1. Introduction
Transient receptor potential melastatin member 4 (TRPM4) and member 5 (TRPM5) channels are Ca
2+-activated nonselective monovalent cation channels. They share 40% homology in their amino-acid sequences, so that they are the closest homologs among eight members of the TRPM family [
1]. TRPM4 is expressed in a broad range of cells such as cardiac myocytes, immune cells, etc. [
1]. Conversely, TRPM5 is expressed in a relatively small number of tissues with the highest expression in type II taste cells, which detect bitter, sweet and umami stimuli [
1]. The activities of TRPM4 and TRPM5 influence the functions of the cells, where they are expressed, by depolarizing the membrane potential when they open by a rise in intracellular Ca
2+ (Ca
2+i) concentration ([Ca
2+]
i). The Ca
2+-sensitivity of TRPM4 has been reported to be maintained by interaction with PI(4,5)P
2, a major phosphoinositide in plasma membrane [
2], and a depletion of PI(4,5)P
2 caused desensitization of TRPM4 [
3,
4]. PI(4,5)P
2 was also reported to have partially restored TRPM5 channel activity after its desensitization [
5]. However, their opening is completely dependent on Ca
2+i because without Ca
2+ they are unable to open even if there is a sufficient amount of PI(4,5)P
2 [
4,
6].
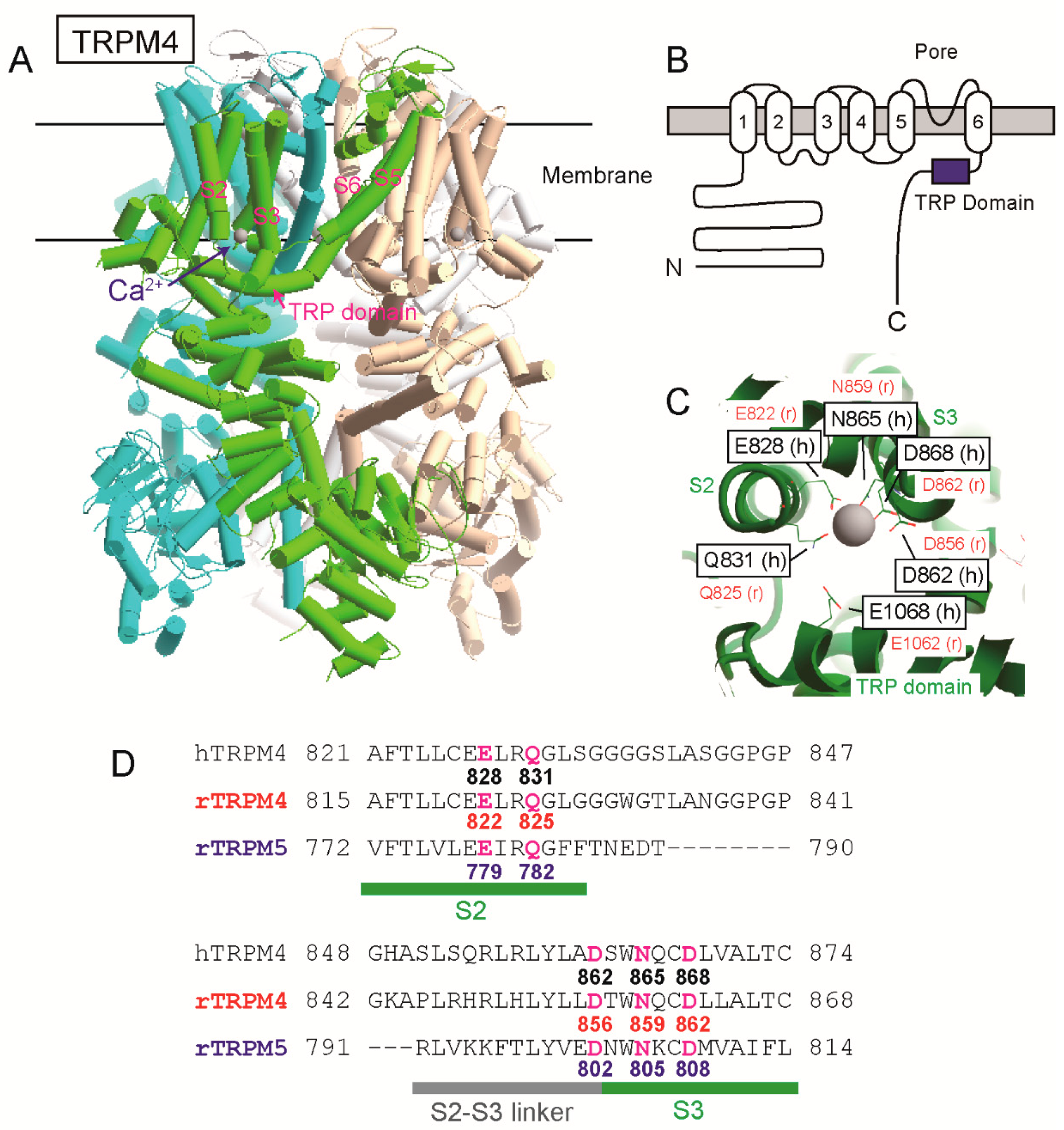
The position of a Ca
2+-binding site of TRPM4 had been unknown for a long time since its cloning. However, as a result of site-directed mutagenesises and patch clamp analyses of rat TRPM4 (rTRPM4), we previously found that negatively charged amino acid residues near and in the TRP domain of the intracellular C-terminal tail are necessary for the normal Ca
2+-sensitivity of rTRPM4 [
6]. They are Asp
1049 and Glu
1062 of rTRPM4 and are conserved in the other Ca
2+-sensitive TRPM channels (TRPM5, TRPM2 and TRPM8) [
6,
7,
8]. More recently, several cryo-electron microscopy (cryo-EM) structures of TRPM4 were reported [
9,
10,
11,
12]. One of them is a Ca
2+-bound structure of human TRPM4 (hTRPM4, [
11]). Ca
2+ was surrounded by four amino acid residues of the transmembrane segment 2 (S2) and 3 (S3) (Glu
828, Gln
831, Asn
865, and Asp
868 of hTRPM4) within a cavity accessible from cytosol (
Figure 1, [
11]). The glutamate in the TRP domain, which we reported, was located at the entrance of the cavity and assumed to enhance access of Ca
2+ to the Ca
2+-binding site through its negative charge (
Figure 1, [
9,
11]). The cryo-EM structures of TRPM2 [
13,
14] and TRPM8 [
15] supported that the four amino acid residues in S2 and S3 form their Ca
2+-binding sites.
Although the proposed Ca
2+-binding site is most likely to be necessary for TRPM4 to open, the possibility has not been fully proven. That is firstly because the Ca
2+-bound structure of hTRPM4 was in a closed state although Ca
2+ bound to the site [
11]. Moreover, that is also because the importance of the four amino acid residues for Ca
2+-sensitivity of TRPM4 has not been evaluated by functional analyses. Concerning TRPM5, although the amino acid residues forming the Ca
2+-binding site of TRPM4 are conserved in TRPM5, the position of a Ca
2+-binding site of TRPM5 has not been experimentally revealed. Therefore, a primary aim of this study is to reveal amino acid residues which form Ca
2+-binding sites of TRPM4 and TRPM5 by functional analyses using mutagenesises, an inside-out patch clamp technique and a whole-cell patch clamp technique.
As a secondary aim of this study, we re-evaluated the effects of PI(4,5)P
2 on rTRPM4 and rTRPM5. The PI(4,5)P
2-binding site of TRPM4 was suggested to be located at the pre-S1 (i.e., just before S1) region in the intracellular N-terminal tail [
16]. However, the similarity of the pre-S1 region between TRPM channels is low and the important amino acid residues for the binding of PI(4,5)P
2 to TRPM4 are not conserved in the pre-S1 region of TPRM5 [
15,
16]. Therefore, the pre-S1 region of TRPM5 may not be able to function as a PI(4,5)P
2 binding site. Additionally, there seems to be a difference between the extent of the effect of PI(4,5)P
2 on TRPM4 and TRPM5 when they are compared in literature. As written in the initial report on the effect of PI(4,5)P
2 on TRPM5, PI(4,5)P
2 “partially” restored the sensitivity of the channel to Ca
2+ [
5]. On the other hand, PI(4,5)P
2 completely restores Ca
2+-sensitivity and channel activity of TRPM4 [
3,
4,
6]. Therefore, in this study, we compared the effects of PI(4,5)P
2 on rTRPM4 and rTRPM5 under the same condition using the same cell line, the same expression system, and the same inside-out patch clamp technique.
3. Discussion
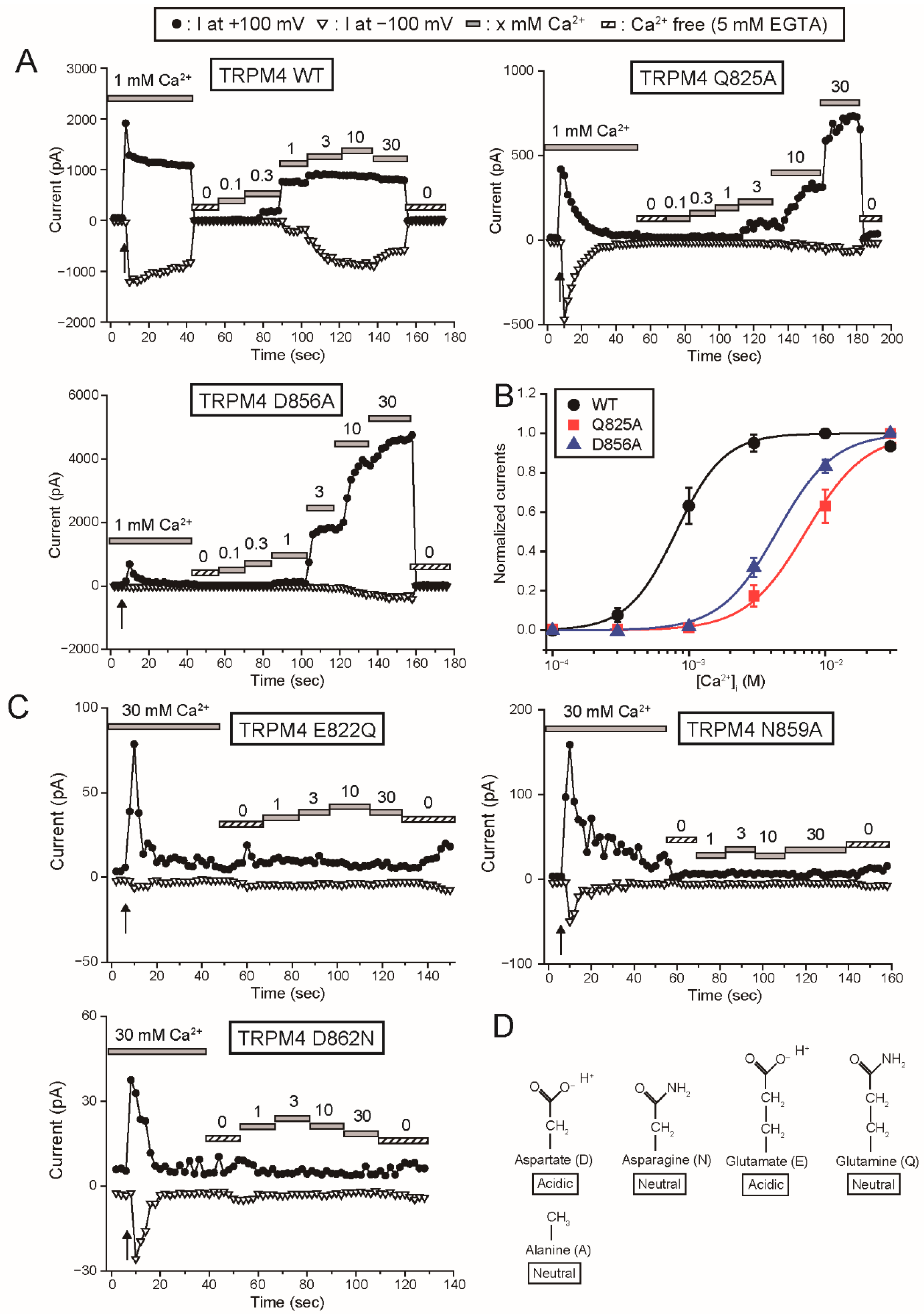
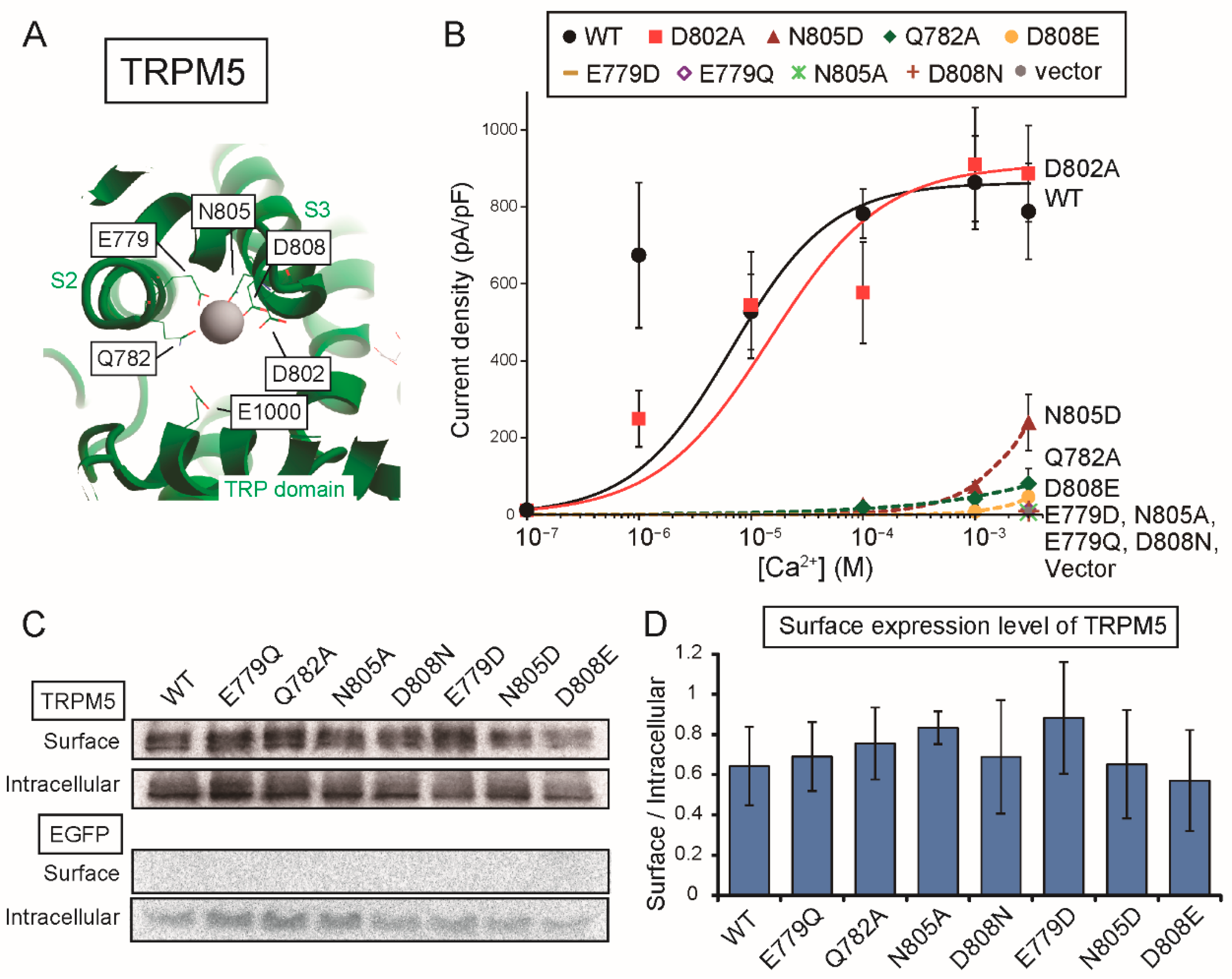
Firstly, the results in this study provide the functional evidence that the four amino acid residues (Glu
822, Gln
825, Asn
859 and Asp
862 of rTRPM4), which bound to Ca
2+ in the cryo-EM structure of hTRPM4 [
11], form the Ca
2+-binding site of rTRPM4. Secondly, although any three-dimensional structures of TRPM5 are not available, the results in this study also indicate that the same amino acid residues (Glu
779, Gln
782, Asn
805 and Asp
808 of rTRPM5) form the Ca
2+-binding site of rTRPM5. The Ca
2+-binding sites of the other Ca
2+-sensitive TRPM channels (TRPM2 and TRPM8) were also reported to be formed by the same four amino acid residues [
13,
14,
15]. Therefore, the Ca
2+-binding site was revealed to be conserved in all the Ca
2+-sensitive TRPM channels as the information on the Ca
2+-binding site of TRPM5 was added by this study.
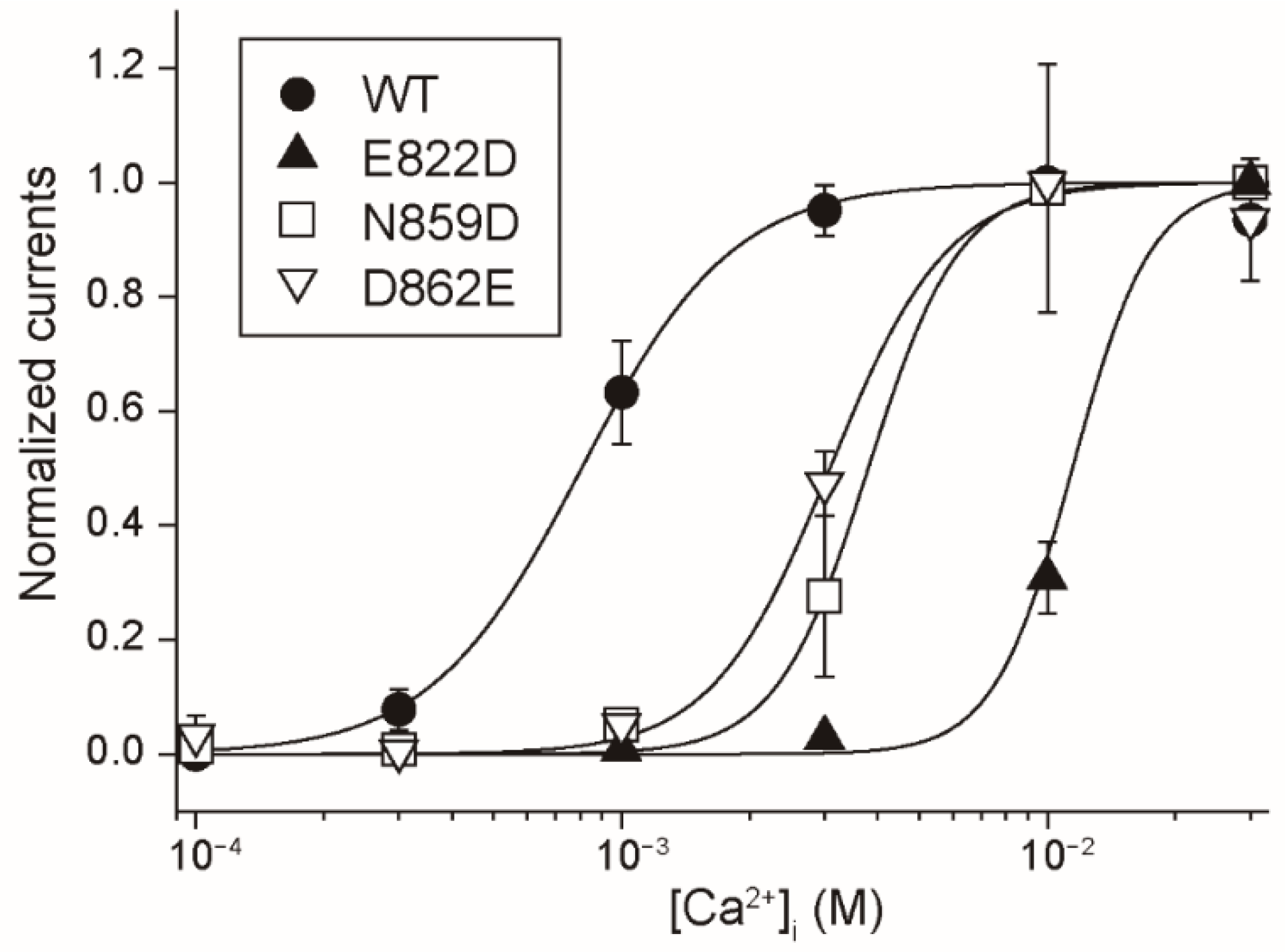
There may be a difference between the Ca
2+-binding site of rTRPM4 and that of rTRPM5 only in the involvement of the aspartate (Asp
856 of rTRPM4 and Asp
802 of rTRPM5) at the entrance of the Ca
2+-binding site. The mutation of the aspartate to alanine reduced the Ca
2+-sensitivity of rTRPM4 (D856A in
Figure 2B), but the same mutation did not have an obvious effect on the Ca
2+-sensitivity of rTRPM5 (D802A in
Figure 6B). In the case of rTRPM4, Asp
856 likely faces the pathway for Ca
2+ as shown in the structure of hTRPM4 [
11] and its negative charge may facilitate the access of Ca
2+ to its binding site. On the other hand, in the case of rTRPM5, Asp
802 might be oriented to a different direction or be covered by other amino acid residues, so that Asp
802 might fail to facilitate the access of Ca
2+. The reason why Asp
802 plays a less important role in the Ca
2+-sensitivity of rTRPM5 will be revealed when the three-dimensional structure of TRPM5 is unveiled.
An important novel finding in this study is that the negatively-charged amino acid residues near and in the TRP domain are also necessary for the normal Ca
2+-sensitivity of rTRPM5. This finding raises the importance of these amino acid residues for understanding the mechanisms of TRPM channel opening by Ca
2+. The glutamate in the TRP domain of the cryo-EM hTRPM4 structure was shown to be located in the pathway leading to the Ca
2+-binding site from the cytoplasmic space [
11]. Therefore, the negative charge of the glutamate is thought to increase the accessibility of Ca
2+ to the site [
11]. However, it should be noted that the corresponding glutamate of human TRPM2 (hTRPM2, Glu
1073) was shown to participate in the coordination of Ca
2+ together with the four amino acid residues in S2 and S3 according to the Ca
2+-bound and “open” structure of hTRPM2 [
14]. The structure of hTRPM4, which was referenced in this study, is a Ca
2+-bound structure but is in a “closed” state [
11]. Therefore, if the Ca
2+-bound and open structures of TRPM4 and TRPM5 are unveiled, the glutamate in TRPM4 and TRPM5 might be revealed to directly participate in the formation of the Ca
2+-binding site in addition to the four amino acid residues in S2 and S3.
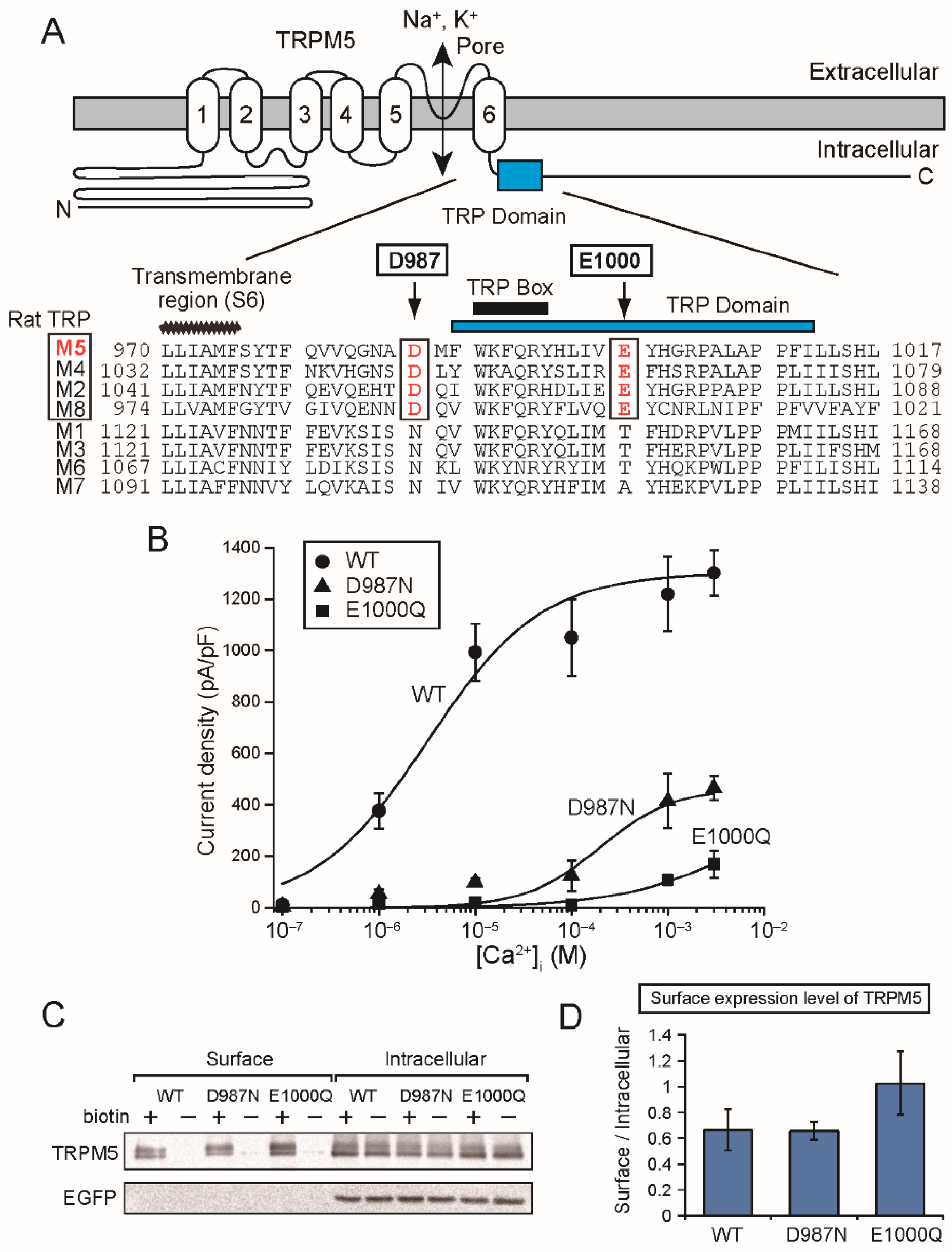
The mutation of the aspartate (Asp
1049 of rTRPM4 and Asp
987 of rTRPM5) just before the TRP domain also reduced Ca
2+-sensitivities of rTRPM4 [
6] and rTRPM5 (
Figure 5B). The aspartate is located at the beginning of the α-helix containing the TRP domain just after the bend below S6 (
Figure 1A and
Figure 5A). Therefore, the mutation of the aspartate might move the direction of the TRP domain, and the consequent displacement of the glutamate in the TRP domain might be a reason for the reduced Ca
2+-sensitivities of the mutants.
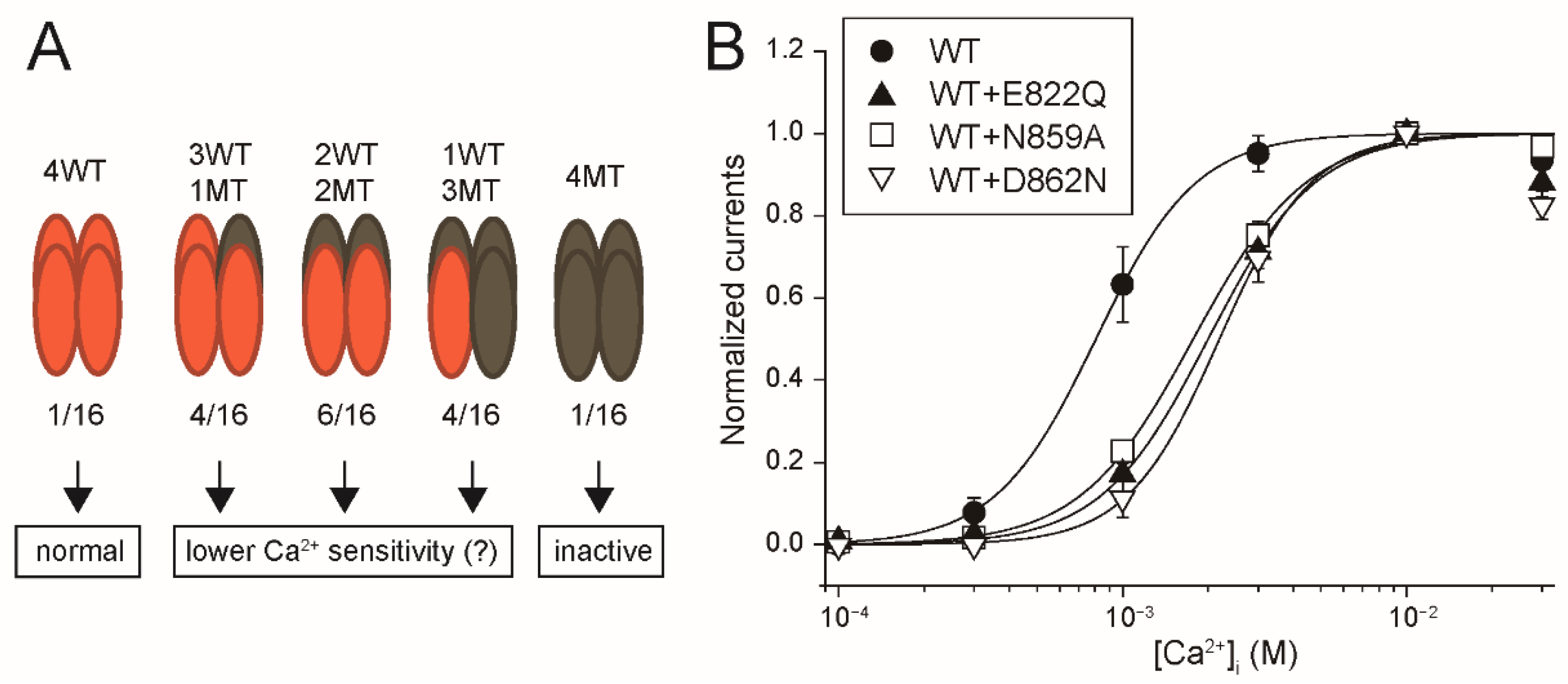
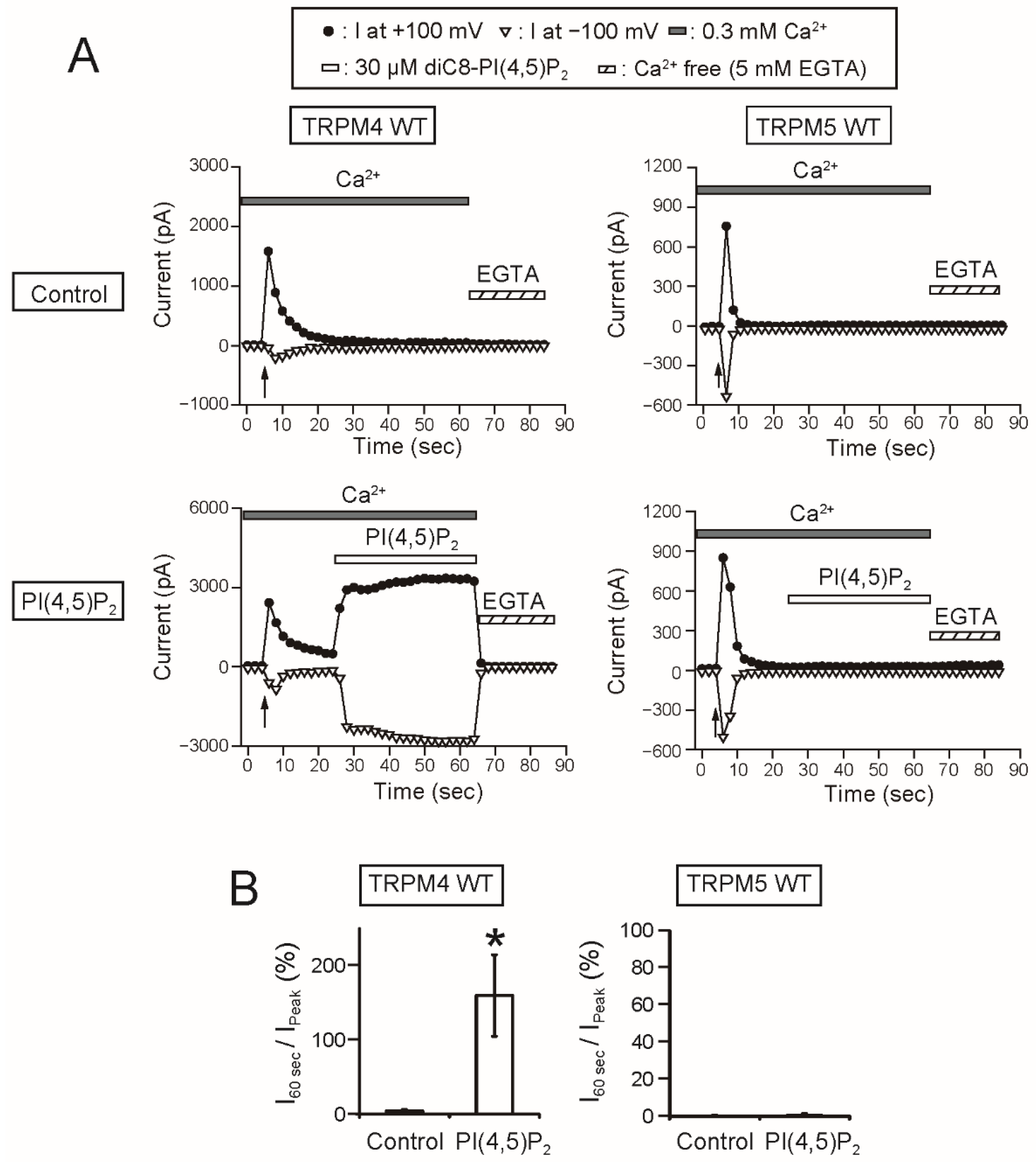
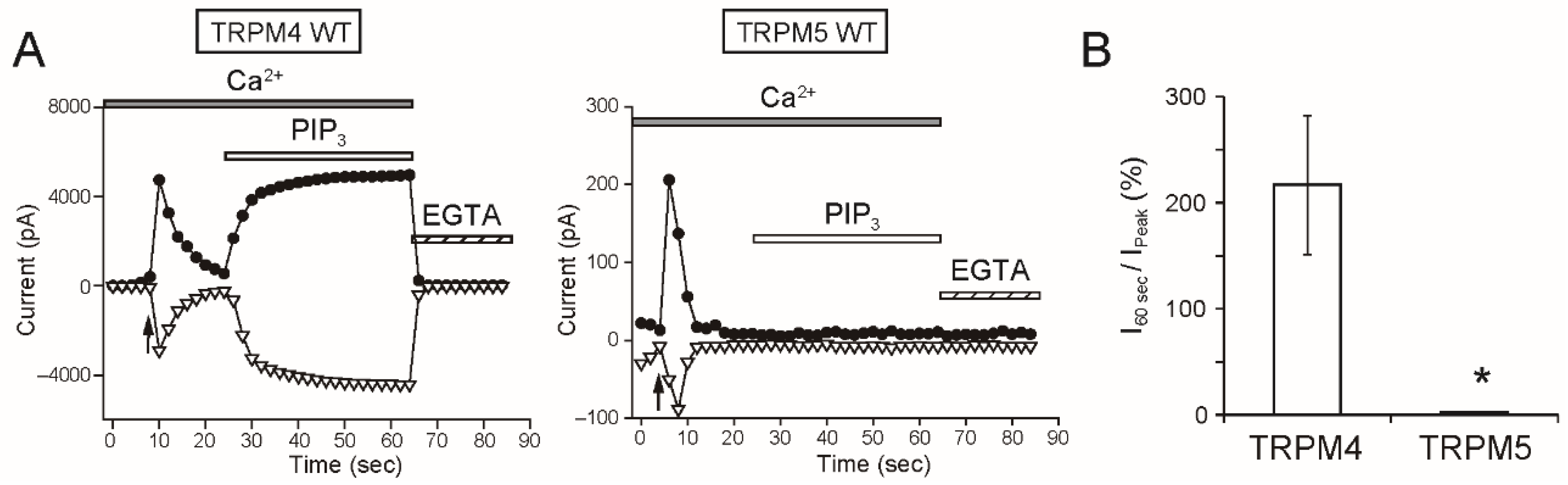
As a remarkable finding, the intracellular applications of PI(4,5)P
2 and PIP
3 did not restore the rTRPM5 currents although they restored the rTRPM4 currents completely (
Figure 7 and
Figure 8). These experiments were conducted under the same conditions. We cannot exclude the possibility that PI(4,5)P
2 and PIP
3 can restore the TRPM5 currents under other experimental conditions. However, from the beginning, the effect of PI(4,5)P
2 on TRPM5 was reported not to be so strong [
5]. Therefore, when we take both observations from elsewhere [
5] and our results into account, we consider it will be most likely that the regulation of the Ca
2+-sensitivity of TRPM5 by PI(4,5)P
2 is limited, if any, in comparison with the case of TRPM4.
The PI(4,5)P
2-binding site of full-length TRPM4 has not been elucidated. Initially, putative pleckstrin homology (PH) domains in its cytosolic C-terminal tail were suggested to be candidates for the PI(4,5)P
2-binding site [
4]. However, the putative PH domains are less likely to be the PI(4,5)P
2-binding site of TRPM4 because the domains appear not to be accessible based on the cryo-EM structure [
9]. A more feasible candidate for the PI(4,5)P
2-binding site of TRPM4 is pre-S1 region in the cytosolic N-terminal region, as PI(4,5)P
2 and PIP
3 were shown to bind to the pre-S1 region fragment of hTRPM4 by surface plasmon resonance measurements [
16]. Arg
755 and Arg
767 of hTRPM4 were shown to be crucial in the interaction with PI(4,5)P
2 and PIP
3. The first arginine is conserved in rTRPM4. However, the second arginine is not conserved in rTRPM4. There is no amino acid residue in the position of the second arginine in rTRPM4 based on an alignment of amino acid sequences of hTRPM4 and rTRPM4. Therefore, it is not certain whether PI(4,5)P
2 binds to the pre-S1 region of rTRPM4. However, the PI(4,5)P
2 binding site of TRPM8 has been reported to be formed by the combination of its pre-S1 region and other regions [
15]. Similarly, in the case of full-length TRPM4, the role of the second arginine, shown in the fragment of hTRPM4, may be played by another basic amino acid residue in another region. Although the pre-S1 region of TRPM4 may be involved in the binding of PI(4,5)P
2, it should be noted that the similarity of the pre-S1 region between TRPM channels is quite low. It has already been pointed out elsewhere that the PI(4,5)P
2 binding site of TRPM8 is not conserved in other TRPM channels [
15]. In the pre-S1 region of TRPM5, the basic amino acid residues which were suggested to be necessary for the interaction with PI(4,5)P
2 in TRPM4 and TRPM8 are not conserved. That might be a reason for the negligible effect of PI(4,5)P
2 on rTRPM5. Conversely, based on the finding that the effect of PI(4,5)P
2 on rTRPM5 was negligible, the comparisons of amino acid sequences or structures between TRPM4 and TRPM5 may lead to revealing of the PI(4,5)P
2 binding site of TRPM4.
In this study, we did not examine the relationship between the activation of TRPM4 by Ca
2+ and the regulation by PI(4,5)P
2. In this study and elsewhere [
11], the amino acid residues forming the Ca
2+-binding site of TRPM4 have been revealed. Therefore, by analyzing PI(4,5)P
2-affinities of their mutants and also the effects of PI(4,5)P
2 on the maximum currents of the mutants, the molecular understanding of the relationship between the activation by Ca
2+ and the regulation by PI(4,5)P
2 will be expanded.
It is difficult to state clearly the physiological meaning of the difference in the effect of PI(4,5)P
2 on TRPM4 and TRPM5. That is because even the physiological meaning of the interaction with PI(4,5)P
2 in TRPM4 in native cells has not yet been clearly explained as far as we know, although it is certain that PI(4,5)P
2 is necessary for the high activity of TRPM4. However, PI(4,5)P
2-insensitivity of TRPM5 might be advantageous for its function in type II taste cells where TRPM5 is natively expressed. In the taste cells, TRPM5 is activated by Ca
2+i, which is released from the endoplasmic reticulum by inositol trisphosphate (IP
3) signaling, and the opening of TRPM5 depolarizes the membrane potential, resulting in release of transmitters [
18]. The IP
3 is produced by hydrolysis of PI(4,5)P
2, which is mediated by phospholipase C (PLC), when tastants bind to G-protein coupled taste receptors. Therefore, due to the low dependency of TRPM5 on PI(4,5)P
2, the Ca
2+-sensitivity of TRPM5 might not be affected by the reduction of PI(4,5)P
2 concentration in the plasma membrane, which might occur due to the PLC-mediated hydrolysis of PI(4,5)P
2. However, in order to prove this theory, it needs to be demonstrated that the PI(4,5)P
2 concentration in the plasma membrane around TRPM5 is actually reduced after the stimulation by tastants.
In conclusion, this study revealed the following three major findings regarding the functional mechanisms of TRPM4 and TRPM5: (1) that both Ca2+-binding sites of TRPM4 and TRPM5 are formed by the same four amino acid residues in S2 and S3; (2) the glutamate in the TRP domain is also necessary for their normal Ca2+-sensitivities; (3) and finally, the regulation of their Ca2+-sensitivities by PI(4,5)P2 may be restricted to TRPM4. These findings based on the functional analyses will be beneficial for the further understanding of the structure-function relationships of TRPM4 and TRPM5 even after the three-dimensional structure of TRPM5 is unveiled.
4. Materials and Methods
4.1. Animal Ethics Approval
Animal experiments were performed in accordance with guidelines and protocols approved by the Institutional Animal Care and Use Committee, Hokkaido University (The project identification code is #13-0212, approved on 27 January 2014).
4.2. Molecular Cloning and Site-Directed Mutagenesis
A male BN/SsNSlc rat (five weeks old) was euthanized by CO
2 inhalation. RNA was extracted from the tongue epithelia containing circumvallate papillae and foliate papillae using NucleoSpin RNA II (Takara Bio, Otsu, Japan). Complementary DNA (cDNA) was synthesized using PrimeScript II Reverse Transcriptase (Takara Bio) and an oligo dT primer. The full-length of the open reading frame of rat
Trpm5 cDNA was amplified by PCR using a high fidelity polymerase (PrimeSTAR GXL, Takara Bio) and the following primers: 5’-GCA AGG GAG GAA CAC AGC CTG AAG TAG-3’ (Forward primer in 5’ UTR) and 5’-GAC GTA AGT AGC CCC ATC CAG GCA G-3’ (Reverse primer in 3’ UTR). These primers were designed based on the predicted
rTrpm5 mRNA sequences (Genbank #XM_017589501.1 and XM_008760160.2) which were derived from a genomic sequence. The PCR product, amplified
rTrpm5 cDNA, was cloned in pGEM T-Easy vector (Promega, Fitchburg, WI, USA) and sequenced. The
rTrpm5 cDNA (Genbank #LC469323) which contained no PCR errors was subcloned into a bicistronic expression vector, pIRES2-EGFP (Takara Bio), from which both EGFP and a protein encoded by the inserted gene can be expressed. The pIRES2-EGFP vectors containing
rTrpm4 cDNA had been made in the previous study [
6]. Site-directed mutagenesis of
rTrpm5 and
rTrpm4 cDNA in pIRES2-EGFP was accomplished using the PrimeSTAR Mutagenesis Basal kit (Takara Bio). The mutations were verified by sequencing.
4.3. Cell Culture and Transfection
HEK 293T cells were obtained from the RIKEN BioResource Research Center through the National Bio-Resource Project of the MEXT, Tokyo, Japan. HEK 293T cells were cultured in DMEM (Dulbecco’s modified Eagle’s medium; Sigma-aldrich, St. Louis, MO, USA) supplemented with 10% FBS (Thermo Fisher Scientific, Waltham, MA, USA) and penicillin/streptomycin (100 U/mL and 100 μg/mL, respectively, Thermo Fisher Scientific) at 37 °C in a 5% CO2 incubator. Cells were transiently transfected with plasmids using TransIT-293 Transfection Reagent (Takara Bio). Two days after the transfection, the cells were used for Western blot analyses and biotinylation assays. For patch clamp experiments, the cells were plated on coverslips the following day of the transfection. After at least 3 h of further culturing, whole-cell patch-clamp recordings were made from EGFP-positive cells. Inside-out patch recordings were performed on the following day of the cell plating on coverslips.
4.4. Electrophysiology
Patch-clamp recordings were performed as described previously [
6,
19]. Briefly, the conditions for inside-out current recordings are as follows. The pipette solution for the inside-out recordings was composed of 145 mM NaCl, 1 mM CaCl
2, 1 mM MgCl
2 and 10 mM HEPES (2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid) (pH = 7.4, with NaOH). Bath solutions contained 145 mM NaCl and appropriate concentrations of CaCl
2 or 5 mM EGTA (Ethylene glycol-bis(2-aminoethylether)-
N,
N,
N′,
N′-tetraacetic acid) for a Ca
2+-free solution. Macroscopic currents under the inside-out configuration were recorded using the ramp pulses. The holding potential was −60 mV (the intracellular side is negative), and the ramp pulses from −100 to +100 mV with durations of 400 ms were applied every 2 s. The currents were filtered at 1 kHz and sampled at 5 kHz. Water soluble phosphoinositides, diC8-PI(4,5)P
2 and diC8-PI(3,4,5)P
3, were obtained from Cellsignals (Columbus, OH, USA) and dissolved in the bath solution.
The whole-cell patch clamp recordings for rTRPM5 current measurements were performed under the conditions described below. The pipette solution was composed of 130 mM CsCl, 10 mM BAPTA (1,2-bis(o-aminophenoxy)ethane-N,N,N’,N’-tetraacetic acid), 4 mM ATP disodium salt, 10 mM HEPES (pH = 7.3, adjusted with CsOH) and appropriate amounts of MgCl2 and CaCl2 in order to make solutions which contained 10−3 M free Mg2+ and 0–10−4 M free Ca2+. The free Mg2+ and Ca2+ concentrations were calculated using the CaBuf software (Guy Droogmans, KU Leuven, Leuven, Belgium). The pipette solutions containing 1 and 3 mM CaCl2 were made without BAPTA. The extracellular solution was composed of 145 mM NaCl, 2 mM MgCl2 and 10 mM HEPES (pH = 7.4, with NaOH). In most experiments, the holding potential was −60 mV and the ramp pulses from −100 to +100 mV with durations of 400 ms were applied every 2 s. The currents were filtered at 1 kHz and sampled at 5 kHz. One minute after the start of the whole-cell recording, 50 μM ZnCl2 was applied through the bath solution. Current densities were calculated by dividing the current amplitudes by the cell capacitances (pA/pF). As an indication of rTRPM5 current densities, the Zn2+-sensitive current densities were calculated by subtracting the current densities in the presence of Zn2+ from the peak current densities. All experiments were conducted at room temperature.
4.5. Biotinylation Assay and Western Blotting
The transfected HEK293T cells were washed twice with ice cold D-PBS-CM (Dulbecco’s phosphate buffered saline with 1 mM MgCl2 and 0.1 mM CaCl2) and incubated 15 min with 1.0 mg/ml EZ-link sulfo-NHS-s-s-biotin (Thermo Fisher Scientific) in cold biotinylation buffer (10 mM triethanolamine, 2 mM CaCl2, 150 mM NaCl, pH 9.0, adjusted with HCl) with gentle agitation at 4 °C. The cells were washed twice with a quenching buffer (100 mM glycine in D-PBS-CM), then rinsed once with D-PBS, scraped in cold D-PBS and pelleted at 290×g for 1 min at 4 °C. The cells were lysed in a lysis buffer (150 mM NaCl, 50 mM HEPES, 1.0 mM EGTA, 1.5 mM MgCl2, pH = 7.4, 1.0% Triton X-100, 10% glycerol and a protease inhibitor cocktail (P8340, Sigma-Aldrich)). The lysates were sonicated using Bioruptor (BM Equipment, Tokyo, Japan) or Ultrasonic homogenizer (UH50, SMT, Tokyo Japan). The lysates were centrifuged for 10 min at 14,000× g at 4 °C. The protein concentrations of the lysates were measured by using DC Protein Assay (Bio-Rad, Hercules, CA, USA) and adjusted to 100 μg in 150 μL with the lysis buffer. Streptavidin-agarose beads (Sigma-Aldrich) were added to the protein extracts. The mixtures were rotated at 20 rpm for 2 h at 4 °C. The beads were pelleted by brief centrifugation (650× g for 1 min). The supernatants were taken as the unbound intracellular fractions, mixed with one fourth volume of 4× Laemmli sample buffer containing 10% β-mercaptoethanol, and heated at 50 °C for 15 min. The beads were washed three times with D-PBS containing 0.5% Triton X-100. The biotinylated proteins were eluted from the beads by mixing the beads with 4× Laemmli sample buffer containing 10% β-mercaptoethanol and heating the mixtures at 50 °C for 15 min (Surface fractions).
The intracellular fractions (10 μg protein = 10% of total protein used for one sample) and the surface fractions were separated by SDS-PAGE and transferred to a PVDF membrane. After incubation in blocking buffer containing 3% skim milk, the blots were treated with the diluted anti-TRPM5 rabbit antibody (1:2000, #ACC-045, Alomone labs, Jerusalem, Israel) or anti-GFP rabbit antibody (1:500, #598, MBL, Nagoya, Japan) and then with horseradish peroxidase-conjugated anti-rabbit IgG antibody (1:1000, GE Healthcare, Buckinghamshire, UK) as the secondary antibody. The chemiluminescent signals were produced by using Immobilon Forte (Merk, Burlington, MA, USA) and detected by a single-lens reflex camera (EOS kiss x7, Canon, Tokyo, Japan).
4.6. Data Analysis
Concentration-response curves were obtained by fitting the averages of current amplitudes or current densities with the Hill equation:
where
Imax is the maximal current amplitude or the maximal current density,
C is the concentration of Ca
2+,
EC50 is the half maximal effective concentration and
n is the Hill coefficient.
All data are expressed as means ± S.E. The statistical analyses were performed using Student’s t test, Welch’s t test or Dunnett’s test as appropriate. A value of p < 0.05 was considered significant.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}