New Targeted Agents in Acute Myeloid Leukemia: New Hope on the Rise

Abstract
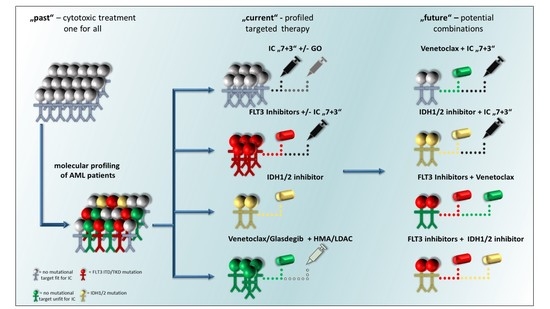
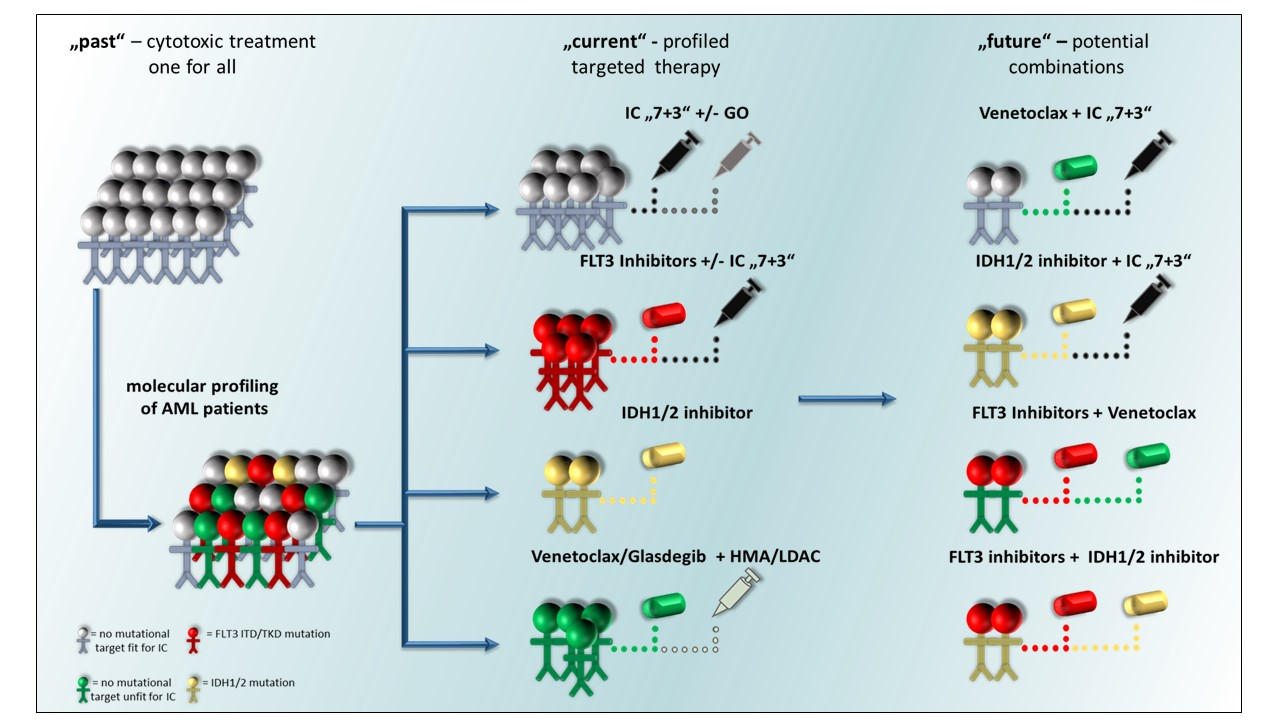
:
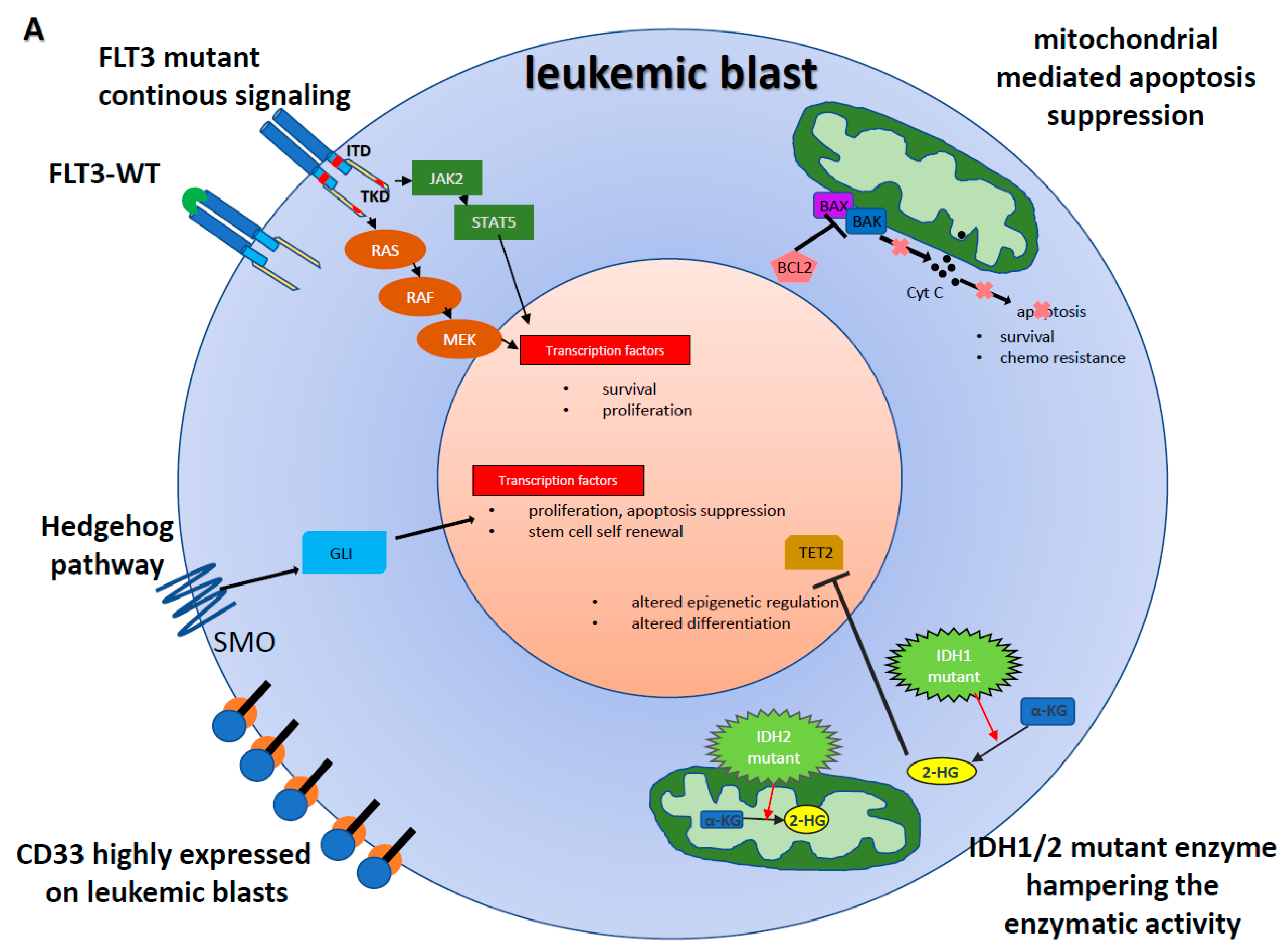
1. Introduction
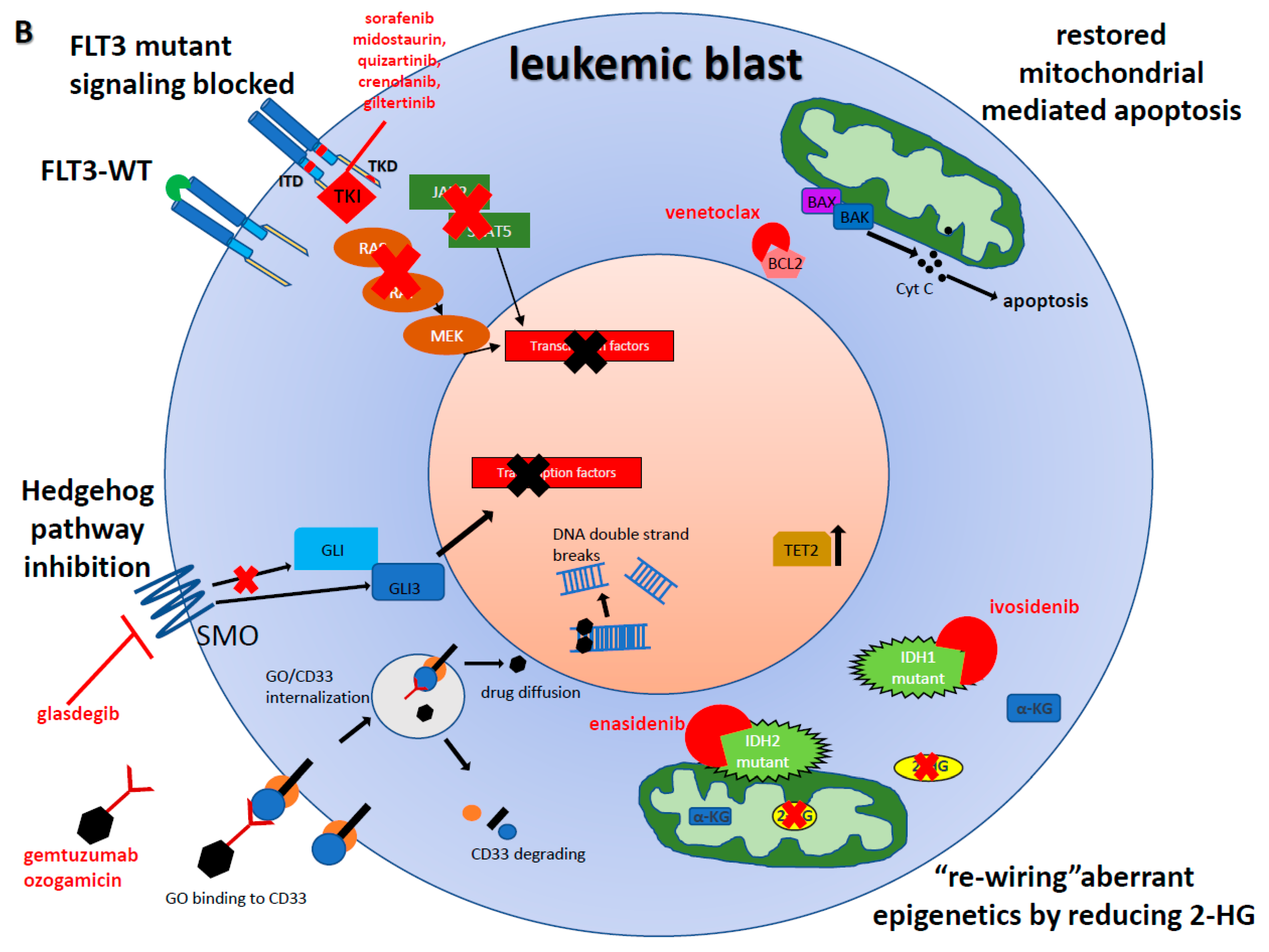
2. Anti-Body Drug Conjugate (ABDC)
Gemtuzumab Ozogamicin (GO)
3. FLT3-Inhibitors
3.1. Sorafenib
3.2. Midostaurin
3.3. Quizartinib
3.4. Crenolanib
3.5. Gilteritinib
4. IDH inhibitors
4.1. Enasidenib
4.2. Ivosidenib
5. Hedgehog Inhibition
Glasdegib
6. BCL2 Inhibition
Venetoclax
7. Upcoming Agents
8. Future Perspectives
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef]
- Perl, A.E. The role of targeted therapy in the management of patients with AML. Blood Adv. 2017, 1, 2281–2294. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [Green Version]
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Dohner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef] [Green Version]
- Perl, A.E.; Cortes, J.E.; Strickland, S.A.; Ritchie, E.K.; Neubauer, A.; Martinelli, G.; Naoe, T.; Pigneux, A.; Rousselot, P.H.; Röllig, C.; et al. An open-label, randomized phase 3 study of gilteritinib versus salvage chemotherapy in relapsed or refractory FLT3 mutation-positive acute myeloid leukemia. J. Clin. Oncol. 2017, 35, TPS7067. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Fathi, A.T.; Pollyea, D.A.; Stone, R.M.; Altman, J.K.; Roboz, G.J.; Patel, M.R.; Collins, R.; Flinn, I.W.; et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood 2019, 133, 676–687. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef]
- Wei, A.H.; Strickland, S.A., Jr.; Hou, J.Z.; Fiedler, W.; Lin, T.L.; Walter, R.B.; Enjeti, A.; Tiong, I.S.; Savona, M.; Lee, S.; et al. Venetoclax Combined with Low-Dose Cytarabine for Previously Untreated Patients with Acute Myeloid Leukemia: Results from a Phase 1b/2 Study. J. Clin. Oncol. 2019, Jco1801600. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef]
- Cortes, J.E.; Heidel, F.H.; Hellmann, A.; Fiedler, W.; Smith, B.D.; Robak, T.; Montesinos, P. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia 2019, 33, 379–389. [Google Scholar] [CrossRef]
- Castaigne, S.; Pautas, C.; Terre, C.; Raffoux, E.; Bordessoule, D.; Bastie, J.N.; Legrand, O.; Thomas, X.; Turlure, P.; Reman, O.; et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukemia (ALFA-0701): A randomised, open-label, phase 3 study. Lancet 2012, 379, 1508–1516. [Google Scholar] [CrossRef]
- Godwin, C.D.; Gale, R.P.; Walter, R.B. Gemtuzumab ozogamicin in acute myeloid leukemia. Leukemia 2017, 31, 1855–1868. [Google Scholar] [CrossRef]
- Raza, A.; Jurcic, J.G.; Roboz, G.J.; Maris, M.; Stephenson, J.J.; Wood, B.L.; Feldman, E.J.; Galili, N.; Grove, L.E.; Drachman, J.G.; et al. Complete remissions observed in acute myeloid leukemia following prolonged exposure to lintuzumab: A phase 1 trial. Leuk. Lymphoma 2009, 50, 1336–1344. [Google Scholar] [CrossRef]
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.A.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 121, 4854–4860. [Google Scholar] [CrossRef] [Green Version]
- Hills, R.K.; Castaigne, S.; Appelbaum, F.R.; Delaunay, J.; Petersdorf, S.; Othus, M.; Estey, E.H.; Dombret, H.; Chevret, S.; Ifrah, N.; et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukemia: A meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014, 15, 986–996. [Google Scholar] [CrossRef]
- Schlenk, R.F.; Paschka, P.; Krzykalla, J.; Weber, D.; Kapp-Schwoerer, S.; Gaidzik, V.I.; Leis, C.; Fiedler, W.; Kindler, T.; Schroeder, T.; et al. Gemtuzumab Ozogamicin in NPM1-Mutated Acute Myeloid Leukemia (AML): Results from the Prospective Randomized AMLSG 09-09 Phase-3 Study. Blood 2018, 132, 81. [Google Scholar] [CrossRef]
- Takeshita, A. Efficacy and resistance of gemtuzumab ozogamicin for acute myeloid leukemia. Int. J. Hematol. 2013, 97, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Lyman, S.D.; James, L.; Vanden Bos, T.; de Vries, P.; Brasel, K.; Gliniak, B.; Hollingsworth, L.T.; Picha, K.S.; McKenna, H.J.; Splett, R.R.; et al. Molecular cloning of a ligand for the flt3/flk-2 tyrosine kinase receptor: A proliferative factor for primitive hematopoietic cells. Cell 1993, 75, 1157–1167. [Google Scholar] [CrossRef]
- Gilliland, D.G.; Griffin, J.D. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002, 100, 1532–1542. [Google Scholar] [CrossRef] [Green Version]
- Bullinger, L.; Dohner, K.; Dohner, H. Genomics of Acute Myeloid Leukemia Diagnosis and Pathways. J. Clin. Oncol. 2017, 35, 934–946. [Google Scholar] [CrossRef]
- Kayser, S.; Schlenk, R.F.; Londono, M.C.; Breitenbuecher, F.; Wittke, K.; Du, J.; Groner, S.; Spath, D.; Krauter, J.; Ganser, A.; et al. Insertion of FLT3 internal tandem duplication in the tyrosine kinase domain-1 is associated with resistance to chemotherapy and inferior outcome. Blood 2009, 114, 2386–2392. [Google Scholar] [CrossRef] [Green Version]
- Schlenk, R.F.; Dohner, K.; Krauter, J.; Frohling, S.; Corbacioglu, A.; Bullinger, L.; Habdank, M.; Spath, D.; Morgan, M.; Benner, A.; et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N. Engl. J. Med. 2008, 358, 1909–1918. [Google Scholar] [CrossRef]
- Grunwald, M.R.; Levis, M.J. FLT3 inhibitors for acute myeloid leukemia: A review of their efficacy and mechanisms of resistance. Int. J. Hematol. 2013, 97, 683–694. [Google Scholar] [CrossRef]
- Levis, M.; Brown, P.; Smith, B.D.; Stine, A.; Pham, R.; Stone, R.; Deangelo, D.; Galinsky, I.; Giles, F.; Estey, E.; et al. Plasma inhibitory activity (PIA): A pharmacodynamic assay reveals insights into the basis for cytotoxic response to FLT3 inhibitors. Blood 2006, 108, 3477–3483. [Google Scholar] [CrossRef]
- Ravandi, F.; Cortes, J.E.; Jones, D.; Faderl, S.; Garcia-Manero, G.; Konopleva, M.Y.; O’Brien, S.; Estrov, Z.; Borthakur, G.; Thomas, D.; et al. Phase 1/2 study of combination therapy with sorafenib, idarubicin and cytarabine in younger patients with acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 1856–1862. [Google Scholar] [CrossRef]
- Rollig, C.; Serve, H.; Huttmann, A.; Noppeney, R.; Muller-Tidow, C.; Krug, U.; Baldus, C.D.; Brandts, C.H.; Kunzmann, V.; Einsele, H.; et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukemia (SORAML): A multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015, 16, 1691–1699. [Google Scholar] [CrossRef]
- Ravandi, F.; Alattar, M.L.; Grunwald, M.R.; Rudek, M.A.; Rajkhowa, T.; Richie, M.A.; Pierce, S.; Daver, N.; Garcia-Manero, G.; Faderl, S.; et al. Phase 2 study of azacitidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood 2013, 121, 4655–4662. [Google Scholar] [CrossRef]
- Ohanian, M.; Garcia-Manero, G.; Levis, M.; Jabbour, E.; Daver, N.; Borthakur, G.; Kadia, T.; Pierce, S.; Burger, J.; Richie, M.A.; et al. Sorafenib Combined with 5-azacitidine in Older Patients with Untreated FLT3-ITD Mutated Acute Myeloid Leukemia. Am. J. Hematol. 2018, 93, 1136–1141. [Google Scholar] [CrossRef]
- Battipaglia, G.; Ruggeri, A.; Massoud, R. Efficacy and feasibility of sorafenib as a maintenance agent after allogeneic hematopoietic stem cell transplantation for Fms-like tyrosine kinase 3-mutated acute myeloid leukemia. Cancer 2017, 123, 2867–2874. [Google Scholar] [CrossRef]
- Chen, Y.B.; Li, S.; Lane, A.A.; Connolly, C.; Del Rio, C.; Valles, B.; Curtis, M.; Ballen, K.; Cutler, C.; Dey, B.R.; et al. Phase 1 trial of maintenance sorafenib after allogeneic hematopoietic stem cell transplantation for fms-like tyrosine kinase 3 internal tandem duplication acute myeloid leukemia. Boil. Blood Marrow Transplant. 2014, 20, 2042–2048. [Google Scholar] [CrossRef]
- Burchert, A.; Bug, G.; Finke, J.; Stelljes, M.; Rollig, C.; Wäsch, R.; Bornhäuser, M.; Berg, T.; Lang, F.; Ehninger, G.; et al. Sorafenib As Maintenance Therapy Post Allogeneic Stem Cell Transplantation for FLT3-ITD Positive AML: Results from the Randomized, Double-Blind, Placebo-Controlled Multicentre Sormain Trial. Blood 2018, 132, 661. [Google Scholar] [CrossRef]
- Fabbro, D.; Buchdunger, E.; Wood, J.; Mestan, J.; Hofmann, F.; Ferrari, S.; Mett, H.; O’Reilly, T.; Meyer, T. Inhibitors of protein kinases: CGP 41251, a protein kinase inhibitor with potential as an anticancer agent. Pharmacol. Ther. 1999, 82, 293–301. [Google Scholar] [CrossRef]
- Weisberg, E.; Boulton, C.; Kelly, L.M.; Manley, P.; Fabbro, D.; Meyer, T.; Gilliland, D.G.; Griffin, J.D. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell 2002, 1, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Stone, R.M.; Fischer, T.; Paquette, R.; Schiller, G.; Schiffer, C.A.; Ehninger, G.; Cortes, J.; Kantarjian, H.M.; DeAngelo, D.J.; Huntsman-Labed, A.; et al. Phase 1b study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukemia. Leukemia 2012, 26, 2061–2068. [Google Scholar] [CrossRef]
- Levis, M.J.; Shi, W.; Chang, K.C.; Laing, C.; Berisha, F.; Adams, E.; Gocke, C.D.; Ding, W.; Nakamaru, K.; Lameh, J.; et al. Development of a Novel Next-Generation Sequencing (NGS)-Based Assay for Measurable Residual Disease (MRD) in FLT3-ITD AML and Its Potential Clinical Application in Patients Treated with Chemotherapy Plus FLT3 Inhibitors. Blood 2018, 132, 1459. [Google Scholar] [CrossRef]
- Levis, M.J.; Perl, A.E.; Altman, J.K.; Cortes, J.E.; Smith, C.C.; Baer, M.R.; Claxton, D.F.; Jurcic, J.G.; Ritchie, E.K.; Strickland, S.A.; et al. Impact of Minimal Residual Disease and Achievement of Complete Remission/Complete Remission with Partial Hematologic Recovery (CR/CRh) on Overall Survival Following Treatment with Gilteritinib in Patients with Relapsed/Refractory (R/R) Acute Myeloid Leukemia (AML) with FLT3 Mutations. Blood 2018, 132, 1458. [Google Scholar] [CrossRef]
- Larson, R.A.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.M.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Dohner, K.; Marcucci, G.; et al. An Analysis of Maintenance Therapy and Post-Midostaurin Outcomes in the International Prospective Randomized, Placebo-Controlled, Double-Blind Trial (CALGB 10603/RATIFY [Alliance]) for Newly Diagnosed Acute Myeloid Leukemia (AML) Patients with FLT3 Mutations. Blood 2017, 130, 145. [Google Scholar]
- Strati, P.; Kantarjian, H.; Ravandi, F.; Nazha, A.; Borthakur, G.; Daver, N.; Kadia, T.; Estrov, Z.; Garcia-Manero, G.; Konopleva, M.; et al. Phase 1/2 trial of the combination of midostaurin (PKC412) and 5-azacitidine for patients with acute myeloid leukemia and myelodysplastic syndrome. Am. J. Hematol. 2015, 90, 276–281. [Google Scholar] [CrossRef]
- Levis, M. Quizartinib for the treatment of FLT3-ITD acute myeloid leukemia. Future Oncol. 2014, 10, 1571–1579. [Google Scholar] [CrossRef]
- Cortes, J.E.; Kantarjian, H.; Foran, J.M.; Ghirdaladze, D.; Zodelava, M.; Borthakur, G.; Gammon, G.; Trone, D.; Armstrong, R.C.; James, J.; et al. Phase 1 study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J. Clin. Oncol. 2013, 31, 3681–3687. [Google Scholar] [CrossRef]
- Schiller, G.J.; Tallman, M.S.; Goldberg, S.L.; Perl, A.E.; Marie, J.-P.; Martinelli, G.; Larson, R.A.; Russell, N.; Trone, D.; Gammon, G.; et al. Final results of a randomized phase 2 study showing the clinical benefit of quizartinib (AC220) in patients with FLT3-ITD positive relapsed or refractory acute myeloid leukemia. J. Clin. Oncol. 2014, 32, 7100. [Google Scholar] [CrossRef]
- Smith, C.C.; Wang, Q.; Chin, C.S.; Salerno, S.; Damon, L.E.; Levis, M.J.; Perl, A.E.; Travers, K.J.; Wang, S.; Hunt, J.P.; et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukemia. Nature 2012, 485, 260–263. [Google Scholar] [CrossRef]
- Cortes, J.; Perl, A.E.; Dohner, H.; Kantarjian, H.; Martinelli, G.; Kovacsovics, T.; Rousselot, P.; Steffen, B.; Dombret, H.; Estey, E.; et al. Quizartinib, an FLT3 inhibitor, as monotherapy in patients with relapsed or refractory acute myeloid leukemia: An open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol. 2018, 19, 889–903. [Google Scholar] [CrossRef]
- Nybakken, G.E.; Canaani, J.; Roy, D.; Morrissette, J.D.; Watt, C.D.; Shah, N.P.; Smith, C.C.; Bagg, A.; Carroll, M.; Perl, A.E. Quizartinib elicits differential responses that correlate with karyotype and genotype of the leukemic clone. Leukemia 2016, 30, 1422–1425. [Google Scholar] [CrossRef]
- Sexauer, A.; Perl, A.; Yang, X.; Borowitz, M.; Gocke, C.; Rajkhowa, T.; Thiede, C.; Frattini, M.; Nybakken, G.E.; Pratz, K.; et al. Terminal myeloid differentiation in vivo is induced by FLT3 inhibition in FLT3-ITD AML. Blood 2012, 120, 4205–4214. [Google Scholar] [CrossRef]
- Cortes, J.E.; Khaled, S.K.; Martinelli, G.; Perl, A.E.; Ganguly, S.; Russell, N.H.; Kramer, A.; Dombret, H.; Hogge, D.; Jonas, B.A.; et al. Efficacy and Safety of Single-Agent Quizartinib (Q), a Potent and Selective FLT3 Inhibitor (FLT3i), in Patients (pts) with FLT3-Internal Tandem Duplication (FLT3-ITD)-Mutated Relapsed/Refractory (R/R) Acute Myeloid Leukemia (AML) Enrolled in the Global, Phase 3, Randomized Controlled Quantum-R Trial. Blood 2018, 132, 563. [Google Scholar] [CrossRef]
- Abdelall, W.; Kantarjian, H.M.; Borthakur, G.; Garcia-Manero, G.; Patel, K.P.; Jabbour, E.J.; Daver, N.G.; Kadia, T.; Gborogen, R.A.; Konopleva, M.; et al. The Combination of Quizartinib with Azacitidine or Low Dose Cytarabine Is Highly Active in Patients (Pts) with FLT3-ITD Mutated Myeloid Leukemias: Interim Report of a Phase 1/2 Trial. Blood 2016, 128, 1642. [Google Scholar]
- Randhawa, J.K.; Kantarjian, H.M.; Borthakur, G.; Thompson, P.A.; Konopleva, M.; Daver, N.; Pemmaraju, N.; Jabbour, E.; Kadia, T.M.; Estrov, Z.; et al. Results of a Phase 2 Study of Crenolanib in Relapsed/Refractory Acute Myeloid Leukemia Patients (Pts) with Activating FLT3 Mutations. Blood 2014, 124, 389. [Google Scholar]
- Cortes, J.E.; Kantarjian, H.M.; Kadia, T.M.; Borthakur, G.; Konopleva, M.; Garcia-Manero, G.; Daver, N.G.; Pemmaraju, N.; Jabbour, E.; Estrov, Z.; et al. Crenolanib besylate, a type I pan-FLT3 inhibitor, to demonstrate clinical activity in multiply relapsed FLT3-ITD and D835 AML. J. Clin. Oncol. 2016, 34, 7008. [Google Scholar] [CrossRef]
- Aboudalle, I.; Kantarjian, H.M.; Ohanian, M.N.; Alvarado, Y.; Jabbour, E.J.; Garcia-Manero, G.; Naqvi, K.; Wierda, W.G.; Daver, N.G.; Burger, J.A.; et al. Phase 1/2 Study of Crenolanib Combined with Standard Salvage Chemotherapy and Crenolanib Combined with 5-Azacitidine in Acute Myeloid Leukemia Patients with FLT3 Activating Mutations. Blood 2018, 132, 2715. [Google Scholar] [CrossRef]
- Wang, E.S.; Tallman, M.S.; Stone, R.M.; Walter, R.B.; Karanes, C.; Jain, V.; Collins, R.H. Low Relapse Rate in Younger Patients ≤ 60 Years Old with Newly Diagnosed FLT3-Mutated Acute Myeloid Leukemia (AML) Treated with Crenolanib and Cytarabine/Anthracycline Chemotherapy. Blood 2017, 130, 566. [Google Scholar]
- Park, I.K.; Mishra, A.; Chandler, J.; Whitman, S.P.; Marcucci, G.; Caligiuri, M.A. Inhibition of the receptor tyrosine kinase Axl impedes activation of the FLT3 internal tandem duplication in human acute myeloid leukemia: Implications for Axl as a potential therapeutic target. Blood 2013, 121, 2064–2073. [Google Scholar] [CrossRef]
- Perl, A.E.; Altman, J.K.; Cortes, J.; Smith, C.; Litzow, M.; Baer, M.R.; Claxton, D.; Erba, H.P.; Gill, S.; Goldberg, S.; et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukemia: A multicentre, first-in-human, open-label, phase 1–2 study. Lancet Oncol. 2017, 18, 1061–1075. [Google Scholar] [CrossRef]
- Ghiaur, G.; Levis, M. Mechanisms of Resistance to FLT3 Inhibitors and the Role of the Bone Marrow Microenvironment. Hematol./Oncol. Clin. N. Am. 2017, 31, 681–692. [Google Scholar] [CrossRef]
- Alonso, S.; Su, M.; Jones, J.W.; Ganguly, S.; Kane, M.A.; Jones, R.J.; Ghiaur, G. Human bone marrow niche chemoprotection mediated by cytochrome P450 enzymes. Oncotarget 2015, 6, 14905–14912. [Google Scholar] [CrossRef]
- Kojima, K.; McQueen, T.; Chen, Y.; Jacamo, R.; Konopleva, M.; Shinojima, N.; Shpall, E.; Huang, X.; Andreeff, M. p53 activation of mesenchymal stromal cells partially abrogates microenvironment-mediated resistance to FLT3 inhibition in AML through HIF-1alpha-mediated down-regulation of CXCL12. Blood 2011, 118, 4431–4439. [Google Scholar] [CrossRef]
- Smith, C.C.; Zhang, C.; Lin, K.C.; Lasater, E.A.; Zhang, Y.; Massi, E.; Damon, L.E.; Pendleton, M.; Bashir, A.; Sebra, R.; et al. Characterizing and Overriding the Structural Mechanism of the Quizartinib-Resistant FLT3 “Gatekeeper” F691L Mutation with PLX3397. Cancer Discov. 2015, 5, 668–679. [Google Scholar] [CrossRef]
- Lindblad, O.; Cordero, E.; Puissant, A.; Macaulay, L.; Ramos, A.; Kabir, N.N.; Sun, J.; Vallon-Christersson, J.; Haraldsson, K.; Hemann, M.T.; et al. Aberrant activation of the PI3K/mTOR pathway promotes resistance to sorafenib in AML. Oncogene 2016, 35, 5119–5131. [Google Scholar] [CrossRef] [Green Version]
- Medeiros, B.C.; Fathi, A.T.; DiNardo, C.D.; Pollyea, D.A.; Chan, S.M.; Swords, R. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia 2017, 31, 272–281. [Google Scholar] [CrossRef]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [PubMed]
- McKerrell, T.; Park, N.; Moreno, T.; Grove, C.S.; Ponstingl, H.; Stephens, J.; Crawley, C.; Craig, J.; Scott, M.A.; Hodkinson, C.; et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep. 2015, 10, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Nassereddine, S.; Lap, C.J.; Haroun, F.; Tabbara, I. The role of mutant IDH1 and IDH2 inhibitors in the treatment of acute myeloid leukemia. Ann. Hematol. 2017, 96, 1983–1991. [Google Scholar] [CrossRef]
- Yen, K.; Travins, J.; Wang, F.; David, M.D.; Artin, E.; Straley, K.; Padyana, A.; Gross, S.; DeLaBarre, B.; Tobin, E.; et al. AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations. Cancer Discov. 2017, 7, 478–493. [Google Scholar] [CrossRef] [PubMed]
- Amatangelo, M.D.; Quek, L.; Shih, A.; Stein, E.M.; Roshal, M.; David, M.D.; Marteyn, B.; Farnoud, N.R.; de Botton, S.; Bernard, O.A.; et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood 2017, 130, 732–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaturvedi, A.; Araujo Cruz, M.M.; Jyotsana, N.; Sharma, A.; Goparaju, R.; Schwarzer, A.; Gorlich, K.; Schottmann, R.; Struys, E.A.; Jansen, E.E.; et al. Enantiomer-specific and paracrine leukemogenicity of mutant IDH metabolite 2-hydroxyglutarate. Leukemia 2016, 30, 1708–1715. [Google Scholar] [CrossRef] [Green Version]
- Stein, E.M.; DiNardo, C.D.; Fathi, A.T.; Mims, A.S.; Pratz, K.W.; Savona, M.R.; Stein, A.S.; Stone, R.M.; Winer, E.S.; Seet, C.S.; et al. Ivosidenib or Enasidenib Combined with Induction and Consolidation Chemotherapy in Patients with Newly Diagnosed AML with an IDH1 or IDH2 Mutation Is Safe, Effective and Leads to MRD-Negative Complete Remissions. Blood 2018, 132, 560. [Google Scholar] [CrossRef]
- Roboz, G.J.; DiNardo, C.D.; Stein, E.M.; de Botton, S.; Mims, A.S.; Prince, G.T.; Altman, J.K.; Arellano, M.L.; Donnellan, W.B.; Erba, H.P.; et al. Ivosidenib (AG-120) Induced Durable Remissions and Transfusion Independence in Patients with IDH1-Mutant Untreated AML: Results from a Phase 1 Dose Escalation and Expansion Study. Blood 2018, 132, 561. [Google Scholar] [CrossRef]
- Quek, L.; David, M.D. Clonal heterogeneity of acute myeloid leukemia treated with the IDH2 inhibitor enasidenib. Nat. Med. 2018, 24, 1167–1177. [Google Scholar] [CrossRef]
- Intlekofer, A.M.; Shih, A.H.; Wang, B.; Nazir, A.; Rustenburg, A.S.; Albanese, S.K.; Patel, M.; Famulare, C.; Correa, F.M.; Takemoto, N.; et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature 2018, 559, 125–129. [Google Scholar] [CrossRef]
- Harding, J.J.; Lowery, M.A.; Shih, A.H.; Schvartzman, J.M. Isoform Switching as a Mechanism of Acquired Resistance to Mutant Isocitrate Dehydrogenase Inhibition. Cancer Discov. 2018, 8, 1540–1547. [Google Scholar] [CrossRef] [PubMed]
- Ok, C.Y.; Singh, R.R.; Vega, F. Aberrant activation of the hedgehog signaling pathway in malignant hematological neoplasms. Am. J. Pathol. 2012, 180, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Armas-Lopez, L.; Zuniga, J.; Arrieta, O.; Avila-Moreno, F. The Hedgehog-GLI pathway in embryonic development and cancer: Implications for pulmonary oncology therapy. Oncotarget 2017, 8, 60684–60703. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting Wnt, Notch and Hedgehog pathways. Nat. Rev. Clin. Oncol. 2011, 8, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Majeti, R. Biology and relevance of human acute myeloid leukemia stem cells. Blood 2017, 129, 1577–1585. [Google Scholar] [CrossRef]
- Wellbrock, J.; Latuske, E.; Kohler, J.; Wagner, K.; Stamm, H.; Vettorazzi, E.; Vohwinkel, G.; Klokow, M.; Uibeleisen, R.; Ehm, P.; et al. Expression of Hedgehog Pathway Mediator GLI Represents a Negative Prognostic Marker in Human Acute Myeloid Leukemia and Its Inhibition Exerts Antileukemic Effects. Clin. Cancer Res. 2015, 21, 2388–2398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Queiroz, K.C.; Ruela-de-Sousa, R.R.; Fuhler, G.M.; Aberson, H.L.; Ferreira, C.V.; Peppelenbosch, M.P.; Spek, C.A. Hedgehog signaling maintains chemoresistance in myeloid leukemic cells. Oncogene 2010, 29, 6314–6322. [Google Scholar] [CrossRef] [Green Version]
- Aberger, F.; Hutterer, E.; Sternberg, C.; Del Burgo, P.J.; Hartmann, T.N. Acute myeloid leukemia—Strategies and challenges for targeting oncogenic Hedgehog/GLI signaling. Cell Commun. Signal. CCS 2017, 15, 8. [Google Scholar] [CrossRef]
- Sadarangani, A.; Pineda, G.; Lennon, K.M.; Chun, H.J.; Shih, A.; Schairer, A.E.; Court, A.C.; Goff, D.J.; Prashad, S.L.; Geron, I.; et al. GLI2 inhibition abrogates human leukemia stem cell dormancy. J. Transl. Med. 2015, 13, 98. [Google Scholar] [CrossRef]
- Fukushima, N.; Minami, Y.; Kakiuchi, S.; Kuwatsuka, Y.; Hayakawa, F.; Jamieson, C.; Kiyoi, H.; Naoe, T. Small-molecule Hedgehog inhibitor attenuates the leukemia-initiation potential of acute myeloid leukemia cells. Cancer Sci. 2016, 107, 1422–1429. [Google Scholar] [CrossRef]
- Martinelli, G.; Oehler, V.G.; Papayannidis, C.; Courtney, R.; Shaik, M.N.; Zhang, X.; O’Connell, A.; McLachlan, K.R.; Zheng, X.; Radich, J.; et al. Treatment with PF-04449913, an oral smoothened antagonist, in patients with myeloid malignancies: A phase 1 safety and pharmacokinetics study. Lancet Haematol. 2015, 2, e339–e346. [Google Scholar] [CrossRef]
- Savona, M.R.; Pollyea, D.A.; Stock, W.; Oehler, V.G.; Schroeder, M.A.; Lancet, J.; McCloskey, J.; Kantarjian, H.M.; Ma, W.W.; Shaik, M.N.; et al. Phase 1b Study of Glasdegib, a Hedgehog Pathway Inhibitor, in Combination with Standard Chemotherapy in Patients with AML or High-Risk MDS. Clin. Cancer Res. 2018, 24, 2294–2303. [Google Scholar] [CrossRef]
- Cortes, J.E.; Papayannidis, C.; Jamieson, C.; Schiller, G.J.; Candoni, A.; Leber, B.; Baldus, C.D.; Pérez-Simón, J.A.; Ma, W.W.; Gallo Stampino, C.; et al. Glasdegib in Addition to Intensive or Non-Intensive Chemotherapy in Patients with Acute Myeloid Leukemia: Safety Analysis of Glasdegib ‘On Target’ Adverse Events. Blood 2018, 132, 2732. [Google Scholar] [CrossRef]
- Chaudhry, P.; Singh, M.; Triche, T.J.; Guzman, M.; Merchant, A.A. GLI3 repressor determines Hedgehog pathway activation and is required for response to SMO antagonist glasdegib in AML. Blood 2017, 129, 3465–3475. [Google Scholar] [CrossRef]
- Vo, T.T.; Ryan, J.; Carrasco, R.; Neuberg, D.; Rossi, D.J.; Stone, R.M.; Deangelo, D.J.; Frattini, M.G.; Letai, A. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell 2012, 151, 344–355. [Google Scholar] [CrossRef]
- Fandy, T.E.; Jiemjit, A.; Thakar, M.; Rhoden, P.; Suarez, L.; Gore, S.D. Decitabine induces delayed reactive oxygen species (ROS) accumulation in leukemia cells and induces the expression of ROS generating enzymes. Clin. Cancer Res. 2014, 20, 1249–1258. [Google Scholar] [CrossRef] [Green Version]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Boil. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Pan, R.; Ruvolo, V.R.; Wei, J.; Konopleva, M.; Reed, J.C.; Pellecchia, M.; Andreeff, M.; Ruvolo, P.P. Inhibition of Mcl-1 with the pan-Bcl-2 family inhibitor (−)BI97D6 overcomes ABT-737 resistance in acute myeloid leukemia. Blood 2015, 126, 363–372. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and Biological Correlates of Response in a Phase 2 Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef] [Green Version]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; Riemondy, K.A. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 2018, 24, 1859–1866. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Albitar, M.; Kadia, T.M.; Naqvi, K.; Vaughan, K.; Cavazos, A.; Pierce, S.A.; Takahashi, K.; Kornblau, S.M.; Ravandi, F.; et al. Venetoclax in Combination with FLAG-IDA Chemotherapy (FLAG-V-I) for Fit, Relapsed/Refractory AML Patients: Interim Results of a Phase 1b/2 Dose Escalation and Expansion Study. Blood 2018, 132, 4048. [Google Scholar] [CrossRef]
- Zhang, Q.; Pan, R.; Han, L.; Shi, C.; Kurtz, S.E.; Mu, H.; Ma, H.; Andreeff, M.; Leverson, J.; Tyner, J.W.; et al. Mechanisms of Acquired Resistance to Venetoclax in Preclinical AML Models. Blood 2015, 126, 328. [Google Scholar]
- Mali, R.S.; Lasater, E.A.; Doyle, K.; Malla, R.; Boghaert, E.; Souers, A.; Leverson, J.D.; Sampath, D. FLT3-ITD Activation Mediates Resistance to the BCL-2 Selective Antagonist, Venetoclax, in FLT3-ITD Mutant AML Models. Blood 2017, 130, 1348. [Google Scholar]
- Blombery, P.; Anderson, M.A.; Gong, J.N.; Thijssen, R.; Birkinshaw, R.W. Acquisition of the Recurrent Gly101Val Mutation in BCL2 Confers Resistance to Venetoclax in Patients with Progressive Chronic Lymphocytic Leukemia. Cancer Discov. 2019, 9, 342–353. [Google Scholar] [CrossRef]
- Caenepeel, S.; Brown, S.P. AMG 176, a Selective MCL1 Inhibitor, Is Effective in Hematologic Cancer Models Alone and in Combination with Established Therapies. Cancer Discov. 2018, 8, 1582–1597. [Google Scholar] [CrossRef]
- Ramsey, H.E.; Fischer, M.A. A Novel MCL1 Inhibitor Combined with Venetoclax Rescues Venetoclax-Resistant Acute Myelogenous Leukemia. Cancer Discov. 2018, 8, 1566–1581. [Google Scholar] [CrossRef]
- Berthon, C.; Raffoux, E.; Thomas, X.; Vey, N.; Gomez-Roca, C.; Yee, K.; Taussig, D.C.; Rezai, K.; Roumier, C.; Herait, P.; et al. Bromodomain inhibitor OTX015 in patients with acute leukemia: A dose-escalation, phase 1 study. Lancet Haematol. 2016, 3, e186–e195. [Google Scholar] [CrossRef]
- Schenk, T.; Chen, W.C.; Gollner, S.; Howell, L.; Jin, L.; Hebestreit, K.; Klein, H.U.; Popescu, A.C.; Burnett, A.; Mills, K.; et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med. 2012, 18, 605–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, M.; Polly, P.; Liu, T. The histone methyltransferase DOT1L: Regulatory functions and a cancer therapy target. Am. J. Cancer Res. 2015, 5, 2823–2837. [Google Scholar] [PubMed]
- Borkin, D.; He, S.; Miao, H.; Kempinska, K.; Pollock, J.; Chase, J.; Purohit, T.; Malik, B.; Zhao, T.; Wang, J.; et al. Pharmacologic inhibition of the Menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell 2015, 27, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; Garcia-Manero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Jongen-Lavrencic, M.; Altman, J.K.; Dohner, H.; Thomson, B.; Blakemore, S.J.; et al. A Phase 1 Study of the DOT1L Inhibitor, Pinometostat (EPZ-5676), in Adults with Relapsed or Refractory Leukemia: Safety, Clinical Activity, Exposure and Target Inhibition. Blood 2015, 126, 2547. [Google Scholar]
- Hoseini, S.S.; Cheung, N.K. Acute myeloid leukemia targets for bispecific antibodies. Blood Cancer J. 2017, 7, e552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravandi, F.; Stein, A.S.; Kantarjian, H.M.; Walter, R.B.; Paschka, P.; Jongen-Lavrencic, M.; Ossenkoppele, G.J.; Yang, Z.; Mehta, B.; Subklewe, M. A Phase 1 First-in-Human Study of AMG 330, an Anti-CD33 Bispecific T-Cell Engager (BiTE®) Antibody Construct, in Relapsed/Refractory Acute Myeloid Leukemia (R/R AML). Blood 2018, 132, 25. [Google Scholar] [CrossRef]
- Uy, G.L.; Godwin, J.; Rettig, M.P.; Vey, N.; Foster, M.; Arellano, M.L.; Rizzieri, D.A.; Topp, M.S.; Huls, G.; Lowenberg, B.; et al. Preliminary Results of a Phase 1 Study of Flotetuzumab, a CD123 x CD3 Bispecific Dart® Protein, in Patients with Relapsed/Refractory Acute Myeloid Leukemia and Myelodysplastic Syndrome. Blood 2017, 130, 637. [Google Scholar]
- Gill, S.; Tasian, S.K.; Ruella, M.; Shestova, O.; Li, Y.; Porter, D.L.; Carroll, M.; Danet-Desnoyers, G.; Scholler, J.; Grupp, S.A.; et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood 2014, 123, 2343–2354. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Cao, Y.; Pinz, K.; Ma, Y.; Wada, M.; Chen, K.; Ma, G.; Shen, J.; Tse, C.O.; Su, Y.; et al. First-in-Human CLL1-CD33 Compound CAR T Cell Therapy Induces Complete Remission in Patients with Refractory Acute Myeloid Leukemia: Update on Phase 1 Clinical Trial. Blood 2018, 132, 901. [Google Scholar] [CrossRef]
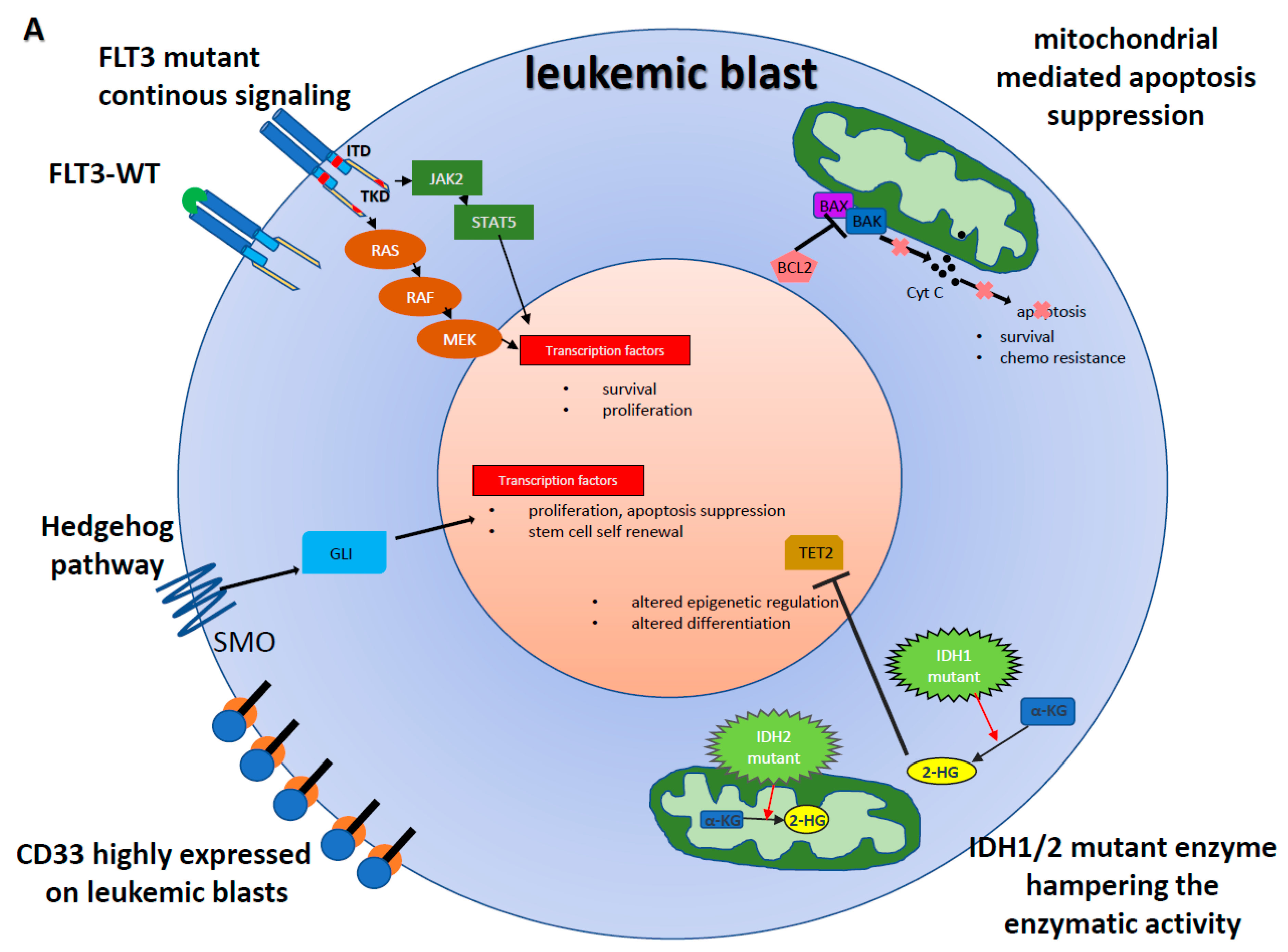
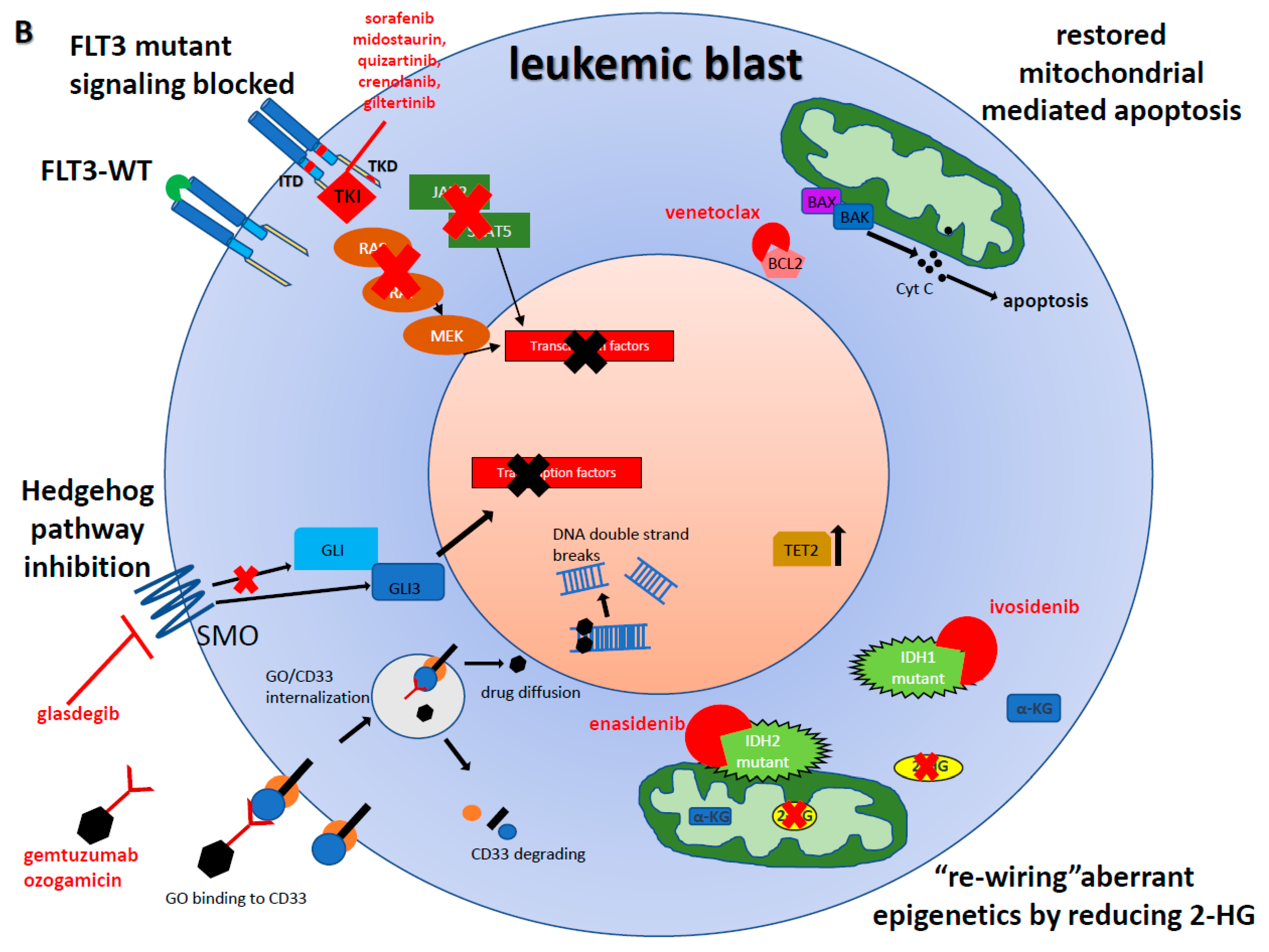

{kind=link}
{kind=link}
{kind=link}
| Agents | Investigation | Phase | Identifier |
|---|---|---|---|
| Gemtuzumab Ozogamicin | Liposome-encapsulated daunorubicin-cytarabine and GO in treating patients with r/r AML or high-risk MDS | 1 | NCT03672539 |
| Fractionated GO in treating MRD measurable residual disease in participants with AML | 2 | NCT03737955 | |
| Sorafenib | Sorafenib + busulfan and fludarabine conditioning in r/r AML undergoing stem cell transplantation | 1/2 | NCT03247088 |
| Sorafenib plus azacitidine in AML/MDS patients with FLT3-ITD mutation | 2 | NCT02196857 | |
| Midostaurin | Midostaurin + chemotherapy in newly diagnosed FLT3-WT AML | 3 | NCT03512197 |
| Crenolanib vs. midostaurin following induction chemotherapy and consolidation therapy in newly diagnosed FLT3 mutant AML | 3 | NCT03258931 | |
| Quizartinib | Quizartinib with standard of care chemotherapy and as continuation therapy in new diagnosed FLT3-ITD AML | 3 | NCT02668653 |
| Quizartinib and venetoclax in r/r FLT3 mutated AML | 1b/2 | NCT03735875 | |
| Crenolanib | Crenolanib combined with chemotherapy in r/r FLT3 mutated AML | 1b/2 | NCT02298166 |
| Crenolanib maintenance following allogeneic stem cell transplantation in FLT3-mutant AML | 2 | NCT02400255 | |
| Gilteritinib | Gilteritinib vs. midostaurin in FLT3 mutant AML during induction and consolidation chemotherapy | 2 | NCT03836209 |
| Gilteritinib as maintenance therapy following induction/consolidation therapy in FLT3-ITD AML in 1.CR | 3 | NCT02927262 | |
| Enasidenib/Ivosidenib | Ivosidenib or Enasidenib combined with induction/consolidation, followed by maintenance therapy in IDH1/IDH2 mutated AML/MDS2-EB2 | 3 | NCT03839771 |
| Enasidenib vs. conventional care regimens in IDH2 mutant elderly AML | 3 | NCT02577406 | |
| Ivosidenib vs. placebo in combination with azacitidine in IDH1 mutant AML | 3 | NCT03173248 | |
| Glasdegib | Intensive chemotherapy +/− glasdegib or azacitidine +/− glasdegib AML patients | 3 | NCT03416179 |
| Immunotherapy combinations for AML for example, glasdegib plus avelumab | 1b/2 | NCT03390296 | |
| Venetoclax | Venetoclax combined with gilteritinib in r/r AML | I | NCT03625505 |
| Venetoclax +/− azacitidine in AML, ineligible for intensive treatment | 3 | NCT02993523 | |
| Venetoclax +/− low dose cytarabine in AML, ineligible for intensive treatment | 3 | NCT03069352 | |
| Venetoclax combined with induction/consolidation chemotherapy in AML | 1b | NCT03709758 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bohl, S.R.; Bullinger, L.; Rücker, F.G. New Targeted Agents in Acute Myeloid Leukemia: New Hope on the Rise. Int. J. Mol. Sci. 2019, 20, 1983. https://doi.org/10.3390/ijms20081983
Bohl SR, Bullinger L, Rücker FG. New Targeted Agents in Acute Myeloid Leukemia: New Hope on the Rise. International Journal of Molecular Sciences. 2019; 20(8):1983. https://doi.org/10.3390/ijms20081983
Chicago/Turabian StyleBohl, Stephan R., Lars Bullinger, and Frank G. Rücker. 2019. "New Targeted Agents in Acute Myeloid Leukemia: New Hope on the Rise" International Journal of Molecular Sciences 20, no. 8: 1983. https://doi.org/10.3390/ijms20081983
APA StyleBohl, S. R., Bullinger, L., & Rücker, F. G. (2019). New Targeted Agents in Acute Myeloid Leukemia: New Hope on the Rise. International Journal of Molecular Sciences, 20(8), 1983. https://doi.org/10.3390/ijms20081983