile [69,70]. This corresponds to the findings of Bogdanova et al. (2015) [39] of a incompatible cross between P. elatius L100 (accD_2/bccp_5) and P. sativum WL12238 (accD_29/bccp_22); a cross between P. elatius JI1794 (accD_25/bccp_27), 721 (accD_5/bccp_22), and P. abyssinicum VIR 2759 (accD_14/bccp_26) were compatible with the cultivated pea P. sativum WL12238 (accD_29/bccp_22) [68]. Moreover, the existence of a second, unlinked, and yet unidentified nuclear scs2 locus also involved in nuclear-cytoplasmic conflict has been proposed [39]. In this study, the authors proposed a model of determinants, based on seven substitutions and three deletions in ACCD and four amino acid substitutions in the biotinyl domain of BCCP protein. The results of our study add to this complexity, as there are far more possible combinations.
3.3. Domestication and Hybrid Incompatibility
In crops, artificial selection and hybridization accelerate the evolutionary process [71]. The majority of economically important crops were isolated from their progenitors through the existence of prezygotic or postzygotic reproductive barriers (or both), even though geographic isolation was absent during the domestication [38]. The reproductive barriers between wild crop progenitors and domesticated crops might be attributed to several mechanisms, including differences in karyotype or chromosomal rearrangements. Such karyotype differences are reported between P. fulvum and P. elatius, P. sativum, and between P. sativum and P. abyssinicum [72,73], and contribute to the partial fertility of the respective hybrids. Much less is known about the interactions between nuclear and cytoplasmic genomes. To date, only a few genes implicated in hybrid incompatibility have been isolated in crops. In maize, Tcb1, Ga1, and Ga2 alleles influence interaction of pollen tubes with silk tissue and confer prezygotic barriers in crosses between cultivated Zea mays and the wild teosinte Z. m. mexicana [74]. About 50 loci controlling postzygotic reproductive barriers between rice subspecies have been identified and molecular products of some genes have been characterized [22]. For example, the S5 locus, a determinant of japonica-indica sterility, is located in proximity to the domestication OsC1 gene [75]. Similarly, the Gn1a gene involved in rice yield formation is linked with S35, which determines pollen sterility of japonica-indica hybrids [76]. Another example was shown in the tomato, where the Cf-2 gene from wild Lycopersicon pimpinellifolium confers resistance to the fungus Cladosporium fulvum in an Rcr3 dependent manner [48]; these two genes interact with each other to induce hybrid necrosis syndrome in the hybrids. Although the occurrence of albino plants in many interspecific crosses in crops is widely documented [77,78], its causes have not been studied in most cases. Notably, crosses between cultivated chickpea (Cicer arietinum) and its progenitor (C. reticulatum) yielded yellow and albino plants and a biparental plastid inheritance [77,78]. We speculate that this was caused by a similar mechanism as in the pea.
The results of this study might be relevant for breeding, particularly using more distant crop wild relatives, as well as hybrid crop breeding [79,80], but it remains to be tested by experimental crosses to identify causal effectors.
4. Material and Methods
4.1. Plant Material
We analyzed 195 previously described pea accessions (Smýkal et al. 2017, 2018, Trněný et al. 2018) [52,53,59], consisting of wild P. elatius (95) and P. fulvum (16) accessions (Table S1). Sixty domesticated P. sativum landraces and 24 domesticated P. abyssinicum accessions were selected to maximize the genetic diversity and to cover the entire range of the wild and landrace pea habitats. This span is approximately 5000 km in longitude from Morocco to Iran, and in latitude from Tunisia to Hungary; altitude ranged from sea level to about 2000 m. This material was previously morphologically described and assessed for its genetic diversity structure [52,53]. Plants were grown in 5 L pots with peat-sand (90:10) substrate mix (Florcom Profi, BB Com Ltd. Letohrad, Czech Republic), in glasshouse conditions (UP campus, Olomouc, Czech Republic).
4.2. DNA and RNA Analysis
Genomic DNA was isolated from a single plant per accession from approximately 100 mg of dry leaf material using the Invisorb Plant Genomic DNA Isolation kit (Invisorb, Berlin, Germany) and standard protocol [52,59]. Total RNA was isolated from young leaves using plant RNA kit (Macherey-Nagel, Düren, Germany). Isolated RNA was treated with DNaseI to remove genomic DNA. The accD gene was amplified directly from genomic DNA using primers (F1—GCATTAGTTTTCATTTTCAGTCC located 27 bp upstream of stop codon, R4—CTTTAATAGGGGTTTAGAATACA, located 94 bp upstream of ATG codon) [39]. We used cDNA as a template to avoid large intron sequences present in the bccp3 gene. One microgram of a total RNA was reversely transcribed with Oligo(dT) primer and AMV reverse transcriptase (Promega, Madison, USA) according to manufacturer´s protocol (Hradilová et al. 2017) [71]. Two step nested PCR amplification was used. After the first PCR (with primers F—CTAATGAAAGTGGCGGAAATC, R—CCTTATTACGCGTCTTAGTGAATG), the product was diluted (1:100) and the second PCR was performed (F33—CCATTCTCTGCACTCCCTTTCGCG, R1113—CAATTATTTCTCAATCTATTCAAAACG), using the conditions as described in Hradilová et al. [71]. PCR products were verified on a 1.5% agarose gel, treated with Exonuclease-Alkaline Phosphatase (Thermo Scientific, Brno, Czech Republic) and sequenced at Macrogene.
4.3. Sequence Analysis
For initial analysis, Geneious 7.1.7 (Biomatters Ltd., Auckland, New Zealand ) was used to edit and align sequences. Due to the presence of large gaps in the accD gene, sequences were translated into protein sequences, which reduced the overall length of the accD nucleotide alignment and partially helped to eliminate large gaps. This procedure reduced the complexity of the accD sequences. Sequences of the bccp gene were treated in the same manner, although these sequences were largely devoid of large indels. The translated protein sequences were aligned in Geneious using the MAFFT algorithm and the final alignment was manually adjusted. From the final alignment, different alleles and their frequencies were identified using the online tool FABOX [81].
To explore possible connections or relationships among the identified alleles, the reduced dataset (including each allele defined only once) was used for the network analysis. Several approaches of network construction were used (based on characters, Median network, Median-joining; based on distances, Neighbor network, Split decomposition) and implemented in SplitsTree [82]. The results were then compared. To compare the results of network analysis with a classically constructed bifurcating tree, a maximum parsimony (MP) tree was built using MEGA 6 with 1000 bootstrap replicates [83]. Because of the complex pattern of gaps within the accD gene, indels were treated as “partially deleted” (pairwise deletion, option implemented in MEGA) during the MP analysis. The final consensus tree was computed from all the equally parsimonious trees found during the analysis and was midpoint rooted. The tree topology was compared against the constructed networks. To simplify or reduce the number of identified alleles, groups of related alleles were inferred based on the constructed networks and the final consensus MP tree for both investigated genes. DnaSP v5.10 was used to determine nucleotide diversity and synonymous/non-synonymous sites ratios [84]. All studied accD and bccp sequences were deposited in the GenBank database under the accession numbers MK619486 to MK619678, and MK644626 to MK644819, respectively.
4.4. Tandem Repeat Analysis
Tandem repeats within DNA and protein sequences were identified in a combination of two algorithms (FastPCR [85] and RADAR [86]). The consensus DNA sequence of accD gene was first scanned by FastPCR at a repeat length ≥20 bp (k-mer = 12 with a tolerance for up to one mismatch within k-mer) with a similarity of above 70%. Potential tandem repeats for consensus protein sequence were further identified by RADAR software. Both methods complemented each other, since the boundaries of some degenerate and mixed tandem repeats were difficult to identify separately.
4.5. Protein Sequence Analysis and Structure Modelling
To identify the domains we used InterPro (www.ebi.ac.uk/interpro) and SMART databases (http://smart.embl-heidelberg.de). To generate molecular models of both proteins, standard sequences of the pea accD (GenBank YP_003587558.1) and bccp (GenBank DR89228.1) were used as queries to identify suitable templates and to perform molecular modelling by threading using Phyre2 in “normal” mode [87]. Only a partial model was generated for each protein, as portions of the sequence predicted to be disordered or lacking a suitable template (including some internal loops) could not be reliably modeled. In the case of ACCD, the structure of Staphylococcus acetyl-CoA carboxylase carboxyltransferase (PDB 2F9I) was identified as the best template. The second best template (PDB 2F9Y, also of bacterial origin) yielded a model of similar coverage and spatial organization. A similar model, also based on the PDB 2F9I template, was obtained for the same part of ACCD using another algorithm, RaptorX [88]. For BCCP, the best template identified by Phyre2 was the pyruvate carboxylase from Methylobacillus flagellatus (PDB 5KS8). The same template was also found by RaptorX as second best; namely, pyruvate carboxylase from Listeria monocytogenes (PDB 4QSH) yielded a spatially similar model. The Phyre2-generated models were subjected to additional refinement in the DeepView environment [89] to eliminate amino acid sidechain clashes. Subsequent evaluation of the resulting models using the WHAT_CHECK tools [90] revealed no critical errors, with scores for some parameters only slightly poorer than observed for the template for both proteins.
4.6. Mapping Protein Sequence Polymorphisms on Predicted Structure
Unique protein sequences encoded by alleles, each of the two loci were identified within aligned protein sequence sets using the ElimDupes tool at the Los Alamos HIV database website (https://www.hiv.lanl.gov/content/sequence/elimdupesv2/elimdupes.html). A map of polymorphisms was then generated manually from the resulting unique sequence alignments. A distribution of the polymorphisms between the modeled and non-modeled portions of the protein was statistically evaluated using the Chi-square test.
Supplementary Materials
The following are available online at https://www.mdpi.com/1422-0067/20/7/1773/s1. Table S1: List and description of analyzed material, Table S2: Table of accD/bccp combinations, Figure S1: The alignment of amino acid sequences of all identified accD alleles, File S1: Theoretical model of pea BCCP protein structure, (co-ordinates in standard PDB format), File S2: Model of BCCP protein structure. Theoretical model of pea ACCD protein structure, (co-ordinates in standard PDB format). File S3: Theoretical model of pea BCCP protein structure, (co-ordinates in standard PDB format).
Author Contributions
Conceptualization, P.S.; methodology, L.M., J.B., F.C., and P.S.; formal analysis, E.N., L.Z., J.B., R.K., V.N., F.C., L.M., and P.H.; resources, P.S.; data curation, P.S.; writing—original draft preparation, P.S., F.C., and L.M.; writing—review and editing, P.S., F.C., and L.M.; funding acquisition, P.S.
Funding
This work received funding from the European Community’s Seventh Framework Programme (FP7/ 2007-2013) under the grant agreement n°FP7-613551, LEGATO project. R.K. was supported by the Science Committee of the Ministry of Education and Science of the Republic of Kazakhstan in the framework of program funding for research (BR05236334 and BR06349586). P.S., L.Z., and E.N. were supported from Grant Agency of Palacký University, IGA 001-2018, IGA 004-2019 projects, F.C. by the Ministry of Education, Youth and Sports of the Czech Republic NPUI LO1417 project.
Acknowledgments
We are thankful to Clarice Coyne, Mike Ambrose, and Bob Redden, genebank curators for stimulating discussion, and sharing their knowledge and selection of pea germplasm.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Coyne, J.A. Genetics and speciation. Nature 1992, 355, 511–515. [Google Scholar] [CrossRef]
- Coyne, J.A.; Orr, H.A. Speciation. Sinauer, Sunderland; Oxford University Press: New York, NY, USA, 2004; pp. 1–545. [Google Scholar]
- Givnish, T.J. Ecology of plant speciation. Taxon 2010, 59, 1326–1366. [Google Scholar] [CrossRef]
- Case, A.L.; Finseth, F.R.; Barr, C.M.; Fishman, L. Selfish evolution of cytonuclear hybrid incompatibility in Mimulus. Proc. Biol. Sci. 2016, 283, 20161493. [Google Scholar] [CrossRef]
- Sambatti, J.B.M.; Ortiz-Barrientos, D.; Baack, E.J.; Rieseberg, L.H. Ecological selection maintains cytonuclear incompatibilities in hybridizing sunflowers. Ecol. Lett. 2008, 11, 1082–1091. [Google Scholar] [CrossRef]
- Bateson, W. Mendel’s Principles of Heredity; Cambridge University Press: Cambridge, UK, 1909. [Google Scholar]
- Dobzhansky, T. Genetics and the Origin of Species. In Columbia Biological Series; Columbia University Press: New York, NY, USA, 1937; Volume 9, pp. 1–364. [Google Scholar]
- Muller, H.J. Isolating mechanisms, evolution, and temperature. Biol. Symp. 1942, 6, 71–125. [Google Scholar]
- Fishman, L.; Sweigart, A.L. When Two Rights Make a Wrong: The Evolutionary Genetics of Plant Hybrid Incompatibilities. Annu. Rev. Plant Biol. 2018, 69, 707–731. [Google Scholar] [CrossRef]
- Barnard-Kubow, K.B.; So, N.; Galloway, L.F. Cytonuclear incompatibility contributes to the early stages of speciation. Evolution 2016, 70, 2752–2766. [Google Scholar] [CrossRef] [PubMed]
- Mayr, E. Systematics and the Origin of Species; Columbia University Press: New York, NY, USA, 1942. [Google Scholar]
- Rieseberg, L.H.; Blackman, B.K. Speciation genes in plants. Ann. Bot. 2010, 106, 439–455. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Lin, H.-X. Evolution and Molecular Control of Hybrid Incompatibility in Plants. Front. Plant Sci. 2016, 7, 1208. [Google Scholar] [CrossRef]
- Orr, H.A.; Masly, J.P.; Presgraves, D.C. Speciation genes. Curr. Opin. Genet. Dev. 2004, 14, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Bomblies, K. Doomed lovers: Mechanisms of isolation and incompatibility in plants. Annu. Rev. Plant Biol. 2010, 61, 109–124. [Google Scholar] [CrossRef]
- Sweigart, A.L.; Willis, J.H. Molecular evolution and genetics of postzygotic reproductive isolation in plants. F1000 Biol. Rep. 2012, 4, 23. [Google Scholar] [CrossRef]
- Baack, E.; Melo, M.C.; Rieseberg, L.H.; Ortiz-Barrientos, D. The origins of reproductive isolation in plants. New Phytol. 2015, 207, 968–984. [Google Scholar] [CrossRef]
- Moison, M.; Roux, F.; Quadrado, M.; Duval, R.; Ekovich, M.; Lê, D.-H.; Verzaux, M.; Budar, F. Cytoplasmic phylogeny and evidence of cyto-nuclear co-adaptation in Arabidopsis thaliana. Plant J. 2010, 63, 728–738. [Google Scholar] [CrossRef]
- Levin, D.A. The cytoplasmic factor in plant speciation. Syst. Bot. 2003, 28, 5–11. [Google Scholar]
- Burton, R.S.; Pereira, R.J.; Barreto, F.S. Cytonuclear Genomic Interactions and Hybrid Breakdown. Annu. Rev. Ecol. Evol. Syst. 2013, 44, 281–302. [Google Scholar] [CrossRef]
- Moyle, L.C.; Nakazato, T. Complex Epistasis for Dobzhansky–Muller Hybrid Incompatibility in Solanum. Genetics 2009, 181, 347–351. [Google Scholar] [CrossRef][Green Version]
- Ouyang, Y.; Liu, Y.-G.; Zhang, Q. Hybrid sterility in plant: Stories from rice. Curr. Opin. Plant Biol. 2010, 13, 186–192. [Google Scholar] [CrossRef]
- Barr, C.M.; Fishman, L. The Nuclear Component of a Cytonuclear Hybrid Incompatibility in Mimulus Maps to a Cluster of Pentatricopeptide Repeat Genes. Genetics 2010, 184, 455–465. [Google Scholar] [CrossRef]
- Greiner, S.; Rauwolf, U.; Meurer, J.; Herrmann, R.G. The role of plastids in plant speciation. Mol. Ecol. 2011, 20, 671–691. [Google Scholar] [CrossRef]
- Leppälä, J.; Savolainen, O. Nuclear-Cytoplasmic Interactions Reduce Male Fertility in Hybrids of Arabidopsis Lyrata Subspecies. Evolution 2011, 65, 2959–2972. [Google Scholar] [CrossRef]
- Törjék, O.; Witucka-Wall, H.; Meyer, R.C.; von Korff, M.; Kusterer, B.; Rautengarten, C.; Altmann, T. Segregation distortion in Arabidopsis C24/Col-0 and Col-0/C24 recombinant inbred line populations is due to reduced fertility caused by epistatic interaction of two loci. Theor. Appl. Genet. 2006, 113, 1551–1561. [Google Scholar] [CrossRef]
- Durand, S.; Bouché, N.; Perez Strand, E.; Loudet, O.; Camilleri, C. Rapid Establishment of Genetic Incompatibility through Natural Epigenetic Variation. Curr. Biol. 2012, 22, 326–331. [Google Scholar] [CrossRef]
- Birky, C.W. The Inheritance of Genes in Mitochondria and Chloroplasts: Laws, Mechanisms, and Models. Annu. Rev. Genet. 2001, 35, 125–148. [Google Scholar] [CrossRef]
- Fishman, L.; Willis, J.H. A cytonuclear incompatibility causes anther sterility in Mimulus hybrids. Evolution 2006, 60, 1372–1381. [Google Scholar] [CrossRef]
- Hanson, M.R.; Bentolila, S. Interactions of mitochondrial and nuclear genes that affect male gametophyte development. Plant Cell 2004, 16 (Suppl. 1), S154–S169. [Google Scholar] [CrossRef]
- Chen, L.; Liu, Y.-G. Male Sterility and Fertility Restoration in Crops. Annu. Rev. Plant Biol. 2014, 65, 579–606. [Google Scholar] [CrossRef]
- Rhoades, M.M. The cytoplasmic inheritance of male sterility in Zea mays. J. Genet. 1931, 27, 71–93. [Google Scholar] [CrossRef]
- Renner, O. Die pflanzlichen Plastiden als selbstandige Elemente der genetischen Konstitution. Ber. Math. Phys. Kl. Sachs. Akad. 1934, 86, 241–266. [Google Scholar]
- Stebbins, G.L. Variation and Evolution in Plants; Columbia University Press: New York, NY, USA, 1950. [Google Scholar]
- Stubbe, W. The role of the plastome in evolution of the genus Oenothera. Genetica 1964, 35, 28–33. [Google Scholar] [CrossRef]
- Crosby, K.; Smith, D.R. Does the mode of plastid inheritance influence plastid genome architecture? PLoS ONE 2012, 7, e46260. [Google Scholar] [CrossRef]
- Roux, F.; Mary-Huard, T.; Barillot, E.; Wenes, E.; Botran, L.; Durand, S.; Villoutreix, R.; Martin-Magniette, M.-L.; Camilleri, C.; Budar, F. Cytonuclear interactions affect adaptive traits of the annual plant Arabidopsis thaliana in the field. Proc. Natl. Acad. Sci. USA 2016, 113, 3687–3692. [Google Scholar] [CrossRef]
- Dempewolf, H.; Hodgins, K.A.; Rummell, S.E.; Ellstrand, N.C.; Rieseberg, L.H. Reproductive isolation during domestication. Plant Cell 2012, 24, 2710–2717. [Google Scholar] [CrossRef]
- Bogdanova, V.S.; Zaytseva, O.O.; Mglinets, A.V.; Shatskaya, N.V.; Kosterin, O.E.; Vasiliev, G.V. Nuclear-cytoplasmic conflict in pea (Pisum sativum L.) is associated with nuclear and plastidic candidate genes encoding acetyl-CoA carboxylase subunits. PLoS ONE 2015, 10, e0119835. [Google Scholar] [CrossRef]
- Sasaki, Y.; Nagano, Y. Plant acetyl-CoA carboxylase: Structure, biosynthesis, regulation, and gene manipulation for plant breeding. Biosci. Biotechnol. Biochem. 2004, 68, 1175–1184. [Google Scholar] [CrossRef]
- Nikolau, B.J.; Ohlrogge, J.B.; Wurtele, E.S. Plant biotin-containing carboxylases. Arch. Biochem. Biophys. 2003, 414, 211–222. [Google Scholar] [CrossRef]
- Szczepaniak, A.; Książkiewicz, M.; Podkowiński, J.; Czyż, K.B.; Figlerowicz, M.; Naganowska, B. Legume Cytosolic and Plastid Acetyl-Coenzyme—A Carboxylase Genes Differ by Evolutionary Patterns and Selection Pressure Schemes Acting before and after Whole-Genome Duplications. Genes 2018, 9, 563. [Google Scholar] [CrossRef]
- Asaf, S.; Khan, A.L.; Aaqil Khan, M.; Muhammad Imran, Q.; Kang, S.-M.; Al-Hosni, K.; Jeong, E.J.; Lee, K.E.; Lee, I.-J. Comparative analysis of complete plastid genomes from wild soybean (Glycine soja) and nine other Glycine species. PLoS ONE 2017, 12, e0182281. [Google Scholar] [CrossRef]
- Gurdon, C.; Maliga, P. Two Distinct Plastid Genome Configurations and Unprecedented Intraspecies Length Variation in the accD Coding Region in Medicago truncatula. DNA Res. 2014, 21, 417–427. [Google Scholar] [CrossRef]
- Magee, A.M.; Aspinall, S.; Rice, D.W.; Cusack, B.P.; Sémon, M.; Perry, A.S.; Stefanović, S.; Milbourne, D.; Barth, S.; Palmer, J.D.; et al. Localized hypermutation and associated gene losses in legume chloroplast genomes. Genome Res. 2010, 20, 1700–1710. [Google Scholar] [CrossRef]
- Ha, Y.-H.; Kim, C.; Choi, K.; Kim, J.-H. Molecular Phylogeny and Dating of Forsythieae (Oleaceae) Provide Insight into the Miocene History of Eurasian Temperate Shrubs. Front. Plant Sci. 2018, 9, 99. [Google Scholar] [CrossRef]
- Rockenbach, K.; Havird, J.C.; Monroe, J.G.; Triant, D.A.; Taylor, D.R.; Sloan, D.B. Positive Selection in Rapidly Evolving Plastid-Nuclear Enzyme Complexes. Genetics 2016, 204, 1507–1522. [Google Scholar] [CrossRef]
- Krüger, J.; Thomas, C.M.; Golstein, C.; Dixon, M.S.; Smoker, M.; Tang, S.; Mulder, L.; Jones, J.D.G. A tomato cysteine protease required for Cf-2-dependent disease resistance and suppression of autonecrosis. Science 2002, 296, 744–747. [Google Scholar] [CrossRef]
- Rooney, H.C.; Van’t Klooster, J.W.; van der Hoorn, R.A.; Joosten, M.H.; Jones, J.D.; de Wit, P.J. Cladosporium Avr2 inhibits tomato Rcr3 protease required for Cf-2-dependent disease resistance. Science 2005, 308, 1783–1786. [Google Scholar] [CrossRef]
- Bomblies, K.; Lempe, J.; Epple, P.; Warthmann, N.; Lanz, C.; Dangl, J.L.; Weigel, D. Autoimmune response as a mechanism for a Dobzhansky-Muller-type incompatibility syndrome in plants. PLoS Biol. 2007, 5, e236. [Google Scholar] [CrossRef]
- Bomblies, K.; Weigel, D. Hybrid necrosis: Autoimmunity as a potential gene-flow barrier in plant species. Nat. Rev. Genet. 2007, 8, 382–393. [Google Scholar] [CrossRef]
- Smýkal, P.; Hradilová, I.; Trněný, O.; Brus, J.; Rathore, A.; Bariotakis, M.; Das, R.R.; Bhattacharyya, D.; Richards, C.; Coyne, C.J.; et al. Genomic diversity and macroecology of the crop wild relatives of domesticated pea. Sci. Rep. 2017, 7, 17384. [Google Scholar] [CrossRef]
- Trněný, O.; Brus, J.; Hradilová, I.; Rathore, A.; Das, R.R.; Kopecký, P.; Coyne, C.J.; Reeves, P.; Richards, C.; Smýkal, P. Molecular Evidence for Two Domestication Events in the Pea Crop. Genes (Basel) 2018, 9, 535. [Google Scholar] [CrossRef]
- D’Agostino, N.; Tamburino, R.; Cantarella, C.; De Carluccio, V.; Sannino, L.; Cozzolino, S.; Cardi, T.; Scotti, N. The Complete Plastome Sequences of Eleven Capsicum Genotypes: Insights into DNA Variation and Molecular Evolution. Genes 2018, 9, 503. [Google Scholar] [CrossRef]
- Greiner, S.; Wang, X.; Herrmann, R.G.; Rauwolf, U.; Mayer, K.; Haberer, G.; Meurer, J. The complete nucleotide sequences of the 5 genetically distinct plastid genomes of Oenothera, subsection Oenothera: II. A microevolutionary view using bioinformatics and formal genetic data. Mol. Biol. Evol. 2008, 25, 2019–2030. [Google Scholar] [CrossRef]
- Sobanski, J.; Giavalisco, P.; Fischer, A.; Kreiner, J.; Walther, D.; Schoettler, M.A.; Pellizzer, T.; Golczyk, H.; Obata, T.; Bock, R.; et al. Chloroplast competition is controlled by lipid biosynthesis in evening primroses. Proc. Natl. Acad. Sci. USA 2019, 116, 5665–5674. [Google Scholar] [CrossRef]
- Li, J.; Su, Y.; Wang, T. The Repeat Sequences and Elevated Substitution Rates of the Chloroplast accD Gene in Cupressophytes. Front. Plant Sci. 2018, 9, 533. [Google Scholar] [CrossRef]
- Ujihara, T.; Hayashi, N.; Kohata, K.; Matsushita, S.; Kitajima, S. Intraspecific Sequence Variation in the rbcL-accD Region of the Chloroplast Genome in Tea (Camellia sinensis). Tea Res. J. 2007, 104, 15–23. [Google Scholar] [CrossRef][Green Version]
- Smýkal, P.; Trněný, O.; Brus, J.; Hanáček, P.; Rathore, A.; Roma, R.D.; Pechanec, V.; Duchoslav, M.; Bhattacharyya, D.; Bariotakis, M.; et al. Genetic structure of wild pea (Pisum sativum subsp. elatius) populations in the northern part of the Fertile Crescent reflects moderate cross-pollination and strong effect of geographic but not environmental distance. PLoS ONE 2018, 13, e0194056. [Google Scholar]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Light, S.; Sagit, R.; Sachenkova, O.; Ekman, D.; Elofsson, A. Protein expansion is primarily due to indels in intrinsically disordered regions. Mol. Biol. Evol. 2013, 30, 2645–2653. [Google Scholar] [CrossRef]
- Simon, M.; Hancock, J.M. Tandem and cryptic amino acid repeats accumulate in disordered regions of proteins. Genome Biol. 2009, 10, R59. [Google Scholar] [CrossRef]
- Kode, V.; Mudd, E.A.; Iamtham, S.; Day, A. The tobacco plastid accD gene is essential and is required for leaf development. Plant J. 2005, 44, 237–244. [Google Scholar] [CrossRef]
- Corriveau, J.L.; Coleman, A.W. Rapid Screening Method to Detect Potential Biparental Inheritance of Plastid DNA and Results for Over 200 Angiosperm Species. Am. J. Bot. 1988, 75, 1443–1458. [Google Scholar] [CrossRef]
- Zhang, Q.; Sodmergen. Why does biparental plastid inheritance revive in angiosperms? J. Plant Res. 2010, 123, 201–206. [Google Scholar] [CrossRef]
- Greiner, S.; Sobanski, J.; Bock, R. Why are most organelle genomes transmitted maternally? Bioessays 2015, 37, 80–94. [Google Scholar] [CrossRef]
- Christie, J.R.; Beekman, M. Uniparental Inheritance Promotes Adaptive Evolution in Cytoplasmic Genomes. Mol. Biol. Evol. 2017, 34, 677–691. [Google Scholar] [CrossRef]
- Bogdanova, V.S.; Kosterin, O.E.; Yadrikhinskiy, A.K. Wild peas vary in their cross-compatibility with cultivated pea (Pisum sativum subsp. sativum L.) depending on alleles of a nuclear-cytoplasmic incompatibility locus. Theor. Appl. Genet. 2014, 127, 1163–1172. [Google Scholar] [CrossRef]
- North, N.; Casey, R.; Domoney, C. Inheritance and mapping of seed lipoxygenase polypeptides in Pisum. Theor. Appl. Genet. 1998, 77, 805–808. [Google Scholar] [CrossRef]
- Hradilová, I.; Trněný, O.; Válková, M.; Cechová, M.; Janská, A.; Prokešová, L.; Aamir, K.; Krezdorn, N.; Rotter, B.; Winter, P.; et al. A Combined Comparative Transcriptomic, Metabolomic, and Anatomical Analyses of Two Key Domestication Traits: Pod Dehiscence and Seed Dormancy in Pea (Pisum sp.). Front. Plant Sci. 2017, 8, 542. [Google Scholar] [CrossRef]
- Meyer, R.S.; Purugganan, M.D. Evolution of crop species: Genetics of domestication and diversification. Nat. Rev. Genet. 2013, 14, 840–852. [Google Scholar] [CrossRef]
- Ben-Ze’Ev, N.; Zohary, D. Species relationships in the genus Pisum L. Isr. J. Bot. 1973, 22, 73–91. [Google Scholar]
- Errico, A.; Conicella, C.; Taliercio, U. Cytological and Morphological Characterization of Pisum sativum and Pisum fulvum Tetraploids. Plant Breed. 1991, 106, 141–148. [Google Scholar] [CrossRef]
- Lu, Y.; Kermicle, J.L.; Evans, M.M.S. Genetic and cellular analysis of cross-incompatibility in Zea mays. Plant Reprod. 2014, 27, 19–29. [Google Scholar] [CrossRef]
- Saitoh, K.; Onishi, K.; Mikami, I.; Thidar, K.; Sano, Y. Allelic diversification at the C (OsC1) locus of wild and cultivated rice: Nucleotide changes associated with phenotypes. Genetics 2004, 168, 997–1007. [Google Scholar] [CrossRef]
- Kubo, T. Genetic mechanisms of postzygotic reproductive isolation: An epistatic network in rice. Breed. Sci. 2013, 63, 359–366. [Google Scholar] [CrossRef]
- Kumari, M.; Clarke, H.J.; Small, I.; Siddique, K.H.M. Albinism in Plants: A Major Bottleneck in Wide Hybridization, Androgenesis and Doubled Haploid Culture. Crit. Rev. Plant Sci. 2009, 28, 393–409. [Google Scholar] [CrossRef]
- Kumari, M.; Clarke, H.J.; des Francs-Small, C.C.; Small, I.; Khan, T.N.; Siddique, K.H.M. Albinism does not correlate with biparental inheritance of plastid DNA in interspecific hybrids in Cicer species. Plant Sci. 2011, 180, 628–633. [Google Scholar] [CrossRef]
- Bohra, A.; Jha, U.C.; Adhimoolam, P. Cytoplasmic male sterility (CMS) in hybrid breeding in field crops. Plant Cell Rep. 2016, 35, 967–993. [Google Scholar] [CrossRef]
- Kim, Y.-J.; Zhang, D. Molecular Control of Male Fertility for Crop Hybrid Breeding. Trends Plant Sci. 2018, 23, 53–65. [Google Scholar] [CrossRef]
- Villesen, P. FaBox: An online toolbox for fasta sequences. Mol. Ecol. Notes 2007, 7, 965–968. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Kalendar, R.; Khassenov, B.; Ramanculov, E.; Samuilova, O.; Ivanov, K.I. FastPCR: An in silico tool for fast primer and probe design and advanced sequence analysis. Genomics 2017, 109, 312–319. [Google Scholar] [CrossRef]
- Heger, A.; Holm, L. Rapid automatic detection and alignment of repeats in protein sequences. Proteins 2000, 41, 224–237. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Källberg, M.; Wang, H.; Wang, S.; Peng, J.; Wang, Z.; Lu, H.; Xu, J. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 2012, 7, 1511–1522. [Google Scholar] [CrossRef]
- Johansson, M.U.; Zoete, V.; Michielin, O.; Guex, N. Defining and searching for structural motifs using DeepView/Swiss-PdbViewer. BMC Bioinform. 2012, 13, 173. [Google Scholar] [CrossRef]
- Hooft, R.W.; Vriend, G.; Sander, C.; Abola, E.E. Errors in protein structures. Nature 1996, 381, 272. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).