Exploiting Gene Expression Profiles for the Automated Prediction of Connectivity between Brain Regions



Abstract
:
1. Introduction
2. Results and Discussion
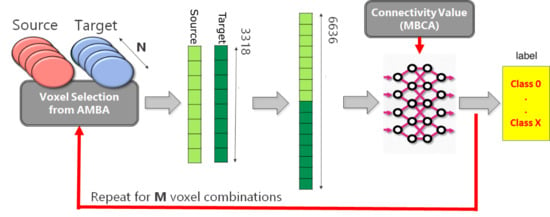
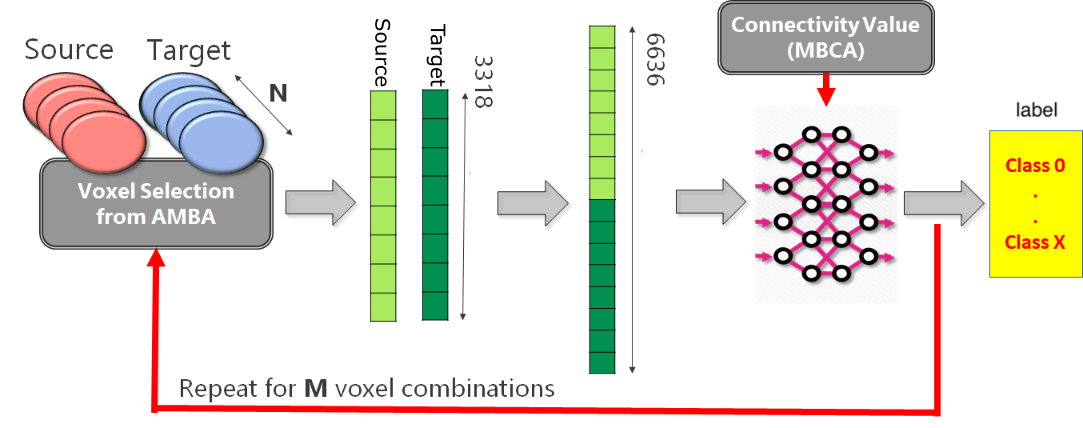
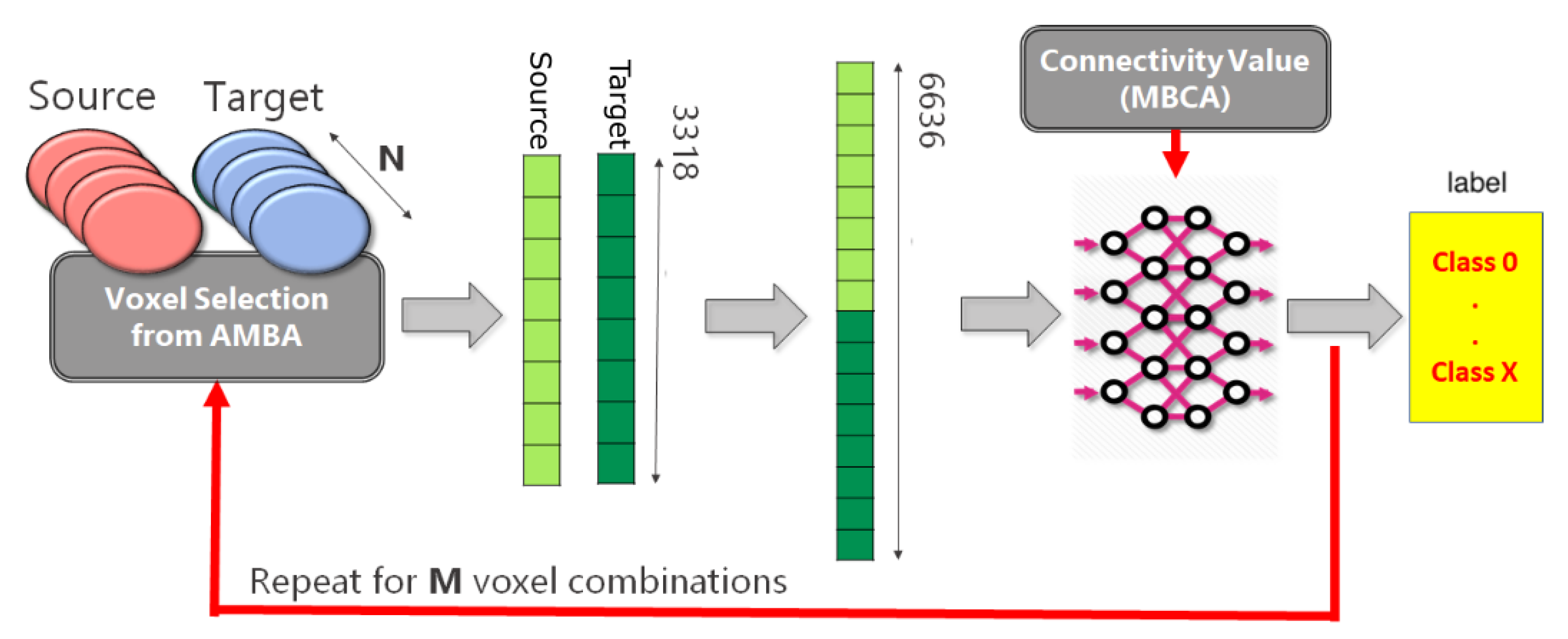
2.1. Analysis Pipeline
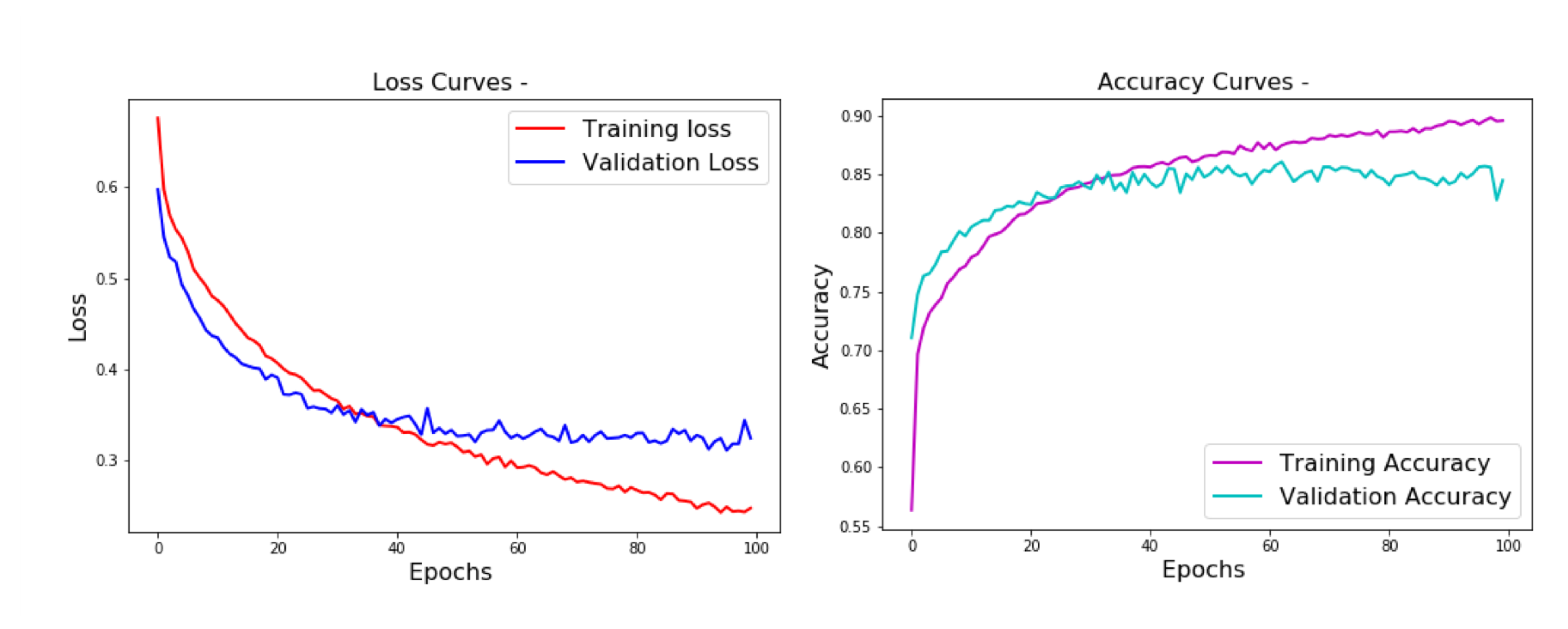
2.2. Classification Performance
2.2.1. Multi-Class Classification Task
- Class label “0” (unconnected): 5000 Source-Target vectors with connectivity equal to 0
- Class label “1” (weekly connected): 5000 Source-Target vectors with connectivity values in the range [0.006, 0.1)
- Class label “2” (strongly connected): 4583 Source-Target vectors with connectivity > 0.1
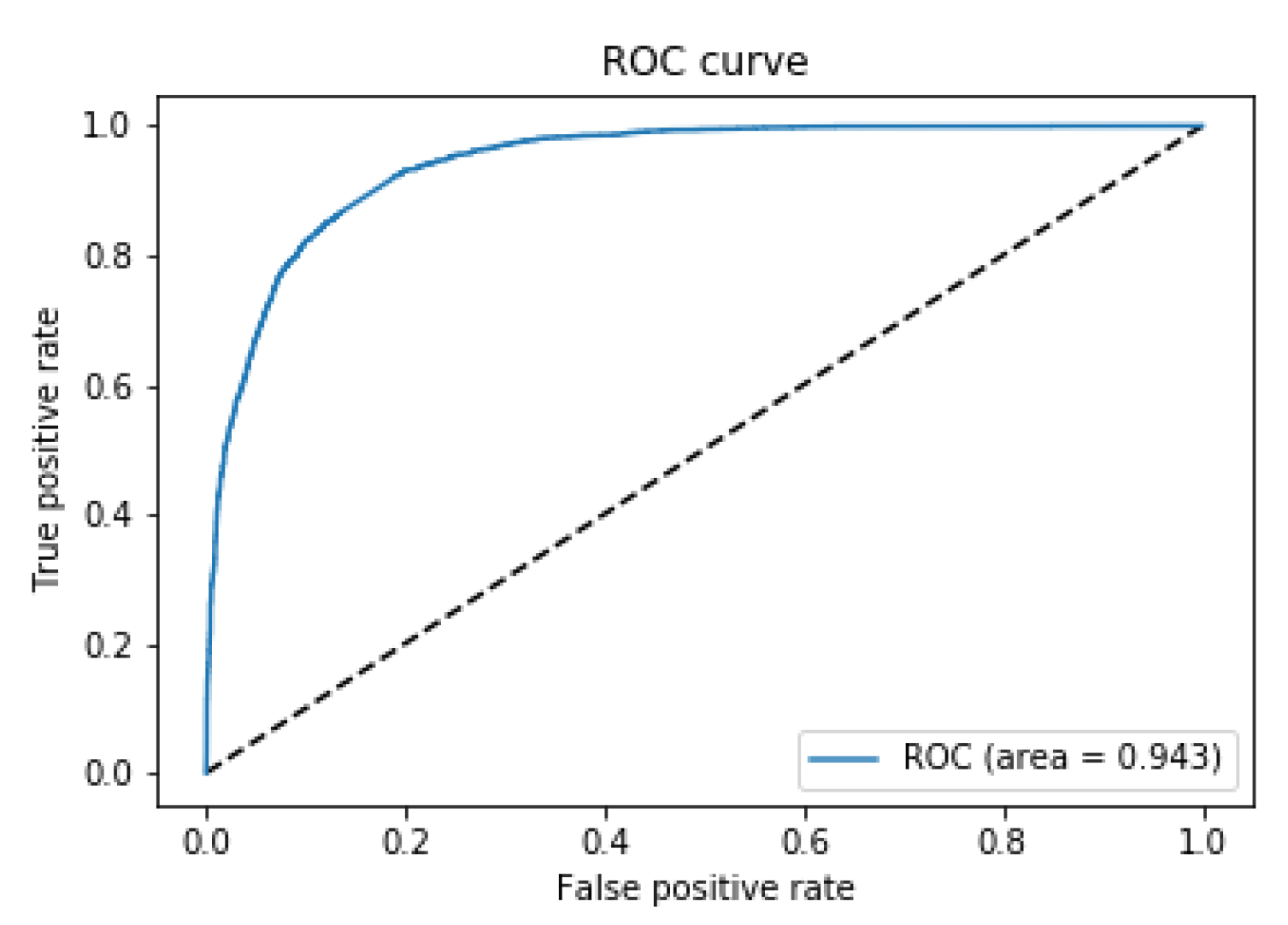
2.2.2. Binary Classification Task
- Class label “0” (unconnected): 20,000 Source-Target vectors with connectivity values equal to 0. This sub-set is composed of gene expression vectors obtained selecting only unconnected Source-Target region pairs.
- Class label “1” (connected): 17,136 Source-Target vectors with connectivity values > 0.006.
3. Materials
Allen Mouse Brain Atlas
4. Methods
- download of grid data from the available data sources;
- processing of the raw grid data to integrate the gene expression and the connectivity information;
- generation of a full and coherent dataset of Source-Target gene expression vectors and corresponding connectivity labels, ready to be cropped into training, validation and test sets for the MLP.
4.1. Download of Grid Data
4.1.1. Gene Expression
4.1.2. Allen Connectivity
4.1.3. Structural Annotation File
4.1.4. Brain Architecture Management System (BAMS)
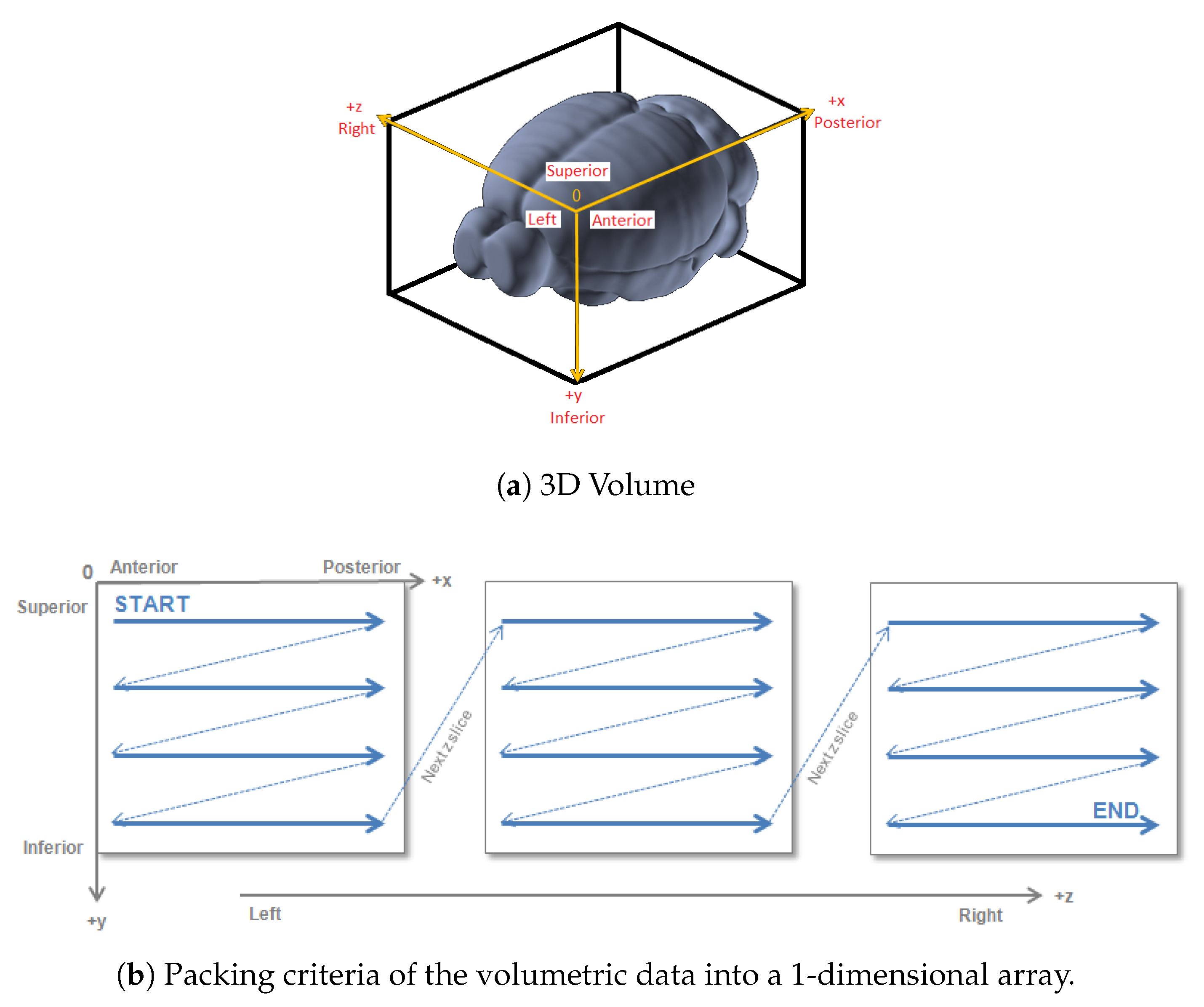
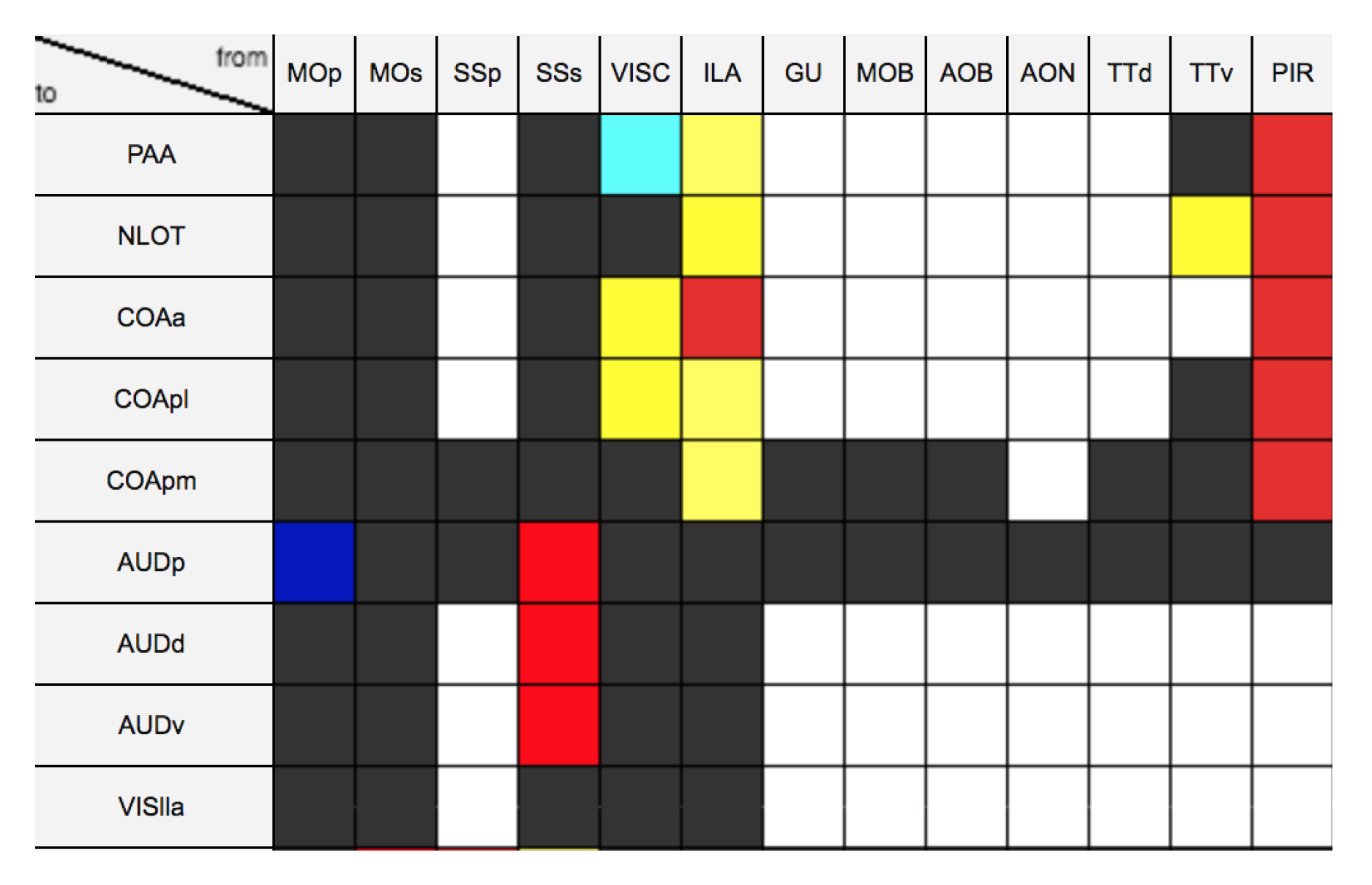
4.2. Generation Source-Target Vectors and Corresponding Connectivity Labels
- Table voxID2Annotation carries the spatial information, and contains the voxel ID and corresponding brain structure annotation.
- Table voxID2GenExpr was obtained by filtering out the voxels with gene expression level value equal to 0. It is made of columns reporting gene expression value, voxel ID and gene ID, respectively.
- Table injection2regionID was obtained by grouping all the voxels by Source and injection ID. Hence, it reports the Source region ID for each injection.
- Table injection2target was obtained by grouping the connection values of each voxels by the Target ID. More specifically, all the voxels belonging to the same Target region were aggregated by the median of the values associated to each of these voxels. Then, the final table is composed of three columns: injection ID, median of the values obtained for a specific Target ID and its annotation ID, respectively.
- for each Source-Target pair, M voxels belonging to the source region and M voxels to the target regions are selected on the expression gene annotation.
- for each selected voxel, a vector composed of 3318 elements is generated, where each element corresponds to the expression level of a specific gene. Hence, M vectors representing the gene expression profile of the Source and M vectors representing the gene expression profile of the Target are obtained.
- A dataset is created by selecting P combinations among all possible Source-Target voxel combinations. More specifically, the gene expression vector corresponding to the Source voxel is concatenated with the gene expression vector corresponding to the Target voxel. Hence, the obtained dataset will be made of P vectors.
- In the end, a unique categorical label representing the Source-Target connectivity is assigned to each combination.
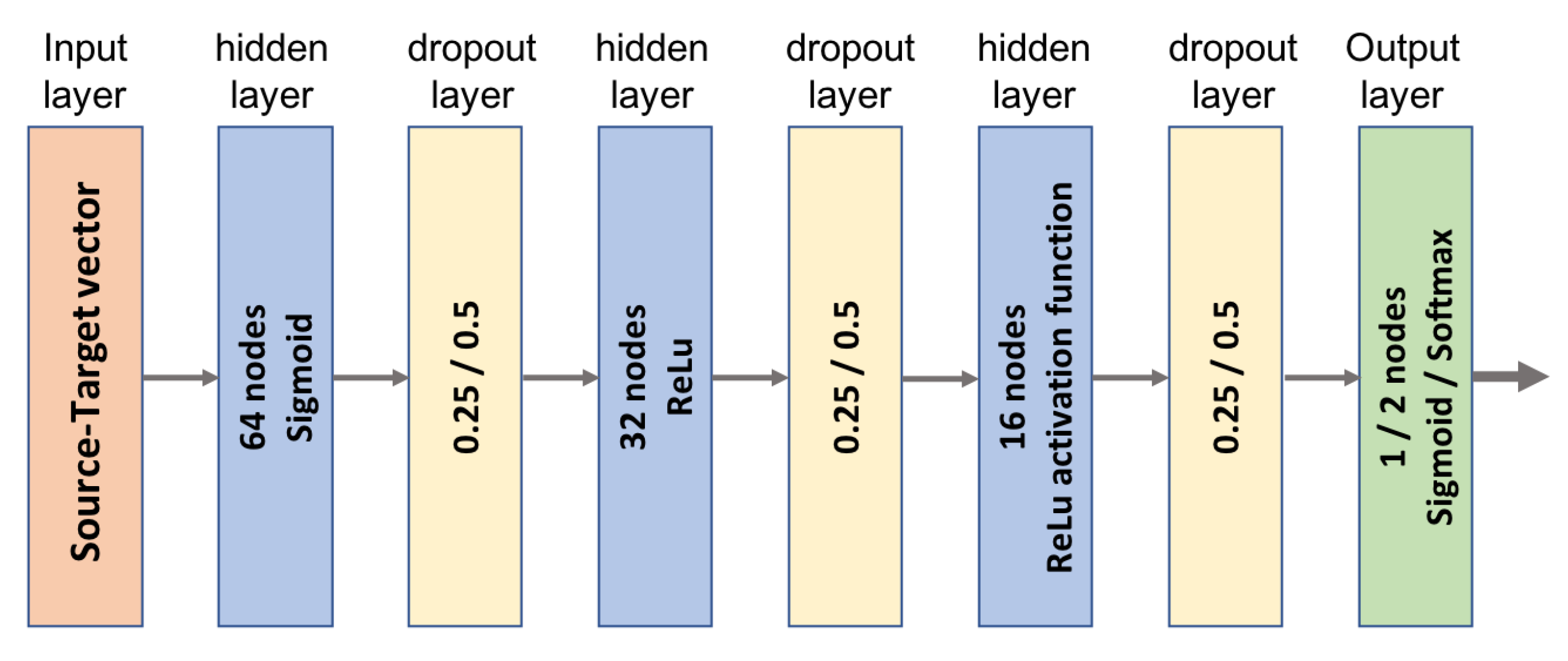
4.3. MLP Predictive Model
5. Conclusions and Future Work
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ISH | In-Situ Hybridization |
| AMBA | Allen Mouse Brain Atlas |
| BAMS | Brain Architecture Management System |
| MBCA | Mouse Brain Connectivity Atlas |
| MLP | Multilayer Perceptron |
References
- Kandel, E.R.; Schwartz, J.H.; Jessell, T.M.; Siegelbaum, S.; Hudspeth, A. Principles of Neural Science; McGraw-Hill: New York, NY, USA, 2000; Volume 4. [Google Scholar]
- Bargmann, C.I.; Marder, E. From the connectome to brain function. Nat. Methods 2013, 10, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Batista-García-Ramó, K.; Fernández-Verdecia, C. What We Know About the Brain Structure–Function Relationship. Behav. Sci. 2018, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Sporns, O.; Tononi, G.; Kötter, R. The human connectome: A structural description of the human brain. PLoS Comput. Biol. 2005, 1, e42. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, A.; Dror, G.; Meilijson, I.; Ruppin, E. Gene expression of Caenorhabditis elegans neurons carries information on their synaptic connectivity. PLoS Comput. Biol. 2006, 2, e167. [Google Scholar] [CrossRef] [PubMed]
- Sieburth, D.; Ch’ng, Q.; Dybbs, M.; Tavazoie, M.; Kennedy, S.; Wang, D.; Dupuy, D.; Rual, J.F.; Hill, D.E.; Vidal, M.; et al. Systematic analysis of genes required for synapse structure and function. Nature 2005, 436, 510–517. [Google Scholar] [CrossRef]
- Levsky, J.M.; Singer, R.H. Gene expression and the myth of the average cell. Trends Cell Biol. 2003, 13, 4–6. [Google Scholar] [PubMed]
- Ke, R.; Mignardi, M.; Hauling, T.; Nilsson, M. Fourth Generation of Next-Generation Sequencing Technologies: Promise and Consequences. Hum. Mutat. 2016, 37, 1363–1367. [Google Scholar] [CrossRef]
- Kuan, L.; Li, Y.; Lau, C.; Feng, D.; Bernard, A.; Sunkin, S.M.; Zeng, H.; Dang, C.; Hawrylycz, M.; Ng, L. Neuroinformatics of the allen mouse brain connectivity atlas. Methods 2015, 73, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Bota, M.; Swanson, L.W. BAMS neuroanatomical ontology: Design and implementation. Front. Neuroinform. 2008, 2, 2. [Google Scholar] [CrossRef]
- Fakhry, A.; Zeng, T.; Peng, H.; Ji, S. Global analysis of gene expression and projection target correlations in the mouse brain. Brain Inform. 2015, 2, 107–117. [Google Scholar] [CrossRef] [Green Version]
- French, L.; Pavlidis, P. Relationships between gene expression and brain wiring in the adult rodent brain. PLoS Comput. Biol. 2011, 7, e1001049. [Google Scholar] [CrossRef]
- Ji, S.; Fakhry, A.; Deng, H. Integrative analysis of the connectivity and gene expression atlases in the mouse brain. NeuroImage 2014, 84, 245–253. [Google Scholar] [CrossRef]
- Strand, A.D.; Aragaki, A.K.; Baquet, Z.C.; Hodges, A.; Cunningham, P.; Holmans, P.; Jones, K.R.; Jones, L.; Kooperberg, C.; Olson, J.M. Conservation of regional gene expression in mouse and human brain. PLoS Genet. 2007, 3, e59. [Google Scholar] [CrossRef]
- Johnson, T.S.; Abrams, Z.B.; Helm, B.R.; Neidecker, P.; Machiraju, R.; Zhang, Y.; Huang, K.; Zhang, J. Integration of Mouse and Human Single-cell RNA Sequencing Infers Spatial Cell-type Composition in Human Brains. bioRxiv 2019. [Google Scholar] [CrossRef]
- Fulcher, B.D.; Fornito, A. A transcriptional signature of hub connectivity in the mouse connectome. Proc. Natl. Acad. Sci. USA 2016, 113, 1435–1440. [Google Scholar] [CrossRef]
- Roy, M.; Sorokina, O.; McLean, C.; Tapia-González, S.; DeFelipe, J.; Armstrong, J.; Grant, S. Regional diversity in the postsynaptic proteome of the mouse brain. Proteomes 2018, 6, 31. [Google Scholar] [CrossRef]
- Richiardi, J.; Altmann, A.; Milazzo, A.C.; Chang, C.; Chakravarty, M.M.; Banaschewski, T.; Barker, G.J.; Bokde, A.L.; Bromberg, U.; Büchel, C.; et al. Correlated gene expression supports synchronous activity in brain networks. Science 2015, 348, 1241–1244. [Google Scholar] [CrossRef] [Green Version]
- Anderson, K.M.; Krienen, F.M.; Choi, E.Y.; Reinen, J.M.; Yeo, B.T.; Holmes, A.J. Gene expression links functional networks across cortex and striatum. Nat. Commun. 2018, 9, 1428. [Google Scholar] [CrossRef]
- Ganglberger, F.; Kaczanowska, J.; Penninger, J.M.; Hess, A.; Buehler, K.; Haubensak, W. Predicting functional neuroanatomical maps from fusing brain networks with genetic information. NeuroImage 2018, 170, 113–120. [Google Scholar] [CrossRef]
- Fakhry, A.; Ji, S. High-resolution prediction of mouse brain connectivity using gene expression patterns. Methods 2015, 73, 71–78. [Google Scholar] [CrossRef]
- John Lu, Z. The elements of statistical learning: Data mining, inference, and prediction. J. R. Stat. Soc. Ser. A Stat. Soc. 2010, 173, 693–694. [Google Scholar] [CrossRef]
- Lein, E.S.; Hawrylycz, M.J.; Ao, N.; Ayres, M.; Bensinger, A.; Bernard, A.; Boe, A.F.; Boguski, M.S.; Brockway, K.S.; Byrnes, E.J.; et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007, 445, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Dozat, T. Incorporating nesterov momentum into adam. In Proceedings of the 4th International Conference on Learning Representations, San Juan, PR, USA, 2–4 May 2016. [Google Scholar]
- Powers, D.M. Evaluation: From precision, recall and F-measure to ROC, informedness, markedness and correlation. J. Mach. Learn. Technol. 2011, 2, 37–63. [Google Scholar]
- Tautz, D.; Pfeifle, C. A non-radioactive in situ hybridization method for the localization of specific RNAs in Drosophila embryos reveals translational control of the segmentation gene hunchback. Chromosoma 1989, 98, 81–85. [Google Scholar] [CrossRef]
- Oh, S.W.; Harris, J.A.; Ng, L.; Winslow, B.; Cain, N.; Mihalas, S.; Wang, Q.; Lau, C.; Kuan, L.; Henry, A.M.; et al. A mesoscale connectome of the mouse brain. Nature 2014, 508, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Thiel, K.; Wiswedel, B. KNIME-the Konstanz information miner: Version 2.0 and beyond. ACM SIGKDD Explor. Newsl. 2009, 11, 26–31. [Google Scholar] [CrossRef]
- Reinert, K.; Dadi, T.H.; Ehrhardt, M.; Hauswedell, H.; Mehringer, S.; Rahn, R.; Kim, J.; Pockrandt, C.; Winkler, J.; Siragusa, E.; et al. The SeqAn C++ template library for efficient sequence analysis: A resource for programmers. J. Biotechnol. 2017, 261, 157–168. [Google Scholar] [CrossRef]
- Allen-Brain-Institute. Allen Institute for Brain Science Documentation; Allen-Brain-Institute: Seattle, WA, USA, 2011. [Google Scholar]
- Allen-Brain-Institute. Allen Institute for Brain Science Documentation; Allen-Brain-Institute: Seattle, WA, USA, 2014. [Google Scholar]
- Agarwal, N.; Xu, X.; Gopi, M. Geometry processing of conventionally produced mouse brain slice images. J. Neurosci. Methods 2018, 306, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Allen-Brain-Institute. Allen Mouse Brain Atlas. 2018. Available online: http://mouse.brain-map.org/static/brainexplorer/ (accessed on 4 October 2018).
- Allen-Brain-Institute. Allen Institute for Brain Science Documentation; Allen-Brain-Institute: Seattle, WA, USA, 2017. [Google Scholar]
- The University of Southern California. The BAMS Rat Connectome Project. 2013. Available online: https://bams2.bams1.org/connections/grid/80/ (accessed on 4 October 2018).
- Hinton, G.E.; Srivastava, N.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R.R. Improving neural networks by preventing co-adaptation of feature detectors. arXiv 2012, arXiv:1207.0580. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Epochs | Learning Rate (Lr) | Decay | Beta1 | Beta9 | Loss Function | Batch Size |
|---|---|---|---|---|---|---|
| 200 | 0.002 | 0.004 | 0.9 | 0.999 | categorical cross entropy | 6 |
| Predicted Class | ||||
|---|---|---|---|---|
| Unconnected | Weakly Connected | Strongly Connected | ||
| Unconnected | 75% | 23% | 2% | |
| Real Class | Weakly Connected | 13% | 81% | 6% |
| Strongly Connected | 2% | 31% | 67% | |
| Quality Metrics | |||||
|---|---|---|---|---|---|
| Recall | Precision | F1_Score | Accuracy | ||
| Unconnected | 75% | 86% | 81% | ||
| Class | Weakly Connected | 81% | 66% | 73% | 76% |
| Strongly Connected | 67% | 83% | 74% | ||
| Epochs | Learning Rate (Lr) | Decay | Beta1 | Beta9 | Loss Function | Batch Size |
|---|---|---|---|---|---|---|
| 100 | 0.002 | 0.004 | 0.9 | 0.999 | binary cross entropy | 32 |
| Quality Metrics | ||||
|---|---|---|---|---|
| Precision | Recall | F1_Score | Accuracy | |
| Unconnected | 94% | 75% | 84% | 85% |
| Connected | 77% | 95% | 85% | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roberti, I.; Lovino, M.; Di Cataldo, S.; Ficarra, E.; Urgese, G. Exploiting Gene Expression Profiles for the Automated Prediction of Connectivity between Brain Regions. Int. J. Mol. Sci. 2019, 20, 2035. https://doi.org/10.3390/ijms20082035
Roberti I, Lovino M, Di Cataldo S, Ficarra E, Urgese G. Exploiting Gene Expression Profiles for the Automated Prediction of Connectivity between Brain Regions. International Journal of Molecular Sciences. 2019; 20(8):2035. https://doi.org/10.3390/ijms20082035
Chicago/Turabian StyleRoberti, Ilaria, Marta Lovino, Santa Di Cataldo, Elisa Ficarra, and Gianvito Urgese. 2019. "Exploiting Gene Expression Profiles for the Automated Prediction of Connectivity between Brain Regions" International Journal of Molecular Sciences 20, no. 8: 2035. https://doi.org/10.3390/ijms20082035
APA StyleRoberti, I., Lovino, M., Di Cataldo, S., Ficarra, E., & Urgese, G. (2019). Exploiting Gene Expression Profiles for the Automated Prediction of Connectivity between Brain Regions. International Journal of Molecular Sciences, 20(8), 2035. https://doi.org/10.3390/ijms20082035