Down-Regulation of MHC Class I Expression in Human Keratinocytes Using Viral Vectors Containing US11 Gene of Human Cytomegalovirus and Cultivation on Bovine Collagen-Elastin Matrix (Matriderm®): Potential Approach for an Immune-Privileged Skin Substitute



Abstract
:
1. Introduction
2. Results
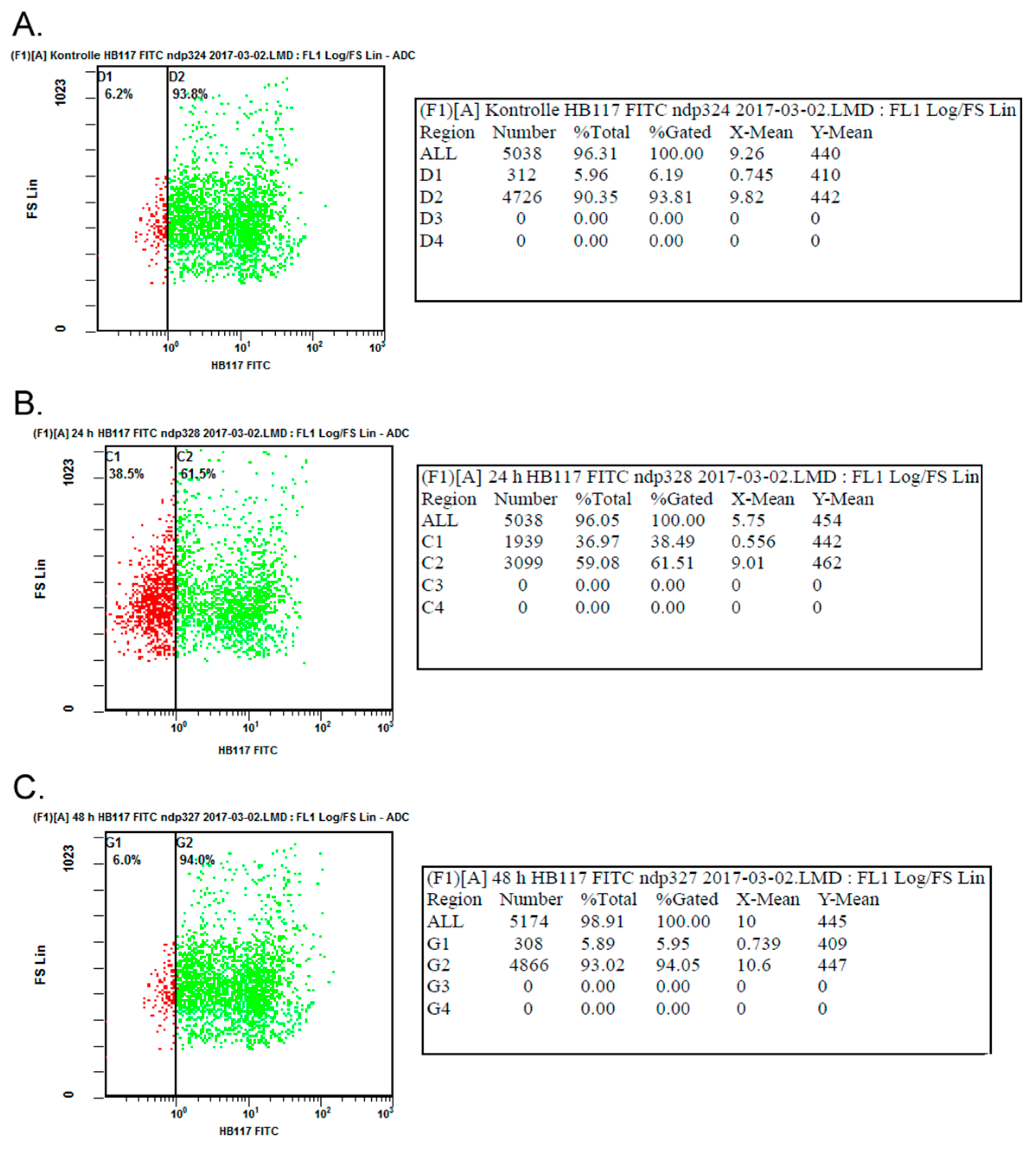
2.1. Transfection with US11 Vectors Induced Transient MHC Class I Down-Regulation
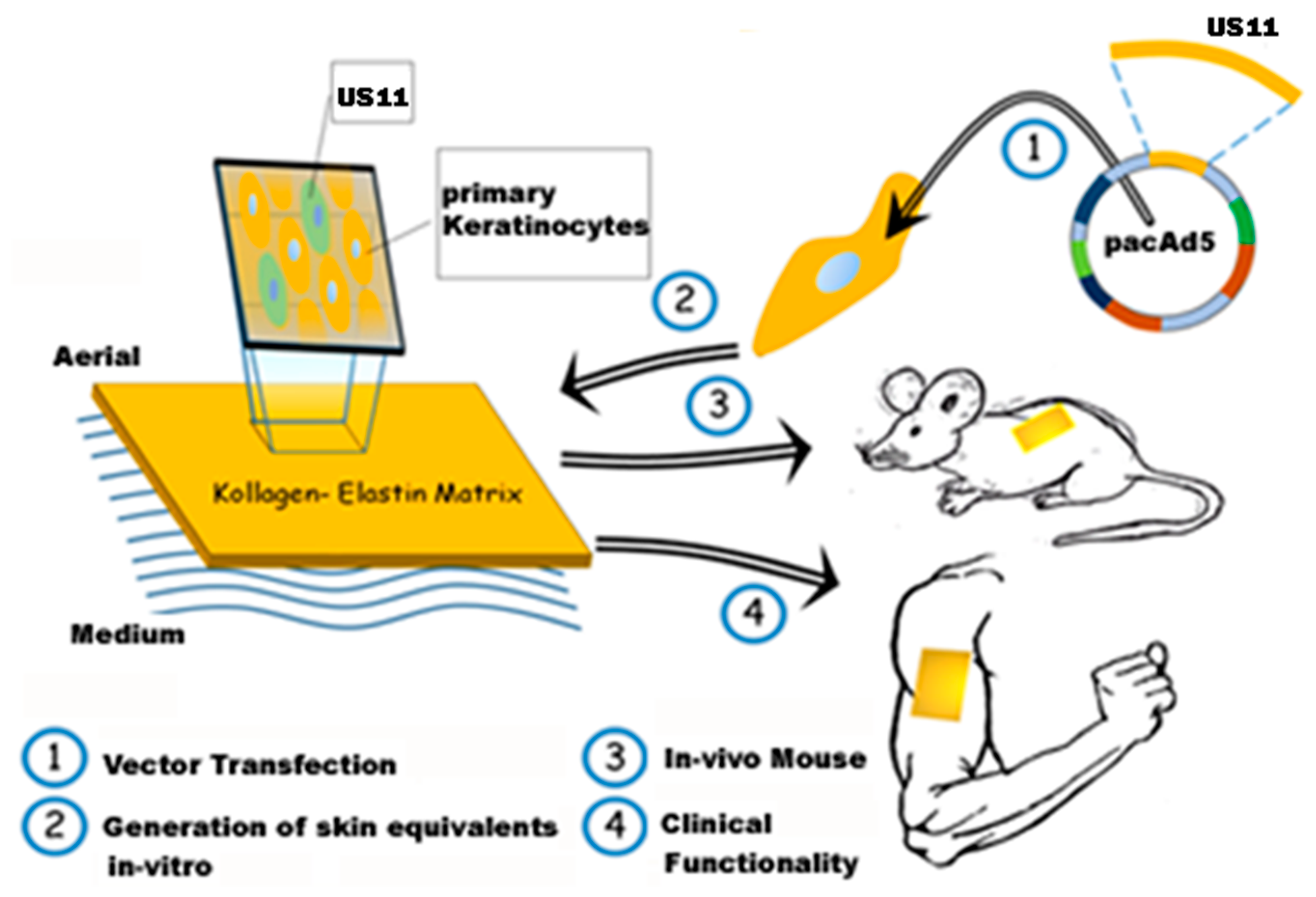
2.2. Seeding of Matriderm® Constructs and Evaluation of Long-Term Cell Survival
2.3. Histological Patterns and Cell Morphology after Seeding of Matriderm® Constructs
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Human Leukocyte Antigen Typing of Primary Keratinocytes
4.3. Vector Transfection
4.4. Immunofluorescence
4.5. Western Blotting
4.6. Flow Cytometry
4.7. Matriderm® Constructs
4.8. Live–Dead Assay
4.9. Histology
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MHC | Major Histocompatibility Complex |
| US | Unique Short Glycoprotein |
| TAP | Transporter-Associated-With-Antigen-Processing |
| β2-M | β2-Microglobulin |
| HLA | Human Leukocyte Antigen |
| HCMV | Human Cytomegalovirus |
| CD | Clusters of Differentiation |
| NK | Natural Killer Cell |
| eGFP | Enhanced Green Fluorescent Protein |
| DAPI | 4′,6-Diamidino-2-Phenylindole |
| FITC | Fluorescein Isothiocyanate |
| LMD | File format provided by Beckman Coulter (Krefeld, Germany) |
| FS | Forward scatter |
| H&E | Haematoxyline and Eosine |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| DMEM | Dulbecco’s Modified Eagle Medium |
| PBS | Phosphate Buffered Saline |
| RIPA | Radioimmunoprecipitation Assay Buffer |
| EDTA | Ethylene Diamine Tetraacetic Acid |
| PMSF | Phenylmethyl Sulfonyl Fluoride |
| PVDF | Polyvinylidene Fluoride |
| FBS | Fetal Bovine Serum |
| BSA | Bovine Serum Albumin |
References
- Welsch, U. Haut. In Sobotta Lehrbuch Histologie: Zytologie, Histologie, Mikroskopische Anatomie, 2nd ed.; Welsch, U., Ed.; Urban & Fischer Verlag/Elsevier GmbH: München & Jena, Germany, 2006; pp. 549–568. [Google Scholar]
- Vogt, P.M.; Kolokythas, P.; Niederbichler, A.; Knobloch, K.; Reimers, K.; Choi, C.Y. Innovative wound therapy and skin substitutes for burns. Chirurg 2007, 78, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Finnerty, C.C.; Capek, K.D.; Voigt, C.; Hundeshagen, G.; Cambiaso-Daniel, J.; Porter, C.; Sousse, L.E.; El Ayadi, A.; Zapata-Sirvent, R.; Guillory, A.N.; et al. The P50 Research Center in Perioperative Sciences. J. Trauma Acute Care Surg. 2017, 83, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Warden, G.D.; Saffle, J.R.; Kravitz, M. A two-stage technique for excision and grafting of burn wounds. J. Trauma 1982, 22, 98–103. [Google Scholar] [CrossRef]
- Alexander, J.W.; MacMillan, B.G.; Law, E.; Kittur, D.S. Treatment of severe burns with widely meshed skin autograft and meshed skin allograft overlay. J. Trauma 1981, 21, 433–438. [Google Scholar]
- Sood, R.; Roggy, D.; Zieger, M.; Balledux, J.; Chaudhari, S.; Koumanis, D.J.; Mir, H.S.; Cohen, A.; Knipe, C.; Gabehart, K.; et al. Cultured Epithelial Autografts for Coverage of Large Burn Wounds in Eighty-Eight Patients: The Indiana University Experience. J. Burn Care Res. 2010, 31, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Gravante, G.; Di Fede, M.C.; Araco, A.; Grimaldi, M.; De Angelis, B.; Arpino, A.; Cervelli, V.; Montone, A. A randomized trial comparing ReCell system of epidermal cells delivery versus classic skin grafts for the treatment of deep partial thickness burns. Burns 2007, 33, 966–972. [Google Scholar] [CrossRef]
- Fletcher, J.L.; Caterson, E.J.; Hale, R.G.; Cancio, L.C.; Renz, E.M.; Chan, R.K. Characterization of Skin Allograft Use in Thermal Injury. J. Burn Care Res. 2013, 34, 168–175. [Google Scholar] [CrossRef]
- Cronin, H.; Goldstein, G. Biologic Skin Substitutes and Their Applications in Dermatology. Dermatol. Surg. 2013, 39, 30–34. [Google Scholar] [CrossRef]
- Wood, F.M.; Giles, N.; Stevenson, A.; Rea, S.; Fear, M. Characterisation of the cell suspension harvested from the dermal epidermal junction using a ReCell® kit. Burns 2012, 38, 44–51. [Google Scholar] [CrossRef]
- Boyce, S.T.; Lalley, A.L. Tissue engineering of skin and regenerative medicine for wound care. Burn. Trauma 2018, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Felder, J.M.; Hechenbleikner, E.; Jordan, M.; Jeng, J. Increasing the Options for Management of Large and Complex Chronic Wounds With a Scalable, Closed-System Dressing for Maggot Therapy. J. Burn Care Res. 2012, 33, e170–e176. [Google Scholar] [CrossRef] [PubMed]
- Atherton, D.D.; Tang, R.; Jones, I.; Jawad, M. Early Excision and Application of Matriderm with Simultaneous Autologous Skin Grafting in Facial Burns. Plast. Reconstr. Surg. 2010, 125, 60e–61e. [Google Scholar] [CrossRef] [PubMed]
- Hartmann-Fritsch, F.; Biedermann, T.; Braziulis, E.; Meuli, M.; Reichmann, E. A new model for preclinical testing of dermal substitutes for human skin reconstruction. Pediatr. Surg. Int. 2013, 29, 479–488. [Google Scholar] [CrossRef]
- Killat, J.; Reimers, K.; Choi, C.; Jahn, S.; Vogt, P.; Radtke, C. Cultivation of Keratinocytes and Fibroblasts in a Three-Dimensional Bovine Collagen-Elastin Matrix (Matriderm®) and Application for Full Thickness Wound Coverage in Vivo. Int. J. Mol. Sci. 2013, 14, 14460–14474. [Google Scholar] [CrossRef] [PubMed]
- Salem, H.; Ciba, P.; Rapoport, D.H.; Egana, J.T.; Reithmayer, K.; Kadry, M.; Machens, H.G.; Kruse, C. The influence of pancreas-derived stem cells on scaffold based skin regeneration. Biomaterials 2009, 30, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Keck, M.; Haluza, D.; Selig, H.F.; Jahl, M.; Lumenta, D.B.; Kamolz, L.-P.; Frey, M. Adipose Tissue Engineering. Ann. Plast. Surg. 2011, 67, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Golinski, P.A.; Zöller, N.; Kippenberger, S.; Menke, H.; Bereiter-Hahn, J.; Bernd, A. Development of an engraftable skin equivalent based on matriderm with human keratinocytes and fibroblasts. Handchir. Mikrochir. Plast. Chir. 2009, 41, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Michael, S.; Sorg, H.; Peck, C.-T.; Reimers, K.; Vogt, P.M. The mouse dorsal skin fold chamber as a means for the analysis of tissue engineered skin. Burns 2013, 39, 82–88. [Google Scholar] [CrossRef]
- Böttcher-Haberzeth, S.; Biedermann, T.; Schiestl, C.; Hartmann-Fritsch, F.; Schneider, J.; Reichmann, E.; Meuli, M. Matriderm® 1 mm versus Integra® Single Layer 1.3 mm for one-step closure of full thickness skin defects: A comparative experimental study in rats. Pediatr. Surg. Int. 2012, 28, 171–177. [Google Scholar]
- Morgan, J.R.; Barrandon, Y.; Green, H.; Mulligan, R.C. Expression of an exogenous growth hormone gene by transplantable human epidermal cells. Science 1987, 237, 1476–1479. [Google Scholar] [CrossRef]
- Grusby, M.J.; Auchincloss, H.; Leet, R.; Johnson:, R.S.; Spencer, J.P.; Zijlstra1ii, M.; Jaenisch, R.; Papaioannou, V.E.; Glimcher, L.H. Mice lacking major histocompatibility complex class I and class II molecules. Immunology 1993, 90, 3913–3917. [Google Scholar] [CrossRef]
- Teumer, J.; Lindahl, A.; Green, H. Human growth hormone in the blood of athymic mice grafted with cultures of hormone-secreting human keratinocytes. FASEB J. 1990, 4, 3245–3250. [Google Scholar] [CrossRef]
- Vogt, P.M.; Thompsont, S.; Andree, C.; Liu, P.; Breuing, K.; Hatzis, D.; Brown, H.; Mulligant, R.C.; Eriksson, E.; Murray, J.E. Genetically modified keratinocytes transplanted to wounds reconstitute the epidermis. Proc. Natl. Acad. Sci. USA 1994, 91, 9307–9311. [Google Scholar] [CrossRef]
- Vogt, P.M.; Eriksson, E. Current aspects of epidermal wound healing. Handchir. Mikrochir. Plast. Chir. 1992, 24, 259–266. [Google Scholar]
- Eming, S.A.; Krieg, T.; Davidson, J.M. Gene transfer in tissue repair: Status, challenges and future directions. Expert Opin. Biol. Ther. 2004, 4, 1373–1386. [Google Scholar] [CrossRef]
- Ljunggren, H.G.; Van Kaer, L.; Sabatine, M.S.; Auchincloss, H.; Tonegawa, S.; Ploegh, H.L. MHC class I expression and CD8+ T cell development in TAP1/beta 2-microglobulin double mutant mice. Int. Immunol. 1995, 7, 975–984. [Google Scholar] [CrossRef]
- Zijlstra, M.; Auchincloss, H.; Loring, J.M.; Chase, C.M.; Russell, P.S.; Jaenisch, R. Skin graft rejection by beta 2-microglobulin-deficient mice. J. Exp. Med. 1992, 175, 885–893. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.S.; Grusby, M.J.; Laufer, T.M.; Colvin, R.; Glimcher, L.H.; Auchincloss, H. CD8+ effector cells responding to residual class I antigens, with help from CD4+ cells stimulated indirectly, cause rejection of “major histocompatibility complex-deficient” skin grafts. Transplantation 1997, 63, 1123–1133. [Google Scholar] [CrossRef]
- Markmann, J.F.; Bassiri, H.; Desai, N.M.; Odorico, J.S.; Kim, J.I.; Koller, B.H.; Smithies, O.; Barker, C.F. Indefinite survival of MHC class I-deficient murine pancreatic islet allografts. Transplantation 1992, 54, 1085–1089. [Google Scholar] [CrossRef]
- Qian, S.; Fu, F.; Li, Y.; Lu, L.; Rao, A.S.; Starzl, T.E.; Thomson, A.W.; Fung, J.J. Impact of donor MHC class I or class II antigen deficiency on first-and second-set rejection of mouse heart or liver allografts. Immunology 1996, 88, 124–129. [Google Scholar] [CrossRef]
- Mhashilkar, A.M.; Doebis, C.; Seifert, M.; Busch, A.; Zani, C.; Soo Hoo, J.; Nagy, M.; Ritter, T.; Volk, H.-D.; Marasco, W.A. Intrabody-mediated phenotypic knockout of major histocompatibility complex class I expression in human and monkey cell lines and in primary human keratinocytes. Gene Ther. 2002, 9, 307–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, A.; Ploegh, H. Getting the inside out: The transporter associated with antigen processing (TAP) and the presentation of viral antigen. Proc. Natl. Acad. Sci. USA 1995, 92, 341–343. [Google Scholar] [CrossRef]
- Powers, C.; DeFilippis, V.; Malouli, D.; Früh, K. Cytomegalovirus immune evasion. Curr. Top. Microbiol. Immunol. 2008, 325, 333–359. [Google Scholar]
- Tortorella, D.; Gewurz, B.E.; Furman, M.H.; Schust, D.J.; Ploegh, H.L. Viral subversion of the immune system. Annu. Rev. Immunol. 2000, 18, 861–926. [Google Scholar] [CrossRef]
- Loenen, W.A.; Bruggeman, C.A.; Wiertz, E.J. Immune evasion by human cytomegalovirus: Lessons in immunology and cell biology. Semin. Immunol. 2001, 13, 41–49. [Google Scholar] [CrossRef]
- Jackson, S.E.; Mason, G.M.; Wills, M.R. Human cytomegalovirus immunity and immune evasion. Virus Res. 2011, 157, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.D.; Grundy, J.E. Down-regulation of the class I HLA heterodimer and beta 2-microglobulin on the surface of cells infected with cytomegalovirus. J. Gen. Virol. 1992, 73, 2395–2403. [Google Scholar] [CrossRef]
- Del Val, M.; Münch, K.; Reddehase, M.J.; Koszinowski, U.H. Presentation of CMV immediate-early antigen to cytolytic T lymphocytes is selectively prevented by viral genes expressed in the early phase. Cell 1989, 58, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, R.H. T-lymphocyte recognition of antigen in association with gene products of the major histocompatibility complex. Annu. Rev. Immunol. 1985, 3, 237–261. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.; Angulot, A.; Ghazalt, P.; Peterson, P.A.; Yang, Y.; Froh, K.; The, T.; Johnson, R.W. Human cytomegalovirus inhibits antigen presentation by a sequential multistep process. Immunology 1996, 93, 10990–10995. [Google Scholar] [CrossRef]
- Jones, T.R.; Hanson, L.K.; Sun, L.; Slater, J.S.; Stenberg, R.M.; Campbell, A.E. Multiple independent loci within the human cytomegalovirus unique short region down-regulate expression of major histocompatibility complex class I heavy chains. J. Virol. 1995, 69, 4830–4841. [Google Scholar] [PubMed]
- Wiertz, E.J.; Jones, T.R.; Sun, L.; Bogyo, M.; Geuze, H.J.; Ploegh, H.L. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell 1996, 84, 769–779. [Google Scholar] [CrossRef]
- Shamu, C.E.; Story, C.M.; Rapoport, T.A.; Ploegh, H.L. The pathway of US11-dependent degradation of MHC class I heavy chains involves a ubiquitin-conjugated intermediate. J. Cell Biol. 1999, 147, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Story, C.M.; Furman, M.H.; Ploegh, H.L. The cytosolic tail of class I MHC heavy chain is required for its dislocation by the human cytomegalovirus US2 and US11 gene products. Proc. Natl. Acad. Sci. USA 1999, 96, 8516–8521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hengel, H.; Flohr, T.; Hämmerling, G.J.; Koszinowski, U.H.; Momburg, F. Human cytomegalovirus inhibits peptide translocation into the endoplasmic reticulum for MHC class I assembly. J. Gen. Virol. 1996, 77, 2287–2296. [Google Scholar] [CrossRef] [PubMed]
- Basta, S.; Chen, W.; Bennink, J.R.; Yewdell, J.W. Inhibitory effects of cytomegalovirus proteins US2 and US11 point to contributions from direct priming and cross-priming in induction of vaccinia virus-specific CD8(+) T cells. J. Immunol. 2002, 168, 5403–5408. [Google Scholar] [CrossRef]
- Ahn, K.; Gruhler, A.; Galocha, B.; Jones, T.R.; Wiertz, E.J.; Ploegh, H.L.; Peterson, P.A.; Yang, Y.; Früh, K. The ER-luminal domain of the HCMV glycoprotein US6 inhibits peptide translocation by TAP. Immunity 1997, 6, 613–621. [Google Scholar] [CrossRef]
- Townsend, A.; Bodmer, H. Antigen recognition by class I-restricted T lymphocytes. Annu. Rev. Immunol. 1989, 7, 601–624. [Google Scholar] [CrossRef]
- Lee, E.M.; Kim, J.Y.; Cho, B.R.; Chung, W.K.; Yoon, B.-W.; Kim, S.U.; Lee, B.C.; Hwang, W.S.; Moon, S.-Y.; Lee, J.S.; et al. Down-regulation of MHC class I expression in human neuronal stem cells using viral stealth mechanism. Biochem. Biophys. Res. Commun. 2005, 326, 825–835. [Google Scholar] [CrossRef] [Green Version]
- Lanier, L.L. Natural killer cell receptors and MHC class I interactions. Curr. Opin. Immunol. 1997, 9, 126–131. [Google Scholar] [CrossRef]
- Farrell, H.E.; Vally, H.; Lynch, D.M.; Fleming, P.; Shellam, G.R.; Scalzo, A.A.; Davis-Poynter, N.J. Inhibition of natural killer cells by a cytomegalovirus MHC class I homologue in vivo. Nature 1997, 386, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Park, B.; Oh, H.; Lee, S.; Song, Y.; Shin, J.; Sung, Y.C.; Hwang, S.-Y.; Ahn, K. The MHC class I homolog of human cytomegalovirus is resistant to down-regulation mediated by the unique short region protein (US)2, US3, US6, and US11 gene products. J. Immunol. 2002, 168, 3464–3469. [Google Scholar] [CrossRef]
- Falk, C.S.; Mach, M.; Schendel, D.J.; Weiss, E.H.; Hilgert, I.; Hahn, G. NK cell activity during human cytomegalovirus infection is dominated by US2-11-mediated HLA class I down-regulation. J. Immunol. 2002, 169, 3257–3266. [Google Scholar] [CrossRef] [PubMed]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef] [Green Version]
- Besold, K.; Wills, M.; Plachter, B. Immune evasion proteins gpUS2 and gpUS11 of human cytomegalovirus incompletely protect infected cells from CD8 T cell recognition. Virology 2009, 391, 5–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barel, M.T.; Ressing, M.; Pizzato, N.; van Leeuwen, D.; Le Bouteiller, P.; Lenfant, F.; Wiertz, E.J.H.J. Human cytomegalovirus-encoded US2 differentially affects surface expression of MHC class I locus products and targets membrane-bound, but not soluble HLA-G1 for degradation. J. Immunol. 2003, 171, 6757–6765. [Google Scholar] [CrossRef] [PubMed]
- Barel, M.T.; Pizzato, N.; Le Bouteiller, P.; Wiertz, E.J.H.J.; Lenfant, F. Subtle sequence variation among MHC class I locus products greatly influences sensitivity to HCMV US2- and US11-mediated degradation. Int. Immunol. 2006, 18, 173–182. [Google Scholar] [CrossRef]
- Barel, M.T.; Hassink, G.C.; van Voorden, S.; Wiertz, E.J.H.J. Human cytomegalovirus-encoded US2 and US11 target unassembled MHC class I heavy chains for degradation. Mol. Immunol. 2006, 43, 1258–1266. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, D.; Choi, I.; Yang, J.S.; Lee, D.-S.; Lee, J.R.; Kang, K.; Kim, S.; Hwang, W.S.; Lee, J.S.; et al. MHC expression in a human adult stem cell line and its down-regulation by hCMV US gene transfection. Int. J. Biochem. Cell Biol. 2005, 37, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Radosevich, T.J.; Seregina, T.; Link, C.J. Effective suppression of class I major histocompatibility complex expression by the US11 or ICP47 genes can be limited by cell type or interferon-gamma exposure. Hum. Gene Ther. 2003, 14, 1765–1775. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef]
- Alemany, R.; Balagué, C.; Curiel, D.T. Replicative adenoviruses for cancer therapy. Nat. Biotechnol. 2000, 18, 723–727. [Google Scholar] [CrossRef]
- Isner, J.M.; Asahara, T. Angiogenesis and vasculogenesis as therapeutic strategies for postnatal neovascularization. J. Clin. Invest. 1999, 103, 1231–1236. [Google Scholar] [CrossRef] [Green Version]
- Gerdes, C.A.; Castro, M.G.; Löwenstein, P.R. Strong promoters are the key to highly efficient, noninflammatory and noncytotoxic adenoviral-mediated transgene delivery into the brain in vivo. Mol. Ther. 2000, 2, 330–338. [Google Scholar] [CrossRef]
- Lusky, M.; Christ, M.; Rittner, K.; Dieterle, A.; Dreyer, D.; Mourot, B.; Schultz, H.; Stoeckel, F.; Pavirani, A.; Mehtali, M. In vitro and in vivo biology of recombinant adenovirus vectors with E1, E1/E2A, or E1/E4 deleted. J. Virol. 1998, 72, 2022–2032. [Google Scholar]
- Andrews, J.L.; Kadan, M.J.; Gorziglia, M.I.; Kaleko, M.; Connelly, S. Generation and characterization of E1/E2a/E3/E4-deficient adenoviral vectors encoding human factor VIII. Mol. Ther. 2001, 3, 329–336. [Google Scholar] [CrossRef]
- Naldini, L.; Blömer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef]
- Naldini, L.; Blömer, U.; Gage, F.H.; Trono, D.; Verma, I.M. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc. Natl. Acad. Sci. USA 1996, 93, 11382–11388. [Google Scholar] [CrossRef]
- Zufferey, R.; Dull, T.; Mandel, R.J.; Bukovsky, A.; Quiroz, D.; Naldini, L.; Trono, D. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J. Virol. 1998, 72, 9873–9880. [Google Scholar]
- Samaniego, L.A.; Wu, N.; DeLuca, N.A. The herpes simplex virus immediate-early protein ICP0 affects transcription from the viral genome and infected-cell survival in the absence of ICP4 and ICP27. J. Virol. 1997, 71, 4614–4625. [Google Scholar]
- Samaniego, L.A.; Neiderhiser, L.; DeLuca, N.A. Persistence and expression of the herpes simplex virus genome in the absence of immediate-early proteins. J. Virol. 1998, 72, 3307–3320. [Google Scholar]
- Palmer, J.A.; Branston, R.H.; Lilley, C.E.; Robinson, M.J.; Groutsi, F.; Smith, J.; Latchman, D.S.; Coffin, R.S. Development and optimization of herpes simplex virus vectors for multiple long-term gene delivery to the peripheral nervous system. J. Virol. 2000, 74, 5604–5618. [Google Scholar] [CrossRef]
- Christinck, E.R.; Luscher, M.A.; Barber, B.H.; Williams, D.B. Peptide binding to class I MHC on living cells and quantitation of complexes required for CTL lysis. Nature 1991, 352, 67–70. [Google Scholar] [CrossRef]
- Sykulev, Y.; Joo, M.; Vturina, I.; Tsomides, T.J.; Eisen, H.N. Evidence that a single peptide-MHC complex on a target cell can elicit a cytolytic T cell response. Immunity 1996, 4, 565–571. [Google Scholar] [CrossRef]
- Timonen, T.; Helander, T.S. Natural killer cell-target cell interactions. Curr. Opin. Cell Biol. 1997, 9, 667–673. [Google Scholar] [CrossRef]
- Shimizu, Y.; DeMars, R. Demonstration by class I gene transfer that reduced susceptibility of human cells to natural killer cell-mediated lysis is inversely correlated with HLA class I antigen expression. Eur. J. Immunol. 1989, 19, 447–451. [Google Scholar] [CrossRef]
- Noriega, V.M.; Hesse, J.; Gardner, T.J.; Besold, K.; Plachter, B.; Tortorella, D. Human cytomegalovirus US3 modulates destruction of MHC class I molecules. Mol. Immunol. 2012, 51, 245–253. [Google Scholar] [CrossRef]
- Raval, A.; Puri, N.; Rath, P.C.; Saxena, R.K. Cytokine regulation of expression of class I MHC antigens. Exp. Mol. Med. 1998, 30, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Collins, K.L.; Chen, B.K.; Kalams, S.A.; Walker, B.D.; Baltimore, D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 1998, 391, 397–401. [Google Scholar] [CrossRef]
- Cohen, G.B.; Gandhi, R.T.; Davis, D.M.; Mandelboim, O.; Chen, B.K.; Strominger, J.L.; Baltimore, D. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 1999, 10, 661–671. [Google Scholar] [CrossRef]
- Berger, C.; Xuereb, S.; Johnson, D.C.; Watanabe, K.S.; Kiem, H.P.; Greenberg, P.D.; Riddell, S.R. Expression of herpes simplex virus ICP47 and human cytomegalovirus US11 prevents recognition of transgene products by CD8(+) cytotoxic T lymphocytes. J. Virol. 2000, 74, 4465–4473. [Google Scholar] [CrossRef]
- Komor, A.C.; Badran, A.H.; Liu, D.R. CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 2017, 168, 20–36. [Google Scholar] [CrossRef]
- Go, D.E.; Stottmann, R.W. The Impact of CRISPR/Cas9-Based Genomic Engineering on Biomedical Research and Medicine. Curr. Mol. Med. 2016, 16, 343–352. [Google Scholar] [CrossRef]
- Peng, R.; Lin, G.; Li, J. Potential pitfalls of CRISPR/Cas9-mediated genome editing. FEBS J. 2016, 283, 1218–1231. [Google Scholar] [CrossRef]
- Yang, S.-W.; Geng, Z.-J.; Ma, K.; Sun, X.-Y.; Fu, X.-B. Comparison of the histological morphology between normal skin and scar tissue. J. Huazhong Univ. Sci. Technol. Med. Sci. 2016, 36, 265–269. [Google Scholar] [CrossRef]
- Elias, P.M. Epidermal lipids, barrier function, and desquamation. J. Invest. Dermatol. 1983, 80, 44s–49s. [Google Scholar] [CrossRef]
- Boyce, S.T. Design principles for composition and performance of cultured skin substitutes. Burns 2001, 27, 523–533. [Google Scholar] [CrossRef]
- Shaw, T.J.; Martin, P. Wound repair at a glance. J. Cell Sci. 2009, 122, 3209–3213. [Google Scholar] [CrossRef] [Green Version]
- Singer, A.J.; Clark, R.A. Cutaneous wound healing. N. Engl. J. Med. 1999, 341, 738–746. [Google Scholar] [CrossRef]
- Seifert, A.W.; Monaghan, J.R.; Voss, S.R.; Maden, M. Skin regeneration in adult axolotls: A blueprint for scar-free healing in vertebrates. PLoS ONE 2012, 7, e32875. [Google Scholar] [CrossRef]
- Takeo, M.; Lee, W.; Ito, M. Wound healing and skin regeneration. Cold Spring Harb. Perspect. Med. 2015, 5, a023267. [Google Scholar] [CrossRef]
- Galiano, R.D.; Michaels, V.J.; Dobryansky, M.; Levine, J.P.; Gurtner, G.C. Quantitative and reproducible murine model of excisional wound healing. Wound Repair Regen. 2004, 12, 485–492. [Google Scholar] [CrossRef]
- Michael, S.; Sorg, H.; Peck, C.-T.; Koch, L.; Deiwick, A.; Chichkov, B.; Vogt, P.M.; Reimers, K. Tissue engineered skin substitutes created by laser-assisted bioprinting form skin-like structures in the dorsal skin fold chamber in mice. PLoS ONE 2013, 8, e57741. [Google Scholar] [CrossRef]
- Frueh, F.S.; Sanchez-Macedo, N.; Calcagni, M.; Giovanoli, P.; Lindenblatt, N. The Crucial Role of Vascularization and Lymphangiogenesis in Skin Reconstruction. Eur. Surg. Res. 2018, 59, 242–254. [Google Scholar] [CrossRef]
- Zarem, H.A. Development of the microcirculation in full thickness autogenous skin grafts in mice. Surg. Forum 1965, 16, 469–470. [Google Scholar] [CrossRef]
- Converse, J.M.; Smahel, J.; Ballantyne, D.L.; Harper, A.D. Inosculation of vessels of skin graft and host bed: A fortuitous encounter. Br. J. Plast. Surg. 1975, 28, 274–282. [Google Scholar] [CrossRef]
- O’Ceallaigh, S.; Herrick, S.E.; Bluff, J.E.; McGrouther, D.A.; Ferguson, M.W.J. Quantification of total and perfused blood vessels in murine skin autografts using a fluorescent double-labeling technique. Plast. Reconstr. Surg. 2006, 117, 140–151. [Google Scholar] [CrossRef]
- Capla, J.M.; Ceradini, D.J.; Tepper, O.M.; Callaghan, M.J.; Bhatt, K.A.; Galiano, R.D.; Levine, J.P.; Gurtner, G.C. Skin graft vascularization involves precisely regulated regression and replacement of endothelial cells through both angiogenesis and vasculogenesis. Plast. Reconstr. Surg. 2006, 117, 836–844. [Google Scholar] [CrossRef]
- Frueh, F.S.; Menger, M.D.; Lindenblatt, N.; Giovanoli, P.; Laschke, M.W. Current and emerging vascularization strategies in skin tissue engineering. Crit. Rev. Biotechnol. 2017, 37, 613–625. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human Leukocyte Antigen | Subtypes |
|---|---|
| HLA-A | *02, *11 |
| HLA-B | *15:01:01G, *27 |
| HLA-C | *02, *03 |
| HLA-DRB1 | *13, *16 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schlottmann, F.; Strauss, S.; Hake, K.; Vogt, P.M.; Bucan, V. Down-Regulation of MHC Class I Expression in Human Keratinocytes Using Viral Vectors Containing US11 Gene of Human Cytomegalovirus and Cultivation on Bovine Collagen-Elastin Matrix (Matriderm®): Potential Approach for an Immune-Privileged Skin Substitute. Int. J. Mol. Sci. 2019, 20, 2056. https://doi.org/10.3390/ijms20092056
Schlottmann F, Strauss S, Hake K, Vogt PM, Bucan V. Down-Regulation of MHC Class I Expression in Human Keratinocytes Using Viral Vectors Containing US11 Gene of Human Cytomegalovirus and Cultivation on Bovine Collagen-Elastin Matrix (Matriderm®): Potential Approach for an Immune-Privileged Skin Substitute. International Journal of Molecular Sciences. 2019; 20(9):2056. https://doi.org/10.3390/ijms20092056
Chicago/Turabian StyleSchlottmann, Frederik, Sarah Strauss, Kevin Hake, Peter M. Vogt, and Vesna Bucan. 2019. "Down-Regulation of MHC Class I Expression in Human Keratinocytes Using Viral Vectors Containing US11 Gene of Human Cytomegalovirus and Cultivation on Bovine Collagen-Elastin Matrix (Matriderm®): Potential Approach for an Immune-Privileged Skin Substitute" International Journal of Molecular Sciences 20, no. 9: 2056. https://doi.org/10.3390/ijms20092056
APA StyleSchlottmann, F., Strauss, S., Hake, K., Vogt, P. M., & Bucan, V. (2019). Down-Regulation of MHC Class I Expression in Human Keratinocytes Using Viral Vectors Containing US11 Gene of Human Cytomegalovirus and Cultivation on Bovine Collagen-Elastin Matrix (Matriderm®): Potential Approach for an Immune-Privileged Skin Substitute. International Journal of Molecular Sciences, 20(9), 2056. https://doi.org/10.3390/ijms20092056