Abstract
Glioblastoma multiforme and medulloblastoma are the most frequent high-grade brain tumors in adults and children, respectively. Standard therapies for these cancers are mainly based on surgical resection, radiotherapy, and chemotherapy. However, intrinsic or acquired resistance to treatment occurs almost invariably in the first case, and side effects are unacceptable in the second. Therefore, the development of new, effective drugs is a very important unmet medical need. A critical requirement for developing such agents is to identify druggable targets required for the proliferation or survival of tumor cells, but not of other cell types. Under this perspective, genes mutated in congenital microcephaly represent interesting candidates. Congenital microcephaly comprises a heterogeneous group of disorders in which brain volume is reduced, in the absence or presence of variable syndromic features. Genetic studies have clarified that most microcephaly genes encode ubiquitous proteins involved in mitosis and in maintenance of genomic stability, but the effects of their inactivation are particularly strong in neural progenitors. It is therefore conceivable that the inhibition of the function of these genes may specifically affect the proliferation and survival of brain tumor cells. Microcephaly genes encode for a few kinases, including CITK, PLK4, AKT3, DYRK1A, and TRIO. In this review, we summarize the evidence indicating that the inhibition of these molecules could exert beneficial effects on different aspects of brain cancer treatment.
1. Background
1.1. The Hurdles of High-Grade Brain Tumor Precision Medicine
High-grade brain tumors (HGBTs) are very aggressive cancers that represent an important unmet medical challenge. Medulloblastoma (MB) is the most common pediatric HGBT, and it also occurs in adults, although less frequently. Based on microarray and genomic sequencing technologies, MB has been classified into four biological subgroups (WNT, SHH, group 3, and group 4) [1,2]. MB is currently treated with surgery, followed by irradiation of the entire neuraxis and high-dose multiagent chemotherapy. The long-term survival rate can be as high as 90% in the rare WNT subgroup, but it is usually around 50% in most of the other cases, with the worst prognosis in group 3 and 4 patients [3,4]. Thus, many patients still die despite treatment, and those who survive suffer from neurological, cognitive, and endocrine disorders caused by the aggressive therapy [3,4].
In adulthood, the most frequent HGBTs are gliomas. Among them, glioblastoma multiforme (GBM) is one of the deadliest human cancers. According to the WHO, GBM accounts for approximately 12%–15% of all brain tumors, and 60%–70% of astrocytic tumors [5]. The standard therapy for GBM is mainly based on surgical resection in combination with radiotherapy and chemotherapy with alkylating agents, such as temozolomide (TMZ). Gene expression profiling has allowed for the classification of GBM into four distinct subtypes (i.e., proneural, neural, classical, and mesenchymal) associated with distinct genomic abnormalities and different responses to aggressive therapy [6]. Nevertheless, the longest median survival obtained in GBM patients treated with combined therapy has been 14 months [7].
For these reasons, more effective and specific therapies are urgently needed for HGBTs.
A common assumption is that the most straightforward strategy to develop new anticancer therapies is to directly target driver mutations, as well as molecular pathways connected to them. A paradigm for this approach is the dramatic improvement in therapy of chronic myeloid leukemia, determined by the introduction of ABL1 tyrosine kinase inhibitors [8], but the extension of this approach to other tumors faces many issues.
In the case of MB, targeted therapy has been developed for the SHH subtype. This subgroup, which represents approximately 30% of MB patients in children and more than 50% in adults, could take advantage of Vismodegib and other smoothened (SMO) inhibitors [9,10]. However, only a subgroup of these patients respond to treatment and, even in these cases, resistance rapidly develops [9,11]. As with many other pediatric cancers, MB is characterized by a low mutation burden [12], leading to a paucity of recurrent alterations. In addition, the recurrent mutations found in groups 3 and 4 involve NMYC amplification, CTNNB1, PRDM6, and TERT variants, which are difficult to target pharmacologically [13].
The current state of precision approaches is not better for GBM. In these tumors, many recurrent mutations are routinely identified, such as those involving growth factor receptors, MAPK, and PI3K/mTOR signaling pathways or inhibitors of cell cycle progression [6,14]. However, these variants have not been associated with clear prognostic and predictive results, challenging the assumption that they are strong cancer drivers. Even more disappointingly, no therapy against these targets has shown significant efficacy in clinical trials, probably as a consequence of cancer cell plasticity, redundancy among alterations, and intratumor genomic heterogeneity [15].
The application of immune checkpoint blockade strategies in HGBTs does not appear to provide much better perspectives. MB is not expected to be very responsive to these treatments, because of the low mutation burden and scarce inflammatory infiltrate [13]. On the other hand, despite encouraging preclinical results, clinical trials with PD1-PDL1 inhibitors have not shown a significant benefit in GBM, probably due to the strong immunosuppressive environment of these tumors [16].
A relatively unexplored alternative is to target molecules and mechanisms that, despite not being mutated, are nevertheless required for tumor growth, progression, and invasiveness [17]. Screening-based strategies have been proposed to identify cancer vulnerabilities in specific patients [18], but the time required for deploying such strategies is a strong barrier to their efficient practical implementation.
1.2. Congenital Microcephaly: A Tissue-Specific Phenotype of Ubiquitously Expressed Genes
A major problem for precision medicine is to understand whether and how the effects of tumor-driving mutations, as well as tumor responses to therapeutic agents, are rooted in the biology of cells that have undergone malignant transformation [19]. Specific epigenomic landscapes and local proteome composition may render a particular tissue or cell type permissive to particular oncogenic mutations, but may also result in tissue-specific vulnerabilities that could be exploited therapeutically [19].
On this ground, genes mutated in congenital microcephaly (CM) syndromes have been proposed as attractive targets for HGBT-directed drug development [20,21,22]. HGBTs originate from different types of neural progenitors. Although it is still debated from which precursors the different cancers originate, it is established that MB and GBM tumor cells share many molecular features with cerebellar granule progenitors and cortical radial glia cells, respectively [23,24]. The inactivation of genes associated with congenital microcephaly leads to specific alterations of proliferation and survival of such cells.
CM is a heterogeneous group of disorders characterized by reduced head circumference at birth, to at least 3 standard deviations below the mean [25,26]. CM can be the result of nongenetic conditions, such as viral infections and toxic exposure, or it can be generated by rare genetic disorders [25]. Primary hereditary microcephaly (MCPH) is the simplest form of genetic CM, in which brain size reduction is accompanied by grossly normal brain architecture and mild to moderate intellectual disability [25,27]. The association of severe microcephaly and proportionate body growth reduction is instead characteristic of Seckel syndrome (SCKS). Pure MCPH and SCKS are rare conditions, since genetic CM is more often associated with syndromic features and comorbidities [25,26,28]. In the Online Mendelian Inheritance in Man (OMIM) database (https://www.omim.org), approximately 450 loci are linked to mendelian phenotypes in which microcephaly is a strong hallmark.
A striking common feature of these genes is that, during development, they are selectively required for proliferation and genomic stability of neural progenitors, despite being expressed in all proliferating cell types [29]. The biological basis of this specificity is only partially understood. In many cases, CM proteins are associated with centrosomes, and their loss leads to cell cycle and mitosis delay, mitotic failure, and randomization of spindle orientation [30]. These alterations may tilt the balance between symmetric and asymmetric divisions of neural stem cells, decreasing the pool of proliferating neural progenitors and/or increasing the frequency of premature commitment or terminal differentiation [30]. However, it has also been shown that the loss of many MCPH proteins leads to the accumulation of DNA damage and apoptosis [31,32,33]. Moreover, there is evidence that nongenetic insults associated with microcephaly could impinge on the same tissue-specific vulnerabilities [34]. Accordingly, oncolytic activity by the ZIKA virus has been proposed as a therapeutic strategy for GBM [35,36,37].
Regardless of the precise mechanisms, the inactivation of CM genes may specifically reduce the expansion of HGBT cells. As in normal neural progenitors, CM gene inhibition could impair cancer cell cycle progression, promote differentiation, and induce apoptosis, with marginal effects on normal cell types. Moreover, the inactivation of CM genes may sensitize HGBT cells to radiotherapy and chemotherapy [20].
Proof of concept about the suitability of MCPH genes as possible therapeutic targets has already been reported for ASPM, the gene mutated at the highest frequency in MCPH patients, as well as for KIF14 and CDK6. ASPM loss has been found to arrest the proliferation of glioma stem cells [38], to radiosensitize GBM cell lines [39], and to reduce tumor growth in MB mouse models [32] by increasing DNA double strand break (DSB) accumulation [32,39]. Similar results have been obtained by inducing KIF14 depletion [40,41]. Finally, CDK4/6 inhibitors have displayed significant antineoplastic activity in pro-neural GBM cells in xenograft assays [42], and new inhibitors of these kinases capable of crossing the blood–brain barrier are actively being developed [43]. However, very little is known about the potential of other CM genes, as well as about the mechanisms that could influence their specificity.
CM-associated genes encode for some protein kinases. Considering their druggability, these proteins could represent a very interesting group of targets for HGBTs. Among them, CDK6 and CITK are involved in MCPH. On the other hand, mutations in ATR, PLK4, AKT3, DYRK1A, and TRIO have been associated with genetic disorders in which brain development is less specifically affected, leading to syndromic forms of CM. The potential of CDK6 and ATR as targets for MB and GBM has been deeply addressed in a recent review [22]. We therefore concentrate our survey on the remaining kinases (Figure 1).
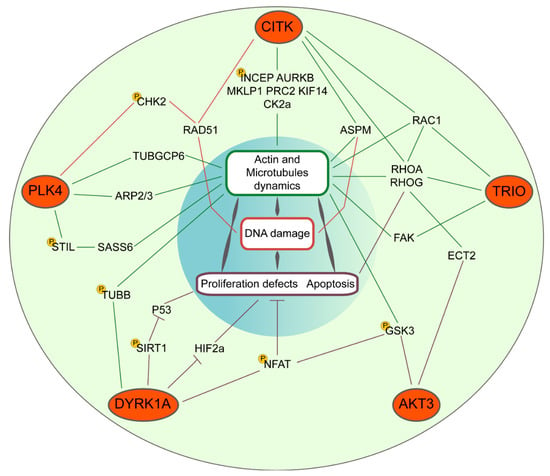
Figure 1.
Convergent molecular pathways of microcephaly kinases CITK, PLK4, DYRK1A, AKT3, and TRIO. Their signaling cascades impinge, throughout the indicated common genes, on cytoskeletal dynamics and DNA damage responses or directly on proliferation and apoptosis. The indicated phosphorylation events (yellow P circles) have been experimentally proven.
1.3. Citron Kinase (CITK)
CITK is a conserved AGC-type serine/threonine kinase. In mammals, it is the largest product of the CIT gene, with a molecular mass of 230 kD [44,45]. It displays a modular organization very similar to other members of the myotonic dystrophy kinase subfamily, comprising Rho-kinases (ROCKs) and CDC42BPA/CDC42BPB kinases (also known as MRCKs) [46]. These proteins share an amino-terminal kinase domain, followed by an extended coiled-coil region, a type 2 zinc finger, and a Pleckstrin homology domain (PH). CITK and MRCKs are characterized by a Citron-Nik1 homology (CNH) domain [45].
CITK is ubiquitously expressed in proliferating cells, with the highest levels in the G2/M phase of the cell cycle [47]. It is enriched at spindle poles before anaphase [48] and concentrates at cleavage furrows and midbody during cytokinesis [45]. The best studied function of CITK is to regulate midbody maturation and abscission at the end of cytokinesis [45,48,49,50,51], in concert with anillin (ANLN) [48], microtubule-binding proteins MKLP1, PRC2, and KIF14 (encoded by microcephaly gene MCPH20) [49], as well as chromosomal passenger complex (CPC) [52] and Casein kinase 2 [53]. CITK is also required for localization of F-actin at the abscission sites for the final cut of the midbody [51].
The only validated substrate of CITK is CPC component INCENP, whose phosphorylation by CITK regulates midbody organization by mediating a positive feedback loop between local CITK recruitment and AURKB activation [52]. CITK also regulates mitotic spindle orientation by interacting with ASPM (encoded by the microcephaly gene MCPH5) [48]. Finally, CITK prevents the accumulation of DNA double strand breaks independently of its role in cytokinesis and affects recruitment of RAD51 at DNA-damage foci [31].
Despite ubiquitous expression, CITK is functionally required in vivo only in a few cell types, including neural progenitors [54,55] and male germ cells [56]. Consequently, CITK loss leads to severe microcephaly in rodents [54,55] and humans [57,58,59,60]. Cells in the affected tissues display cytokinesis failure, apoptosis, and the accumulation of DNA damage [31,54,57,58]. The syndrome associated with CITK mutations is a particularly severe form of CM, known as MCPH17. Head circumference can be as low as 8 standard deviations below the mean, with moderate or severe intellectual disability. Many patients show a short stature and spasticity, and a few cases also display renal agenesis. Notably, in approximately half of MCPH17 patients, homozygous missense mutations in the kinase domain have been found, resulting in a loss of catalytic activity [58]. Together with the other data, the latter evidence underscores that CITK activity is at the center of a complex interaction network essential for normal proliferation and survival of neural progenitors, comprising many other CM-associated proteins.
Concerning its possible role as a cancer drug target, CITK has long been a “neglected” protein [61], probably due to its highly specific developmental role.
As with most microcephaly proteins, CITK is expressed at high levels in tumors [62,63,64], but this could likely be a reflection of cell-cycle regulated expression. Despite its strong tissue-specific requirement in normal cells, CITK knockdown negatively impacts the proliferation of tumor cell lines of different origins, in which it consistently induces cytokinesis failure, resulting in the accumulation of multinucleated cells [65,66].
The possible usefulness of CITK as a target for CNS tumor treatment has recently been explored in MB models [67]. CITK depletion by RNAi impairs in vitro expansion of MB cell lines and limits the growth of xenograft tumors. Moreover, temporally controlled deletion of CITK in tumors arising in the transgenic SmoA1 MB model reduces tumor growth and increases survival. In all models, CITK loss has been accompanied by cytokinesis failure, as well as by DNA damage and the induction of cell senescence. Interestingly, similar effects were obtained both in P53-proficient and P53-deficient cells [67]. At the moment, no data are available on the possible requirement for CITK in GBM cells, and no specific inhibitors of CITK have been reported.
1.4. Polo-Like Kinase 4 (PLK4)
PLK4 is one of the members of the Polo-like proteins, a kinase subfamily that plays a pivotal role in cell cycle progression and cytokinesis [68]. It is characterized by an N-terminal kinase domain, closely related to other PLKs. However, it shows a divergent carboxy-terminal domain, containing a single polo-box domain (PBD) instead of two tandem PBDs [68]. Polo boxes of PLK4 are involved in protein–protein interactions and control kinase activation, protein localization, and substrate specificity [69]. The PDB functions as a phosphoserine/threonine-binding module that has the highest affinity for Ser-[pSer/pThr]-[Pro/X], suggesting that PLK4 binds to docking sites primed by CDKs, MAP kinases, and other mitotic kinases [70].
In vivo expression of PLK4 is correlated with proliferation, and is very high in embryonic tissues and adult testes, moderate in the spleen and thymus, and not detectable in the brain, lungs, kidneys, breasts, heart, ovaries, and liver [71]. Expression peaks during the S, G2, and M phases of the cell cycle, while kinase activity is induced in the S phase and doubles from S to G2 [72].
PLK4 is localized in the nucleolus during G2, becomes enriched at centrosomes in the M phase, and concentrates at the midbody in cytokinesis [73]. In the centrosome, PLK4 is specifically concentrated at the proximal ends of the centriole outer wall and has also been observed close to the distal appendages of the mother centriole [74].
Consistent with its localization, PLK4 plays a fundamental role in centriole duplication [75]. PLK4 knockdown leads to centriole loss [75], while PLK4 overexpression increases centrioles’ number [74], producing abnormal spindle and mitotic abnormalities in both cases [76,77]. In mammals, centriole biogenesis is initiated by CEP192 and CEP152 (MCPH9), which recruit PLK4 to the proximal end of the mother centriole [78,79]. An interaction between PLK4 and STIL (MCPH7) results in STIL phosphorylation [80] and subsequent recruitment of SASS6 (MCPH14), which initiates nine-fold symmetric cartwheel nucleation and γ-tubulin assembly [81]. PLK4 also phosphorylates CHK2, a key transducer of ATM and ATR in the DNA damage response [82]. Therefore, PLK4 physically and functionally interacts with the products of many CM genes.
Homozygous deletion of PLK4 in mice is embryonically lethal at the postgastrulation stage, with a marked increase in mitotic and apoptotic cells [73]. PLK4 +/− mouse embryonic fibroblasts have demonstrated a high rate of primary cytokinesis failure, associated with aberrant acto-myosin ring formation, reduced RHOA activation, and failure to localize the RHOA guanine nucleotide exchange factor ECT2 to the midbody [83].
Although these data suggest a direct involvement of PLK4 in cytokinesis, further studies have excluded this possibility, indicating that cytokinesis failure from decreased PLK4 function is only an indirect consequence of the spindle abnormalities caused by centrioles [84].
Despite PLK4′s house-keeping functions in mitosis, the identification of PLK4 mutations in microcephaly patients has provided evidence that neural progenitors are particularly sensitive to its levels. Homozygous truncating mutations in PLK4 have been identified in seven affected members of a consanguineous family with autosomal recessive microcephaly, short statures, and chorioretinopathy, as well as in another two unrelated families [85]. A similar phenotype resulted from mutations in the PLK4 substrate TUBGCP6 [85]. Patient fibroblasts showed reduced centriole numbers, abnormal spindle formation, and decreased numbers of ciliated cells, correlating with the absence of basal bodies. However, patients did not show a ciliopathy phenotype [85]. In addition, patients’ cells showed genomic instability and altered DNA damage response [86].
There is abundant evidence that the alteration of PLK4 levels and activity may play a general driver role in cancer and that PLK4 expression is deregulated in many cancer types [69]. Notably, PLK4 acts as a tumor suppressor in haplo-insufficiency conditions by causing mitotic infidelity [87] and as an oncogene in overexpression conditions [88]. Centrosome amplification associated with PLK4 overexpression and its correlation with poor prognosis has been reported in many cancer types [69]. Moreover, the promotion of actin nucleation and invasiveness through phosphorylation of the Arp2/3 complex may contribute to PLK4 pro-metastatic potential [89].
These findings have led to rising interest in PLK4 as a promising and feasible target for cancer therapy and to the consequent development of PLK4 inhibitors. The first of such compounds was CFI-400945, which is capable of inducing mitotic defects and cell death in epithelial tumor cells [90]. In particular, CFI-400945 was reported to inhibit the growth of patient-derived pancreatic xenografts [91] and to induce polyploidy and cell death in lung cancer cells [92]. However, controversy exists about the specificity of this compound, because it leads to centrosome amplification, rather than the centrosome loss that would be expected from PLK4 inhibition [93]. This phenotype could be explained in part by the fact that CFI-400945 is also able to inhibit AURKB, leading to cytokinesis inhibition, in part by a feedback loop derived from partial PLK4 inhibition, preventing the degradation of autophosphorylated protein [94]. The latter mechanism could promote a paradoxical increase in PLK4 levels, with actual PLK4-dependent centrosome amplification [93].
A more specific compound is Centrinone, whose administration leads to total but reversible centrosome loss [95]. This compound has allowed for demonstrating that centrosomes are essential to the proliferation of normal cells, which undergo a permanent P53-dependent growth arrest upon Centrinone treatment [96]. In contrast, cancer cells continue to proliferate after centrosome loss, suggesting that centrosome depletion must be combined with other perturbations to selectively target them [93].
PLK4 is upregulated in embryonal CNS cancers, such as brain rhabdoid tumors [97], medulloblastoma [97,98], and neuroblastoma [99]. Rhabdoid cells within which PLK4 was targeted by CRISPR/CAS9 demonstrated significantly decreased proliferation, viability, and survival [100]. The PLK4 inhibitor CFI-400945 showed cytotoxic effects on rhabdoid tumor cell lines, while sparing non-neoplastic human fibroblasts and developing zebrafish larvae [100].
PLK4 inhibition has induced apoptosis, senescence, and polyploidy in MB cells, thereby increasing the susceptibility of cancer cells to DNA-damaging agents [97]. In malignant gliomas, elevated PLK4 levels were associated with poor prognosis and enhanced radio-resistance, while PLK4 knockdown significantly increased the radio-sensitivity of GBM cells [101]. The sensitivity of GBM cells to TMZ was also decreased by ectopic expression of PLK4 and was enhanced by PLK4 depletion and CFI-400945 treatment [102]. No reports are available on the effects of Centrinone in HGBT cells.
1.5. AKT Serine/Threonine Kinase 3 (AKT3)
AKT3 is one of three closely related serine/threonine-protein kinases, also called PKB, belonging to the family of AGC ser/thr protein kinases. These proteins share a conserved structure that includes three functional domains: an N-terminal PH domain, a central kinase domain, and a C-terminal regulatory domain containing the hydrophobic motif phosphorylation site [FxxF(S/T)Y] [103,104]. AKT1 and AKT2 play partially redundant roles in many processes of normal and cancer cells, including metabolism, proliferation, cell survival, growth, and angiogenesis [103,105,106].
AKT3 mRNA is expressed in many tissues, with the highest levels in the brain, testes, lungs, heart, kidneys, mammary glands, and fat [107]. It is activated by insulin through a PI3K-dependent mechanism requiring the PH domain and thr-305 phosphorylation [107,108]. AKT3 is the most represented AKT paralog in the brain during neurogenesis, and levels of phosphorylated pan-AKT are abundant in cortical progenitor cells during cortical development [109].
Akt3 -/- mice show a selective 20% decrease in brain volume and hypoplasia of the corpus callosum, resulting from the reduction of both cell size and cell numbers [110,111]. This phenotype differs from Akt1 -/- mice, in which brain size is reduced in the context of global body size decrease [110].
In consideration of the mouse knockout phenotype and its chromosomal localization (1q23-24), AKT3 is considered the strongest candidate gene for 1q22-24 deletion syndrome, characterized by microcephaly and corpus callosum agenesis [112]. Identification of a balanced reciprocal t(1;13)(q44;q32) translocation in a patient with a similar phenotype, with a breakpoint close to the AKT3 promoter, further supported this association [112]. Conversely, activating germline and somatic AKT3 mutations have been identified as a rare cause of megalencephaly and hemimegalencephaly, respectively [109,113,114].
The brain-specific role of AKT3 is at least partially explained by its expression pattern, since this kinase shares the same activation mechanisms and most substrates with its paralogs [115]. Thus, microcephaly could be explained by pro-proliferative and antiapoptotic effects on neural progenitors. However, it is possible that subtler mechanisms exist [115].
The AKT pathway is hyperactive in a large fraction of human cancers, including brain tumors [116]. Selective activation of AKT3 through overexpression or copy number increase is a recurrent event in nonfamilial melanomas [117]. AKT3 amplification has also been found in GBM [118] and MB [119]. AKT3 is required in transformed astrocytes and human glioma cells for anchorage-independent growth, and its loss has inhibited transformed cell invasion [120,121]. AKT3-expressing human GBM cells have shown enhanced activation of DNA repair proteins, leading to increased DNA repair and subsequent resistance to radiation and TMZ [122]. Accordingly, AKT3 knockdown had synergistic effects with TMZ and BCNU [123]. The development of therapeutic strategies based on selective and nonselective inhibitors of AKT3 and other AKTs represents an area of intensive investigation [116].
1.6. Dual Specificity Tyrosine Phosphorylation Regulated Kinase 1A (DYRK1A)
DYRK1A is a dual-specificity kinase regulated by tyrosine phosphorylation, belonging to the CMGC protein kinase family [124,125]. Human DYRK1A is a multidomain protein containing a highly conserved kinase domain preceded by a DH domain, which is characteristic of the DYRK kinase subfamily. In addition, it also contains two different nuclear localization signals, a PEST region, a histidine-rich region, and an extreme C-terminal region rich in serine and threonine. DYRK1A is capable of intermolecular activating autophosphorylation on tyrosine residues and phosphorylates its substrates on serine/threonine [124,126].
DYRK proteins are homologous to the Drosophila minibrain gene, and DYRK1A is widely known for its role in Down syndrome (DS). It is one of the more actively studied genes of the Down critical region, a relatively small part of human Chromosome 21 that plays a paramount role in DS-associated intellectual disability [125,127].
DYRK1A is expressed in most tissues, but during embryogenesis it is most abundant in the brain, spinal cord, and retina [128]. Homozygous deletion of Dyrk1a in mice is embryonically lethal, leading to general growth delay and death during midgestation. Heterozygous mice have shown decreased neonatal viability, reduced body size from birth to adulthood, and region-specific brain size decrease [129].
Accordingly, heterozygous DYRK1A deletion [130], as well as truncating and missense variants [131], have been found in patients showing microcephaly, intellectual disability, and autism spectrum symptoms. Both increased and decreased DYRK1A dosages have profound effects on neural progenitor biology. DYRK1A loss of function severely affects neural lineage specification [132], while the 1.5–2-fold increased expression that characterizes DS slows cell cycle progression, reduces progenitor pools, and impairs neuroblast differentiation in the developing neocortex [133].
Through the identification of substrates and interactors, DYRK1A has been involved in a broad range of cellular processes, including cell cycle regulation, cellular signaling, gene expression, chromatin modulation, alternative splicing, and membrane trafficking [134]. In particular, it has been shown to promote cell survival through different mechanisms, including inhibitory phosphorylation of Caspase 9 [135,136], priming phosphorylation of NFAT in the GSK3 pathway [137,138], and SIRT1 phosphorylation in response to genotoxic stress, in turn inhibiting TP53 activity and apoptosis [139]. Moreover, it may affect microtubule dynamics by phosphorylating β-Tubulin [140] and Tau [141].
Both tumor-suppressive and pro-oncogenic roles have been suggested for DYRK1A [134], especially in relation to brain tumors. In gliomas, it has been reported that DYRK1A may destabilize HIF2-alpha in hypoxic conditions by phosphorylating thr27 of ID2, leading to reduced self-renewal of glioma stem cells, the inhibition of tumor growth, and more favorable outcomes for patients with glioblastoma [142]. On the other hand, DYRK1A inhibition promotes EGFR degradation in primary GBM cells, thus compromising their survival and producing a profound decrease in tumor burden [143]. Accordingly, some novel, potent inhibitors (IC50 ≤50 nM) are capable of significantly decreasing viability, clonogenic survival, migration, and invasion of glioblastoma cells [144]. DYRK1A has also been shown to interfere with Shh/Gli signaling in MB [145].
1.7. Trio Rho Guanine Nucleotide Exchange Factor (TRIO)
The TRIO name derives from its sequence containing three main functional domains: two guanine nucleotide exchange domains for Rho-family small GTPases (GEFD1 and GEFD2), composed of a Dbl homology region followed by a PH domain, as well as one C-terminal ser/thr kinase domain [146]. In addition, TRIO comprises other modular units belonging to different structural classes [147]. TRIO was originally identified as an interactor of receptor tyrosine phosphatase LAR and is ubiquitously expressed with many different isoforms [146].
The principal functional regions of TRIO are the two GEFDs, which are capable of inducing actin remodeling through Rho GTPases. In particular, GEFD1 activates both RAC1 and RHOG, while GEFD2 acts specifically on RHOA [146,148]. The kinase domain is constitutively tyrosine-phosphorylated and interacts with LAR and with FAK. Increased tyrosine phosphorylation after FAK cotransfection increases TRIO association with the cytoskeleton [149].
A complete loss of function of TRIO is embryonically lethal in mice between E15.5 and birth, with a few escapers surviving for no more than one month [150]. Trio -/- mice display abnormal myotube fusion and aberrant organization in several brain regions, including in hippocampal formation and in the olfactory bulb [150].
Exome sequencing has revealed that TRIO is a haplo-insufficient gene, since heterozygous germline or de novo mutations have been found in several patients, characterized by delays in the acquisition of motor and language skills, mild to borderline intellectual disability, neurobehavioral problems, and microcephaly [151,152]. TRIO mediates axon outgrowth and guidance in response to extracellular cues transduced by different receptors. For instance, it may act downstream of the NGF receptor by activating RHOG through GEFD1 [153]. Moreover, it promotes signal transduction through axon-guidance receptor DCC, by favoring its membrane insertion and by becoming phosphorylated by tyrosine kinase FYN when DCC is bound by its ligand netrin-1. FYN-mediated tyrosine phosphorylation enhances the activity of GEFD1 toward RAC1, thereby promoting actin dynamics at the growth cone [154].
A very important function of TRIO for its involvement in cancer is to control cell adhesion and migration, especially downstream of Cadherin proteins [147]. For instance, both GEFD1 and GEFD2 are essential to mediate the collective migration of neural crest cells downstream of Cadherin-11.
Upregulation of TRIO expression, often associated with poor patient survival, is found in different tumor types, including urinary bladder, breast, lung soft tissue sarcoma, and glioblastoma [147]. TRIO mediates glioma cell migration and invasion produced by stimulation of the TWEAK-Fn14 signaling axis by inducing RAC1 activation [155]. Accordingly, TRIO-directed siRNA oligonucleotides suppress glioblastoma cell migration and invasion and also reduce the rate of cell proliferation [156]. Compared to other kinases, TRIO is a more challenging target for pharmacological development, because the kinase domain is not crucial for function. Nevertheless, peptide aptamer-based [157] and small-molecule inhibitors [158], both interfering with the binding of GEF domains to cognate GTPases, are being developed.
2. Remarks and Conclusions
Sufficient evidence exists to support the notion that protein kinases associated with CM are promising targets for HGBT treatment. The results obtained through the inactivation or depletion of these proteins consistently have shown that, by interfering with microcephaly-related mechanisms, it is possible to decrease tumor cell clonal expansion, increase their sensitivity to chemotherapy and radiotherapy, and decrease their invasiveness. In many cases, experiments performed with the available inhibitors in vitro or with heterotopic xenograft models have provided proof of concept about their possible usefulness in therapy. However, many problems remain to be solved. In most of the studied cases, important pharmacological issues exist, especially concerning inhibitor development and/or delivery through the blood–brain barrier. Most inhibitors have not been tested yet in transgenic or orthotopic models, which more closely resemble the in vivo human condition. Since it is difficult to imagine that inhibitors of CM genes could be used as a monotherapy, a big effort is needed to address the effects of combining them with radiotherapy and chemotherapy, both in xenograft and orthotopic models. Moreover, it would be very interesting to study the effects of simultaneously inhibiting CM genes impinging on similar or different mechanisms. On these bases, although the inhibition of ubiquitous kinases may appear to be an old-fashioned approach, we are convinced that the underlying biological complexity may still offer a field with great potential.
Author Contributions
Conceptualization, G.P., G.E.B and F.D.; writing—original draft preparation, G.P., G.E.B; writing—review and editing, F.D., G.P., G.E.B; supervision, F.D.; funding acquisition, F.D., G.P.
Funding
The financial support to the F.D. laboratory by the Associazione Italiana per la Ricerca sul Cancro (AIRC—grant IG17527), by the Telethon Foundation (grant n. GGP12095), by the ‘Consiglio Nazionale delle Ricerche’ through the EPIGEN project, and by the Jérôme Lejeune Foundation is gratefully acknowledged. G.P. is supported by a PhD fellowship from Italian Ministry of University and Research.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.-J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. (Berl.) 2012, 123, 465–472. [Google Scholar] [CrossRef]
- Northcott, P.A.; Shih, D.J.H.; Peacock, J.; Garzia, L.; Morrissy, A.S.; Zichner, T.; Stütz, A.M.; Korshunov, A.; Reimand, J.; Schumacher, S.E.; et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature 2012, 488, 49–56. [Google Scholar] [CrossRef]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Bailey, S.; Clifford, S.C.; Doz, F.; Kool, M.; Dufour, C.; Vassal, G.; Milde, T.; et al. Risk stratification of childhood medulloblastoma in the molecular era: The current consensus. Acta Neuropathol. (Berl.) 2016, 131, 821–831. [Google Scholar] [CrossRef]
- Packer, R.J.; Gajjar, A.; Vezina, G.; Rorke-Adams, L.; Burger, P.C.; Robertson, P.L.; Bayer, L.; LaFond, D.; Donahue, B.R.; Marymont, M.H.; et al. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J. Clin. Oncol. 2006, 24, 4202–4208. [Google Scholar] [PubMed]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A “state of the science” review. Neuro-Oncol. 2014, 16, 896–913. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Delgado-López, P.D.; Corrales-García, E.M. Survival in glioblastoma: A review on the impact of treatment modalities. Clin. Transl. Oncol. 2016, 18, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Soverini, S.; Mancini, M.; Bavaro, L.; Cavo, M.; Martinelli, G. Chronic myeloid leukemia: The paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol. Cancer 2018, 17, 49. [Google Scholar] [CrossRef]
- Robinson, G.W.; Orr, B.A.; Wu, G.; Gururangan, S.; Lin, T.; Qaddoumi, I.; Packer, R.J.; Goldman, S.; Prados, M.D.; Desjardins, A.; et al. Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog-Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J. Clin. Oncol. 2015, 33, 2646–2654. [Google Scholar] [CrossRef]
- Remke, M.; Hielscher, T.; Northcott, P.A.; Witt, H.; Ryzhova, M.; Wittmann, A.; Benner, A.; von Deimling, A.; Scheurlen, W.; Perry, A.; et al. Adult medulloblastoma comprises three major molecular variants. J. Clin. Oncol. 2011, 29, 2717–2723. [Google Scholar]
- Gajjar, A.; Stewart, C.F.; Ellison, D.W.; Kaste, S.; Kun, L.E.; Packer, R.J.; Goldman, S.; Chintagumpala, M.; Wallace, D.; Takebe, N.; et al. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: A pediatric brain tumor consortium study. Clin. Cancer Res. 2013, 19, 6305–6312. [Google Scholar] [CrossRef]
- Sweet-Cordero, E.A.; Biegel, J.A. The genomic landscape of pediatric cancers: Implications for diagnosis and treatment. Science 2019, 363, 1170–1175. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Penas-Prado, M.; Zhou, S.; Wei, J.; Khatua, S.; Hodges, T.R.; Sanai, N.; Xiu, J.; Gatalica, Z.; Kim, L.; et al. Rethinking medulloblastoma from a targeted therapeutics perspective. J. Neurooncol. 2018, 139, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Touat, M.; Idbaih, A.; Sanson, M.; Ligon, K.L. Glioblastoma targeted therapy: Updated approaches from recent biological insights. Ann. Oncol. 2017, 28, 1457–1472. [Google Scholar] [CrossRef]
- Wang, X.; Guo, G.; Guan, H.; Yu, Y.; Lu, J.; Yu, J. Challenges and potential of PD-1/PD-L1 checkpoint blockade immunotherapy for glioblastoma. J. Exp. Clin. Cancer Res. 2019, 38, 87. [Google Scholar] [CrossRef]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of cancer therapy: Oncogene and non-oncogene addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef]
- Toledo, C.M.; Ding, Y.; Hoellerbauer, P.; Davis, R.J.; Basom, R.; Girard, E.J.; Lee, E.; Corrin, P.; Hart, T.; Bolouri, H.; et al. Genome-wide CRISPR-Cas9 Screens Reveal Loss of Redundancy between PKMYT1 and WEE1 in Glioblastoma Stem-like Cells. Cell Rep. 2015, 13, 2425–2439. [Google Scholar] [CrossRef]
- Haigis, K.M.; Cichowski, K.; Elledge, S.J. Tissue-specificity in cancer: The rule, not the exception. Science 2019, 363, 1150–1151. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, T.; Suresh, P.S. Emerging roles of MCPH1: Expedition from primary microcephaly to cancer. Eur. J. Cell Biol. 2014, 93, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Zhang, B.; Carlson, M.; Lu, K.V.; Zhu, S.; Felciano, R.M.; Laurance, M.F.; Zhao, W.; Qi, S.; Chen, Z.; et al. Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc. Natl. Acad. Sci. USA 2006, 103, 17402–17407. [Google Scholar]
- Lang, P.Y.; Gershon, T.R. A New Way to Treat Brain Tumors: Targeting Proteins Coded by Microcephaly Genes?: Brain tumors and microcephaly arise from opposing derangements regulating progenitor growth. Drivers of microcephaly could be attractive brain tumor targets. BioEssays 2018, 40, e1700243. [Google Scholar]
- Gibson, P.; Tong, Y.; Robinson, G.; Thompson, M.C.; Currle, D.S.; Eden, C.; Kranenburg, T.A.; Hogg, T.; Poppleton, H.; Martin, J.; et al. Subtypes of medulloblastoma have distinct developmental origins. Nature 2010, 468, 1095–1099. [Google Scholar]
- Smith, A.W.; Mehta, M.P.; Wernicke, A.G. Neural stem cells, the subventricular zone and radiotherapy: Implications for treating glioblastoma. J. Neurooncol. 2016, 128, 207–216. [Google Scholar]
- Passemard, S.; Kaindl, A.M.; Verloes, A. Microcephaly. Handb. Clin. Neurol. 2013, 111, 129–141. [Google Scholar]
- Woods, C.G.; Parker, A. Investigating microcephaly. Arch. Dis. Child. 2013, 98, 707–713. [Google Scholar] [CrossRef]
- Zaqout, S.; Morris-Rosendahl, D.; Kaindl, A.M. Autosomal Recessive Primary Microcephaly (MCPH): An Update. Neuropediatrics 2017, 48, 135–142. [Google Scholar]
- Abuelo, D. Microcephaly syndromes. Semin. Pediatr. Neurol. 2007, 14, 118–127. [Google Scholar] [CrossRef]
- Faheem, M.; Naseer, M.I.; Rasool, M.; Chaudhary, A.G.; Kumosani, T.A.; Ilyas, A.M.; Pushparaj, P.; Ahmed, F.; Algahtani, H.A.; Al-Qahtani, M.H.; et al. Molecular genetics of human primary microcephaly: An overview. BMC Med. Genomics 2015, 8 (Suppl. 1), S4. [Google Scholar] [CrossRef]
- O’Neill, R.S.; Schoborg, T.A.; Rusan, N.M. Same but different: Pleiotropy in centrosome-related microcephaly. Mol. Biol. Cell 2018, 29, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, F.T.; Tocco, C.; Pallavicini, G.; Liu, Y.; Vernì, F.; Merigliano, C.; Bonaccorsi, S.; El-Assawy, N.; Priano, L.; Gai, M.; et al. Citron Kinase Deficiency Leads to Chromosomal Instability and TP53-Sensitive Microcephaly. Cell Rep. 2017, 18, 1674–1686. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.E.; Garcia, I.; Crowther, A.J.; Li, S.; Stewart, A.; Liu, H.; Lough, K.J.; O’Neill, S.; Veleta, K.; Oyarzabal, E.A.; et al. Aspm sustains postnatal cerebellar neurogenesis and medulloblastoma growth in mice. Dev. Camb. Engl. 2015, 142, 3921–3932. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-W.; Tapias, A.; Bruhn, C.; Gruber, R.; Sukchev, M.; Wang, Z.-Q. DNA damage response in microcephaly development of MCPH1 mouse model. DNA Repair 2013, 12, 645–655. [Google Scholar] [CrossRef] [PubMed]
- El-Ghouzzi, V.; Bianchi, F.T.; Molineris, I.; Mounce, B.C.; Berto, G.E.; Rak, M.; Lebon, S.; Aubry, L.; Tocco, C.; Gai, M.; et al. ZIKA virus elicits P53 activation and genotoxic stress in human neural progenitors similar to mutations involved in severe forms of genetic microcephaly. Cell Death Dis. 2016, 7, e2440. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, Q.; Jiang, Y.; Ye, Q.; Xu, D.; Gao, F.; Xu, J.W.; Wang, R.; Zhu, X.; Shi, L.; et al. Disruption of glial cell development by Zika virus contributes to severe microcephalic newborn mice. Cell Discov. 2018, 4, 43. [Google Scholar] [CrossRef]
- Lubin, J.A.; Zhang, R.R.; Kuo, J.S. Zika Virus has Oncolytic Activity Against Glioblastoma Stem Cells. Neurosurgery 2018, 82, E113–E114. [Google Scholar] [CrossRef] [PubMed]
- Wood, H. Neuro-oncology: A new role for Zika virus in glioblastoma therapy? Nat. Rev. Neurol. 2017, 13, 640–641. [Google Scholar] [CrossRef]
- Bikeye, S.-N.N.; Colin, C.; Marie, Y.; Vampouille, R.; Ravassard, P.; Rousseau, A.; Boisselier, B.; Idbaih, A.; Calvo, C.F.; Leuraud, P.; et al. ASPM-associated stem cell proliferation is involved in malignant progression of gliomas and constitutes an attractive therapeutic target. Cancer Cell Int. 2010, 10, 1. [Google Scholar] [CrossRef]
- Kato, T.A.; Okayasu, R.; Jeggo, P.A.; Fujimori, A. ASPM influences DNA double-strand break repair and represents a potential target for radiotherapy. Int. J. Radiat. Biol. 2011, 87, 1189–1195. [Google Scholar] [CrossRef]
- Huang, W.; Wang, J.; Zhang, D.; Chen, W.; Hou, L.; Wu, X.; Lu, Y. Inhibition of KIF14 Suppresses Tumor Cell Growth and Promotes Apoptosis in Human Glioblastoma. Cell. Physiol. Biochem. 2015, 37, 1659–1670. [Google Scholar] [CrossRef]
- Li, K.K.-W.; Qi, Y.; Xia, T.; Chan, A.K.-Y.; Zhang, Z.-Y.; Aibaidula, A.; Zhang, R.; Zhou, L.; Yao, Y.; Ng, H.-K. The kinesin KIF14 is overexpressed in medulloblastoma and downregulation of KIF14 suppressed tumor proliferation and induced apoptosis. Lab. Investig. 2017, 97, 946–961. [Google Scholar] [CrossRef]
- Li, M.; Xiao, A.; Floyd, D.; Olmez, I.; Lee, J.; Godlewski, J.; Bronisz, A.; Bhat, K.P.L.; Sulman, E.P.; Nakano, I.; et al. CDK4/6 inhibition is more active against the glioblastoma proneural subtype. Oncotarget 2017, 8, 55319–55331. [Google Scholar] [CrossRef]
- Yin, L.; Li, H.; Liu, W.; Yao, Z.; Cheng, Z.; Zhang, H.; Zou, H. A highly potent CDK4/6 inhibitor was rationally designed to overcome blood brain barrier in gliobastoma therapy. Eur. J. Med. Chem. 2018, 144, 1–28. [Google Scholar] [CrossRef]
- Di Cunto, F.; Calautti, E.; Hsiao, J.; Ong, L.; Topley, G.; Turco, E.; Dotto, G.P. Citron rho-interacting kinase, a novel tissue-specific ser/thr kinase encompassing the Rho-Rac-binding protein Citron. J. Biol. Chem. 1998, 273, 29706–29711. [Google Scholar] [CrossRef]
- Madaule, P.; Eda, M.; Watanabe, N.; Fujisawa, K.; Matsuoka, T.; Bito, H.; Ishizaki, T.; Narumiya, S. Role of citron kinase as a target of the small GTPase Rho in cytokinesis. Nature 1998, 394, 491–494. [Google Scholar] [CrossRef]
- Naim, V.; Imarisio, S.; Di Cunto, F.; Gatti, M.; Bonaccorsi, S. Drosophila citron kinase is required for the final steps of cytokinesis. Mol. Biol. Cell 2004, 15, 5053–5063. [Google Scholar] [CrossRef]
- Liu, H.; Di Cunto, F.; Imarisio, S.; Reid, L.M. Citron kinase is a cell cycle-dependent, nuclear protein required for G2/M transition of hepatocytes. J. Biol. Chem. 2003, 278, 2541–2548. [Google Scholar] [CrossRef]
- Gai, M.; Bianchi, F.T.; Vagnoni, C.; Vernì, F.; Bonaccorsi, S.; Pasquero, S.; Berto, G.E.; Sgrò, F.; Chiotto, A.M.; Annaratone, L.; et al. ASPM and CITK regulate spindle orientation by affecting the dynamics of astral microtubules. EMBO Rep. 2016, 17, 1396–1409. [Google Scholar] [CrossRef]
- Bassi, Z.I.; Audusseau, M.; Riparbelli, M.G.; Callaini, G.; D’Avino, P.P. Citron kinase controls a molecular network required for midbody formation in cytokinesis. Proc. Natl. Acad. Sci. USA 2013, 110, 9782–9787. [Google Scholar] [CrossRef]
- Bassi, Z.I.; Verbrugghe, K.J.; Capalbo, L.; Gregory, S.; Montembault, E.; Glover, D.M.; D’Avino, P.P. Sticky/Citron kinase maintains proper RhoA localization at the cleavage site during cytokinesis. J. Cell Biol. 2011, 195, 595–603. [Google Scholar] [CrossRef]
- Dema, A.; Macaluso, F.; Sgrò, F.; Berto, G.E.; Bianchi, F.T.; Chiotto, A.A.; Pallavicini, G.; Di Cunto, F.; Gai, M. Citron kinase-dependent F-actin maintenance at midbody secondary ingression sites mediates abscission. J. Cell Sci. 2018, 131, jcs209080. [Google Scholar] [CrossRef]
- McKenzie, C.; Bassi, Z.I.; Debski, J.; Gottardo, M.; Callaini, G.; Dadlez, M.; D’Avino, P.P. Cross-regulation between Aurora B and Citron kinase controls midbody architecture in cytokinesis. Open Biol. 2016, 6, 1–15. [Google Scholar] [CrossRef]
- Sgrò, F.; Bianchi, F.T.; Falcone, M.; Pallavicini, G.; Gai, M.; Chiotto, A.M.A.; Berto, G.E.; Turco, E.; Chang, Y.J.; Huttner, W.B.; et al. Tissue-specific control of midbody microtubule stability by Citron kinase through modulation of TUBB3 phosphorylation. Cell Death Differ. 2016, 23, 801–813. [Google Scholar] [CrossRef]
- Di Cunto, F.; Imarisio, S.; Hirsch, E.; Broccoli, V.; Bulfone, A.; Migheli, A.; Atzori, C.; Turco, E.; Triolo, R.; Dotto, G.P.; et al. Defective neurogenesis in citron kinase knockout mice by altered cytokinesis and massive apoptosis. Neuron 2000, 28, 115–127. [Google Scholar] [CrossRef]
- Sarkisian, M.R.; Li, W.; Di Cunto, F.; D’Mello, S.R.; LoTurco, J.J. Citron-kinase, a protein essential to cytokinesis in neuronal progenitors, is deleted in the flathead mutant rat. J. Neurosci. 2002, 22, RC217. [Google Scholar] [CrossRef]
- Di Cunto, F.; Imarisio, S.; Camera, P.; Boitani, C.; Altruda, F.; Silengo, L. Essential role of citron kinase in cytokinesis of spermatogenic precursors. J. Cell Sci. 2002, 115, 4819–4826. [Google Scholar] [CrossRef]
- Harding, B.N.; Moccia, A.; Drunat, S.; Soukarieh, O.; Tubeuf, H.; Chitty, L.S.; Verloes, A.; Gressens, P.; El Ghouzzi, V.; Joriot, S.; et al. Mutations in Citron Kinase Cause Recessive Microlissencephaly with Multinucleated Neurons. Am. J. Hum. Genet. 2016, 99, 511–520. [Google Scholar] [CrossRef]
- Li, H.; Bielas, S.L.; Zaki, M.S.; Ismail, S.; Farfara, D.; Um, K.; Rosti, R.O.; Scott, E.C.; Tu, S.; Chi, N.C.; et al. Biallelic Mutations in Citron Kinase Link Mitotic Cytokinesis to Human Primary Microcephaly. Am. J. Hum. Genet. 2016, 99, 501–510. [Google Scholar] [CrossRef]
- Basit, S.; Al-Harbi, K.M.; Alhijji, S.A.M.; Albalawi, A.M.; Alharby, E.; Eldardear, A.; Samman, M.I. CIT, a gene involved in neurogenic cytokinesis, is mutated in human primary microcephaly. Hum. Genet. 2016, 135, 1199–1207. [Google Scholar] [CrossRef]
- Shaheen, R.; Hashem, A.; Abdel-Salam, G.M.H.; Al-Fadhli, F.; Ewida, N.; Alkuraya, F.S. Mutations in CIT, encoding citron rho-interacting serine/threonine kinase, cause severe primary microcephaly in humans. Hum. Genet. 2016, 135, 1191–1197. [Google Scholar] [CrossRef]
- D’Avino, P.P. Citron kinase - renaissance of a neglected mitotic kinase. J. Cell Sci. 2017, 130, 1701–1708. [Google Scholar] [CrossRef]
- Fu, Y.; Huang, J.; Wang, K.-S.; Zhang, X.; Han, Z.-G. RNA interference targeting CITRON can significantly inhibit the proliferation of hepatocellular carcinoma cells. Mol. Biol. Rep. 2011, 38, 693–702. [Google Scholar] [CrossRef]
- Ehrlichova, M.; Mohelnikova-Duchonova, B.; Hrdy, J.; Brynychova, V.; Mrhalova, M.; Kodet, R.; Rob, L.; Pluta, M.; Gut, I.; Soucek, P.; et al. The association of taxane resistance genes with the clinical course of ovarian carcinoma. Genomics 2013, 102, 96–101. [Google Scholar] [CrossRef]
- Tong, H.; Wang, J.; Chen, H.; Wang, Z.; Fan, H.; Ni, Z. Transcriptomic analysis of gene expression profiles of stomach carcinoma reveal abnormal expression of mitotic components. Life Sci. 2017, 170, 41–49. [Google Scholar] [CrossRef]
- Gruneberg, U.; Neef, R.; Li, X.; Chan, E.H.Y.; Chalamalasetty, R.B.; Nigg, E.A.; Barr, F.A. KIF14 and citron kinase act together to promote efficient cytokinesis. J. Cell Biol. 2006, 172, 363–372. [Google Scholar] [CrossRef]
- McKenzie, C.; D’Avino, P.P. Investigating cytokinesis failure as a strategy in cancer therapy. Oncotarget 2016, 7, 87323–87341. [Google Scholar] [CrossRef]
- Pallavicini, G.; Sgrò, F.; Garello, F.; Falcone, M.; Bitonto, V.; Berto, G.E.; Bianchi, F.T.; Gai, M.; Chiotto, A.M.A.; Filippi, M.; et al. Inactivation of Citron Kinase Inhibits Medulloblastoma Progression by Inducing Apoptosis and Cell Senescence. Cancer Res. 2018, 78, 4599–4612. [Google Scholar] [CrossRef]
- Lowery, D.M.; Lim, D.; Yaffe, M.B. Structure and function of Polo-like kinases. Oncogene 2005, 24, 248–259. [Google Scholar] [CrossRef]
- Maniswami, R.R.; Prashanth, S.; Karanth, A.V.; Koushik, S.; Govindaraj, H.; Mullangi, R.; Rajagopal, S.; Jegatheesan, S.K. PLK4: A link between centriole biogenesis and cancer. Expert Opin. Ther. Targets 2018, 22, 59–73. [Google Scholar] [CrossRef]
- Elia, A.E.H.; Cantley, L.C.; Yaffe, M.B. Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science 2003, 299, 1228–1231. [Google Scholar] [CrossRef]
- Karn, T.; Holtrich, U.; Wolf, G.; Hock, B.; Strebhardt, K.; Rubsamenwaigmann, H. Human SAK related to the PLK/polo family of cell cycle kinases shows high mRNA expression in testis. Oncol. Rep. 1997, 4, 505–510. [Google Scholar] [CrossRef]
- Sillibourne, J.E.; Tack, F.; Vloemans, N.; Boeckx, A.; Thambirajah, S.; Bonnet, P.; Ramaekers, F.C.S.; Bornens, M.; Grand-Perret, T. Autophosphorylation of polo-like kinase 4 and its role in centriole duplication. Mol. Biol. Cell 2010, 21, 547–561. [Google Scholar] [CrossRef] [PubMed]
- Hudson, J.W.; Kozarova, A.; Cheung, P.; Macmillan, J.C.; Swallow, C.J.; Cross, J.C.; Dennis, J.W. Late mitotic failure in mice lacking Sak, a polo-like kinase. Curr. Biol. 2001, 11, 441–446. [Google Scholar] [CrossRef]
- Kleylein-Sohn, J.; Westendorf, J.; Le Clech, M.; Habedanck, R.; Stierhof, Y.-D.; Nigg, E.A. Plk4-induced centriole biogenesis in human cells. Dev. Cell 2007, 13, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Bettencourt-Dias, M.; Rodrigues-Martins, A.; Carpenter, L.; Riparbelli, M.; Lehmann, L.; Gatt, M.K.; Carmo, N.; Balloux, F.; Callaini, G.; Glover, D.M. SAK/PLK4 is required for centriole duplication and flagella development. Curr. Biol. 2005, 15, 2199–2207. [Google Scholar] [CrossRef]
- Duensing, A.; Liu, Y.; Perdreau, S.A.; Kleylein-Sohn, J.; Nigg, E.A.; Duensing, S. Centriole overduplication through the concurrent formation of multiple daughter centrioles at single maternal templates. Oncogene 2007, 26, 6280–6288. [Google Scholar] [CrossRef] [PubMed]
- Habedanck, R.; Stierhof, Y.-D.; Wilkinson, C.J.; Nigg, E.A. The Polo kinase Plk4 functions in centriole duplication. Nat. Cell Biol. 2005, 7, 1140–1146. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-S.; Park, J.-E.; Shukla, A.; Choi, S.; Murugan, R.N.; Lee, J.H.; Ahn, M.; Rhee, K.; Bang, J.K.; Kim, B.Y.; et al. Hierarchical recruitment of Plk4 and regulation of centriole biogenesis by two centrosomal scaffolds, Cep192 and Cep152. Proc. Natl. Acad. Sci. USA 2013, 110, E4849–E4857. [Google Scholar] [CrossRef]
- Sonnen, K.F.; Gabryjonczyk, A.-M.; Anselm, E.; Stierhof, Y.-D.; Nigg, E.A. Human Cep192 and Cep152 cooperate in Plk4 recruitment and centriole duplication. J. Cell Sci. 2013, 126, 3223–3233. [Google Scholar] [CrossRef]
- Kratz, A.-S.; Bärenz, F.; Richter, K.T.; Hoffmann, I. Plk4-dependent phosphorylation of STIL is required for centriole duplication. Biol. Open 2015, 4, 370–377. [Google Scholar] [CrossRef]
- Ohta, M.; Ashikawa, T.; Nozaki, Y.; Kozuka-Hata, H.; Goto, H.; Inagaki, M.; Oyama, M.; Kitagawa, D. Direct interaction of Plk4 with STIL ensures formation of a single procentriole per parental centriole. Nat. Commun. 2014, 5, 5267. [Google Scholar] [CrossRef]
- Petrinac, S.; Ganuelas, M.L.; Bonni, S.; Nantais, J.; Hudson, J.W. Polo-like kinase 4 phosphorylates Chk2. Cell Cycle 2009, 8, 327–329. [Google Scholar] [CrossRef]
- Rosario, C.O.; Ko, M.A.; Haffani, Y.Z.; Gladdy, R.A.; Paderova, J.; Pollett, A.; Squire, J.A.; Dennis, J.W.; Swallow, C.J. Plk4 is required for cytokinesis and maintenance of chromosomal stability. Proc. Natl. Acad. Sci. USA 2010, 107, 6888–6893. [Google Scholar] [CrossRef]
- Holland, A.J.; Fachinetti, D.; Da Cruz, S.; Zhu, Q.; Vitre, B.; Lince-Faria, M.; Chen, D.; Parish, N.; Verma, I.M.; Bettencourt-Dias, M.; et al. Polo-like kinase 4 controls centriole duplication but does not directly regulate cytokinesis. Mol. Biol. Cell 2012, 23, 1838–1845. [Google Scholar] [CrossRef]
- Martin, C.-A.; Ahmad, I.; Klingseisen, A.; Hussain, M.S.; Bicknell, L.S.; Leitch, A.; Nürnberg, G.; Toliat, M.R.; Murray, J.E.; Hunt, D.; et al. Mutations in PLK4, encoding a master regulator of centriole biogenesis, cause microcephaly, growth failure and retinopathy. Nat. Genet. 2014, 46, 1283–1292. [Google Scholar] [CrossRef]
- Dinçer, T.; Yorgancıoğlu-Budak, G.; Ölmez, A.; Er, İ.; Dodurga, Y.; Özdemir, Ö.M.; Toraman, B.; Yıldırım, A.; Sabir, N.; Akarsu, N.A.; et al. Analysis of centrosome and DNA damage response in PLK4 associated Seckel syndrome. Eur. J. Hum. Genet. 2017, 25, 1118–1125. [Google Scholar] [CrossRef]
- Ko, M.A.; Rosario, C.O.; Hudson, J.W.; Kulkarni, S.; Pollett, A.; Dennis, J.W.; Swallow, C.J. Plk4 haploinsufficiency causes mitotic infidelity and carcinogenesis. Nat. Genet. 2005, 37, 883–888. [Google Scholar] [CrossRef]
- Li, Z.; Dai, K.; Wang, C.; Song, Y.; Gu, F.; Liu, F.; Fu, L. Expression of Polo-Like Kinase 4(PLK4) in Breast Cancer and Its Response to Taxane-Based Neoadjuvant Chemotherapy. J. Cancer 2016, 7, 1125–1132. [Google Scholar] [CrossRef]
- Kazazian, K.; Go, C.; Wu, H.; Brashavitskaya, O.; Xu, R.; Dennis, J.W.; Gingras, A.-C.; Swallow, C.J. Plk4 Promotes Cancer Invasion and Metastasis through Arp2/3 Complex Regulation of the Actin Cytoskeleton. Cancer Res. 2017, 77, 434–447. [Google Scholar] [CrossRef]
- Mason, J.M.; Lin, D.C.-C.; Wei, X.; Che, Y.; Yao, Y.; Kiarash, R.; Cescon, D.W.; Fletcher, G.C.; Awrey, D.E.; Bray, M.R.; et al. Functional characterization of CFI-400945, a Polo-like kinase 4 inhibitor, as a potential anticancer agent. Cancer Cell 2014, 26, 163–176. [Google Scholar] [CrossRef]
- Lohse, I.; Mason, J.; Cao, P.M.; Pintilie, M.; Bray, M.; Hedley, D.W. Activity of the novel polo-like kinase 4 inhibitor CFI-400945 in pancreatic cancer patient-derived xenografts. Oncotarget 2017, 8, 3064–3071. [Google Scholar] [CrossRef]
- Kawakami, M.; Mustachio, L.M.; Zheng, L.; Chen, Y.; Rodriguez-Canales, J.; Mino, B.; Kurie, J.M.; Roszik, J.; Villalobos, P.A.; Thu, K.L.; et al. Polo-like kinase 4 inhibition produces polyploidy and apoptotic death of lung cancers. Proc. Natl. Acad. Sci. USA 2018, 115, 1913–1918. [Google Scholar] [CrossRef]
- Oegema, K.; Davis, R.L.; Lara-Gonzalez, P.; Desai, A.; Shiau, A.K. CFI-400945 is not a selective cellular PLK4 inhibitor. Proc. Natl. Acad. Sci. USA 2018, 115, E10808–E10809. [Google Scholar] [CrossRef]
- Zitouni, S.; Nabais, C.; Jana, S.C.; Guerrero, A.; Bettencourt-Dias, M. Polo-like kinases: Structural variations lead to multiple functions. Nat. Rev. Mol. Cell Biol. 2014, 15, 433–452. [Google Scholar] [CrossRef]
- Wong, Y.L.; Anzola, J.V.; Davis, R.L.; Yoon, M.; Motamedi, A.; Kroll, A.; Seo, C.P.; Hsia, J.E.; Kim, S.K.; Mitchell, J.W.; et al. Cell biology. Reversible centriole depletion with an inhibitor of Polo-like kinase 4. Science 2015, 348, 1155–1160. [Google Scholar] [CrossRef]
- Meitinger, F.; Anzola, J.V.; Kaulich, M.; Richardson, A.; Stender, J.D.; Benner, C.; Glass, C.K.; Dowdy, S.F.; Desai, A.; Shiau, A.K.; et al. 53BP1 and USP28 mediate p53 activation and G1 arrest after centrosome loss or extended mitotic duration. J. Cell Biol. 2016, 214, 155–166. [Google Scholar] [CrossRef]
- Sredni, S.T.; Bailey, A.W.; Suri, A.; Hashizume, R.; He, X.; Louis, N.; Gokirmak, T.; Piper, D.R.; Watterson, D.M.; Tomita, T. Inhibition of polo-like kinase 4 (PLK4): A new therapeutic option for rhabdoid tumors and pediatric medulloblastoma. Oncotarget 2017, 8, 111190–111212. [Google Scholar] [CrossRef]
- Sredni, S.T.; Tomita, T. The polo-like kinase 4 gene (PLK4) is overexpressed in pediatric medulloblastoma. Childs Nerv. Syst. 2017, 33, 1031. [Google Scholar] [CrossRef]
- Bailey, A.W.; Suri, A.; Chou, P.M.; Pundy, T.; Gadd, S.; Raimondi, S.L.; Tomita, T.; Sredni, S.T. Polo-Like Kinase 4 (PLK4) Is Overexpressed in Central Nervous System Neuroblastoma (CNS-NB). Bioengineering 2018, 5, 96. [Google Scholar] [CrossRef]
- Sredni, S.T.; Suzuki, M.; Yang, J.-P.; Topczewski, J.; Bailey, A.W.; Gokirmak, T.; Gross, J.N.; de Andrade, A.; Kondo, A.; Piper, D.R.; et al. A functional screening of the kinome identifies the Polo-like kinase 4 as a potential therapeutic target for malignant rhabdoid tumors, and possibly, other embryonal tumors of the brain. Pediatr. Blood Cancer 2017, 64, e26551. [Google Scholar] [CrossRef]
- Wang, J.; Zuo, J.; Wang, M.; Ma, X.; Gao, K.; Bai, X.; Wang, N.; Xie, W.; Liu, H. Polo-like kinase 4 promotes tumorigenesis and induces resistance to radiotherapy in glioblastoma. Oncol. Rep. 2019, 41, 2159–2167. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Z.; Huang, K.; Liu, Y.; Wei, C.; Zhou, J.; Zhang, W.; Wang, Q.; Liang, H.; Zhang, A.; et al. PLK4 is a determinant of temozolomide sensitivity through phosphorylation of IKBKE in glioblastoma. Cancer Lett. 2019, 443, 91–107. [Google Scholar] [CrossRef]
- Fayard, E.; Tintignac, L.A.; Baudry, A.; Hemmings, B.A. Protein kinase B/Akt at a glance. J. Cell Sci. 2005, 118, 5675–5678. [Google Scholar] [CrossRef]
- Hanada, M.; Feng, J.; Hemmings, B.A. Structure, regulation and function of PKB/AKT--a major therapeutic target. Biochim. Biophys. Acta 2004, 1697, 3–16. [Google Scholar] [CrossRef]
- Yang, Z.-Z.; Tschopp, O.; Baudry, A.; Dümmler, B.; Hynx, D.; Hemmings, B.A. Physiological functions of protein kinase B/Akt. Biochem. Soc. Trans. 2004, 32, 350–354. [Google Scholar] [CrossRef]
- Osaki, M.; Oshimura, M.; Ito, H. PI3K-Akt pathway: Its functions and alterations in human cancer. Apoptosis Int. J. Program. Cell Death 2004, 9, 667–676. [Google Scholar] [CrossRef]
- Brodbeck, D.; Cron, P.; Hemmings, B.A. A human protein kinase Bgamma with regulatory phosphorylation sites in the activation loop and in the C-terminal hydrophobic domain. J. Biol. Chem. 1999, 274, 9133–9136. [Google Scholar] [CrossRef]
- Masure, S.; Haefner, B.; Wesselink, J.J.; Hoefnagel, E.; Mortier, E.; Verhasselt, P.; Tuytelaars, A.; Gordon, R.; Richardson, A. Molecular cloning, expression and characterization of the human serine/threonine kinase Akt-3. Eur. J. Biochem. 1999, 265, 353–360. [Google Scholar] [CrossRef]
- Poduri, A.; Evrony, G.D.; Cai, X.; Elhosary, P.C.; Beroukhim, R.; Lehtinen, M.K.; Hills, L.B.; Heinzen, E.L.; Hill, A.; Hill, R.S.; et al. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron 2012, 74, 41–48. [Google Scholar] [CrossRef]
- Easton, R.M.; Cho, H.; Roovers, K.; Shineman, D.W.; Mizrahi, M.; Forman, M.S.; Lee, V.M.-Y.; Szabolcs, M.; de Jong, R.; Oltersdorf, T.; et al. Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol. Cell. Biol. 2005, 25, 1869–1878. [Google Scholar] [CrossRef]
- Tschopp, O.; Yang, Z.-Z.; Brodbeck, D.; Dummler, B.A.; Hemmings-Mieszczak, M.; Watanabe, T.; Michaelis, T.; Frahm, J.; Hemmings, B.A. Essential role of protein kinase B gamma (PKB gamma/Akt3) in postnatal brain development but not in glucose homeostasis. Dev. Camb. Engl. 2005, 132, 2943–2954. [Google Scholar]
- Boland, E.; Clayton-Smith, J.; Woo, V.G.; McKee, S.; Manson, F.D.C.; Medne, L.; Zackai, E.; Swanson, E.A.; Fitzpatrick, D.; Millen, K.J.; et al. Mapping of deletion and translocation breakpoints in 1q44 implicates the serine/threonine kinase AKT3 in postnatal microcephaly and agenesis of the corpus callosum. Am. J. Hum. Genet. 2007, 81, 292–303. [Google Scholar] [CrossRef]
- Rivière, J.-B.; Mirzaa, G.M.; O’Roak, B.J.; Beddaoui, M.; Alcantara, D.; Conway, R.L.; St-Onge, J.; Schwartzentruber, J.A.; Gripp, K.W.; Nikkel, S.M.; et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat. Genet. 2012, 44, 934–940. [Google Scholar] [CrossRef]
- Lee, J.H.; Huynh, M.; Silhavy, J.L.; Kim, S.; Dixon-Salazar, T.; Heiberg, A.; Scott, E.; Bafna, V.; Hill, K.J.; Collazo, A.; et al. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat. Genet. 2012, 44, 941–945. [Google Scholar] [CrossRef]
- Toker, A. Achieving specificity in Akt signaling in cancer. Adv. Biol. Regul. 2012, 52, 78–87. [Google Scholar] [CrossRef]
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.-H. AKT as a Therapeutic Target for Cancer. Cancer Res. 2019, 79, 1019–1031. [Google Scholar] [CrossRef]
- Stahl, J.M.; Sharma, A.; Cheung, M.; Zimmerman, M.; Cheng, J.Q.; Bosenberg, M.W.; Kester, M.; Sandirasegarane, L.; Robertson, G.P. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res. 2004, 64, 7002–7010. [Google Scholar] [CrossRef]
- Ichimura, K.; Vogazianou, A.P.; Liu, L.; Pearson, D.M.; Bäcklund, L.M.; Plant, K.; Baird, K.; Langford, C.F.; Gregory, S.G.; Collins, V.P. 1p36 is a preferential target of chromosome 1 deletions in astrocytic tumours and homozygously deleted in a subset of glioblastomas. Oncogene 2008, 27, 2097–2108. [Google Scholar] [CrossRef]
- Kagawa, N.; Maruno, M.; Suzuki, T.; Hashiba, T.; Hashimoto, N.; Izumoto, S.; Yoshimine, T. Detection of genetic and chromosomal aberrations in medulloblastomas and primitive neuroectodermal tumors with DNA microarrays. Brain Tumor Pathol. 2006, 23, 41–47. [Google Scholar] [CrossRef]
- Endersby, R.; Zhu, X.; Hay, N.; Ellison, D.W.; Baker, S.J. Nonredundant functions for Akt isoforms in astrocyte growth and gliomagenesis in an orthotopic transplantation model. Cancer Res. 2011, 71, 4106–4116. [Google Scholar] [CrossRef]
- Paul-Samojedny, M.; Pudełko, A.; Suchanek-Raif, R.; Kowalczyk, M.; Fila-Daniłow, A.; Borkowska, P.; Kowalski, J. Knockdown of the AKT3 (PKBγ), PI3KCA, and VEGFR2 genes by RNA interference suppresses glioblastoma multiforme T98G cells invasiveness in vitro. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 3263–3277. [Google Scholar] [CrossRef]
- Turner, K.M.; Sun, Y.; Ji, P.; Granberg, K.J.; Bernard, B.; Hu, L.; Cogdell, D.E.; Zhou, X.; Yli-Harja, O.; Nykter, M.; et al. Genomically amplified Akt3 activates DNA repair pathway and promotes glioma progression. Proc. Natl. Acad. Sci. USA 2015, 112, 3421–3426. [Google Scholar] [CrossRef]
- Paul-Samojedny, M.; Pudełko, A.; Kowalczyk, M.; Fila-Daniłow, A.; Suchanek-Raif, R.; Borkowska, P.; Kowalski, J. Combination Therapy with AKT3 and PI3KCA siRNA Enhances the Antitumor Effect of Temozolomide and Carmustine in T98G Glioblastoma Multiforme Cells. BioDrugs 2016, 30, 129–144. [Google Scholar] [CrossRef]
- Becker, W.; Joost, H.G. Structural and functional characteristics of Dyrk, a novel subfamily of protein kinases with dual specificity. Prog. Nucleic Acid Res. Mol. Biol. 1999, 62, 1–17. [Google Scholar]
- Kay, L.J.; Smulders-Srinivasan, T.K.; Soundararajan, M. Understanding the Multifaceted Role of Human Down Syndrome Kinase DYRK1A. Adv. Protein Chem. Struct. Biol. 2016, 105, 127–171. [Google Scholar]
- Galceran, J.; de Graaf, K.; Tejedor, F.J.; Becker, W. The MNB/DYRK1A protein kinase: Genetic and biochemical properties. J. Neural Transm. Suppl. 2003, 67, 139–148. [Google Scholar]
- Feki, A.; Hibaoui, Y. DYRK1A Protein, A Promising Therapeutic Target to Improve Cognitive Deficits in Down Syndrome. Brain Sci. 2018, 8, 187. [Google Scholar] [CrossRef]
- Song, W.J.; Sternberg, L.R.; Kasten-Sportès, C.; Keuren, M.L.; Chung, S.H.; Slack, A.C.; Miller, D.E.; Glover, T.W.; Chiang, P.W.; Lou, L.; et al. Isolation of human and murine homologues of the Drosophila minibrain gene: Human homologue maps to 21q22.2 in the Down syndrome “critical region”. Genomics 1996, 38, 331–339. [Google Scholar]
- Fotaki, V.; Dierssen, M.; Alcántara, S.; Martínez, S.; Martí, E.; Casas, C.; Visa, J.; Soriano, E.; Estivill, X.; Arbonés, M.L. Dyrk1A haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Mol. Cell. Biol. 2002, 22, 6636–6647. [Google Scholar] [CrossRef]
- Van Bon, B.W.M.; Hoischen, A.; Hehir-Kwa, J.; de Brouwer, A.P.M.; Ruivenkamp, C.; Gijsbers, A.C.J.; Marcelis, C.L.; de Leeuw, N.; Veltman, J.A.; Brunner, H.G.; et al. Intragenic deletion in DYRK1A leads to mental retardation and primary microcephaly. Clin. Genet. 2011, 79, 296–299. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Vives, L.; Fu, W.; Egertson, J.D.; Stanaway, I.B.; Phelps, I.G.; Carvill, G.; Kumar, A.; Lee, C.; Ankenman, K.; et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 2012, 338, 1619–1622. [Google Scholar] [CrossRef]
- Bellmaine, S.F.; Ovchinnikov, D.A.; Manallack, D.T.; Cuddy, C.E.; Elefanty, A.G.; Stanley, E.G.; Wolvetang, E.J.; Williams, S.J.; Pera, M. Inhibition of DYRK1A disrupts neural lineage specificationin human pluripotent stem cells. eLife 2017, 6, e24502. [Google Scholar] [CrossRef]
- Kurabayashi, N.; Nguyen, M.D.; Sanada, K. Triple play of DYRK1A kinase in cortical progenitor cells of Trisomy 21. Neurosci. Res. 2019, 138, 19–25. [Google Scholar] [CrossRef]
- Abbassi, R.; Johns, T.G.; Kassiou, M.; Munoz, L. DYRK1A in neurodegeneration and cancer: Molecular basis and clinical implications. Pharmacol. Ther. 2015, 151, 87–98. [Google Scholar] [CrossRef]
- Seifert, A.; Allan, L.A.; Clarke, P.R. DYRK1A phosphorylates caspase 9 at an inhibitory site and is potently inhibited in human cells by harmine. FEBS J. 2008, 275, 6268–6280. [Google Scholar] [CrossRef]
- Laguna, A.; Aranda, S.; Barallobre, M.J.; Barhoum, R.; Fernández, E.; Fotaki, V.; Delabar, J.M.; de la Luna, S.; de la Villa, P.; Arbonés, M.L. The protein kinase DYRK1A regulates caspase-9-mediated apoptosis during retina development. Dev. Cell 2008, 15, 841–853. [Google Scholar] [CrossRef]
- Arron, J.R.; Winslow, M.M.; Polleri, A.; Chang, C.-P.; Wu, H.; Gao, X.; Neilson, J.R.; Chen, L.; Heit, J.J.; Kim, S.K.; et al. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature 2006, 441, 595–600. [Google Scholar] [CrossRef]
- Gwack, Y.; Sharma, S.; Nardone, J.; Tanasa, B.; Iuga, A.; Srikanth, S.; Okamura, H.; Bolton, D.; Feske, S.; Hogan, P.G.; et al. A genome-wide Drosophila RNAi screen identifies DYRK-family kinases as regulators of NFAT. Nature 2006, 441, 646–650. [Google Scholar] [CrossRef]
- Guo, X.; Williams, J.G.; Schug, T.T.; Li, X. DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. J. Biol. Chem. 2010, 285, 13223–13232. [Google Scholar] [CrossRef]
- Ori-McKenney, K.M.; McKenney, R.J.; Huang, H.H.; Li, T.; Meltzer, S.; Jan, L.Y.; Vale, R.D.; Wiita, A.P.; Jan, Y.N. Phosphorylation of β-Tubulin by the Down Syndrome Kinase, Minibrain/DYRK1a, Regulates Microtubule Dynamics and Dendrite Morphogenesis. Neuron 2016, 90, 551–563. [Google Scholar] [CrossRef]
- Stotani, S.; Giordanetto, F.; Medda, F. DYRK1A inhibition as potential treatment for Alzheimer’s disease. Future Med. Chem. 2016, 8, 681–696. [Google Scholar] [CrossRef]
- Lee, S.B.; Frattini, V.; Bansal, M.; Castano, A.M.; Sherman, D.; Hutchinson, K.; Bruce, J.N.; Califano, A.; Liu, G.; Cardozo, T.; et al. An ID2-dependent mechanism for VHL inactivation in cancer. Nature 2016, 529, 172–177. [Google Scholar] [CrossRef]
- Pozo, N.; Zahonero, C.; Fernández, P.; Liñares, J.M.; Ayuso, A.; Hagiwara, M.; Pérez, A.; Ricoy, J.R.; Hernández-Laín, A.; Sepúlveda, J.M.; et al. Inhibition of DYRK1A destabilizes EGFR and reduces EGFR-dependent glioblastoma growth. J. Clin. Invest. 2013, 123, 2475–2487. [Google Scholar] [CrossRef]
- Zhou, Q.; Phoa, A.F.; Abbassi, R.H.; Hoque, M.; Reekie, T.A.; Font, J.S.; Ryan, R.M.; Stringer, B.W.; Day, B.W.; Johns, T.G.; et al. Structural Optimization and Pharmacological Evaluation of Inhibitors Targeting Dual-Specificity Tyrosine Phosphorylation-Regulated Kinases (DYRK) and CDC-like kinases (CLK) in Glioblastoma. J. Med. Chem. 2017, 60, 2052–2070. [Google Scholar] [CrossRef]
- Shimokawa, T.; Rahman, M.F.-U.; Tostar, U.; Sonkoly, E.; Ståhle, M.; Pivarcsi, A.; Palaniswamy, R.; Zaphiropoulos, P.G. RNA editing of the GLI1 transcription factor modulates the output of Hedgehog signaling. RNA Biol. 2013, 10, 321–333. [Google Scholar] [CrossRef]
- Debant, A.; Serra-Pagès, C.; Seipel, K.; O’Brien, S.; Tang, M.; Park, S.H.; Streuli, M. The multidomain protein Trio binds the LAR transmembrane tyrosine phosphatase, contains a protein kinase domain, and has separate rac-specific and rho-specific guanine nucleotide exchange factor domains. Proc. Natl. Acad. Sci. USA 1996, 93, 5466–5471. [Google Scholar] [CrossRef]
- Schmidt, S.; Debant, A. Function and regulation of the Rho guanine nucleotide exchange factor Trio. Small GTPases 2014, 5, e29769. [Google Scholar] [CrossRef]
- Blangy, A.; Vignal, E.; Schmidt, S.; Debant, A.; Gauthier-Rouvière, C.; Fort, P. TrioGEF1 controls Rac- and Cdc42-dependent cell structures through the direct activation of rhoG. J. Cell Sci. 2000, 113, 729–739. [Google Scholar]
- Medley, Q.G.; Buchbinder, E.G.; Tachibana, K.; Ngo, H.; Serra-Pagès, C.; Streuli, M. Signaling between focal adhesion kinase and trio. J. Biol. Chem. 2003, 278, 13265–13270. [Google Scholar] [CrossRef]
- O’Brien, S.P.; Seipel, K.; Medley, Q.G.; Bronson, R.; Segal, R.; Streuli, M. Skeletal muscle deformity and neuronal disorder in Trio exchange factor-deficient mouse embryos. Proc. Natl. Acad. Sci. USA 2000, 97, 12074–12078. [Google Scholar] [CrossRef]
- Varvagiannis, K.; Vissers, L.E.; Baralle, D.; de Vries, B.B. TRIO-Related Intellectual Disability. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Ba, W.; Yan, Y.; Reijnders, M.R.F.; Schuurs-Hoeijmakers, J.H.M.; Feenstra, I.; Bongers, E.M.H.F.; Bosch, D.G.M.; De Leeuw, N.; Pfundt, R.; Gilissen, C.; et al. TRIO loss of function is associated with mild intellectual disability and affects dendritic branching and synapse function. Hum. Mol. Genet. 2016, 25, 892–902. [Google Scholar] [CrossRef]
- Estrach, S.; Schmidt, S.; Diriong, S.; Penna, A.; Blangy, A.; Fort, P.; Debant, A. The Human Rho-GEF trio and its target GTPase RhoG are involved in the NGF pathway, leading to neurite outgrowth. Curr. Biol. CB 2002, 12, 307–312. [Google Scholar] [CrossRef]
- DeGeer, J.; Boudeau, J.; Schmidt, S.; Bedford, F.; Lamarche-Vane, N.; Debant, A. Tyrosine phosphorylation of the Rho guanine nucleotide exchange factor Trio regulates netrin-1/DCC-mediated cortical axon outgrowth. Mol. Cell. Biol. 2013, 33, 739–751. [Google Scholar] [CrossRef]
- Fortin, S.P.; Ennis, M.J.; Schumacher, C.A.; Zylstra-Diegel, C.R.; Williams, B.O.; Ross, J.T.D.; Winkles, J.A.; Loftus, J.C.; Symons, M.H.; Tran, N.L. Cdc42 and the guanine nucleotide exchange factors Ect2 and trio mediate Fn14-induced migration and invasion of glioblastoma cells. Mol. Cancer Res. MCR 2012, 10, 958–968. [Google Scholar] [CrossRef]
- Salhia, B.; Tran, N.L.; Chan, A.; Wolf, A.; Nakada, M.; Rutka, F.; Ennis, M.; McDonough, W.S.; Berens, M.E.; Symons, M.; et al. The guanine nucleotide exchange factors trio, Ect2, and Vav3 mediate the invasive behavior of glioblastoma. Am. J. Pathol. 2008, 173, 1828–1838. [Google Scholar] [CrossRef]
- Bouquier, N.; Fromont, S.; Debant, A.; Schmidt, S. Random mutagenesis of peptide aptamers as an optimization strategy for inhibitor screening. Methods Mol. Biol. 2012, 928, 97–118. [Google Scholar]
- Yano, T.; Yamazaki, Y.; Adachi, M.; Okawa, K.; Fort, P.; Uji, M.; Tsukita, S.; Tsukita, S. Tara up-regulates E-cadherin transcription by binding to the Trio RhoGEF and inhibiting Rac signaling. J. Cell Biol. 2011, 193, 319–332. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).