Fructose-1,6-Bisphosphate Protects Hippocampal Rat Slices from NMDA Excitotoxicity

 ,
, 
 ,
,  , ,
, , 
 ,
,  ,
, 
Abstract
:1. Introduction
2. Results
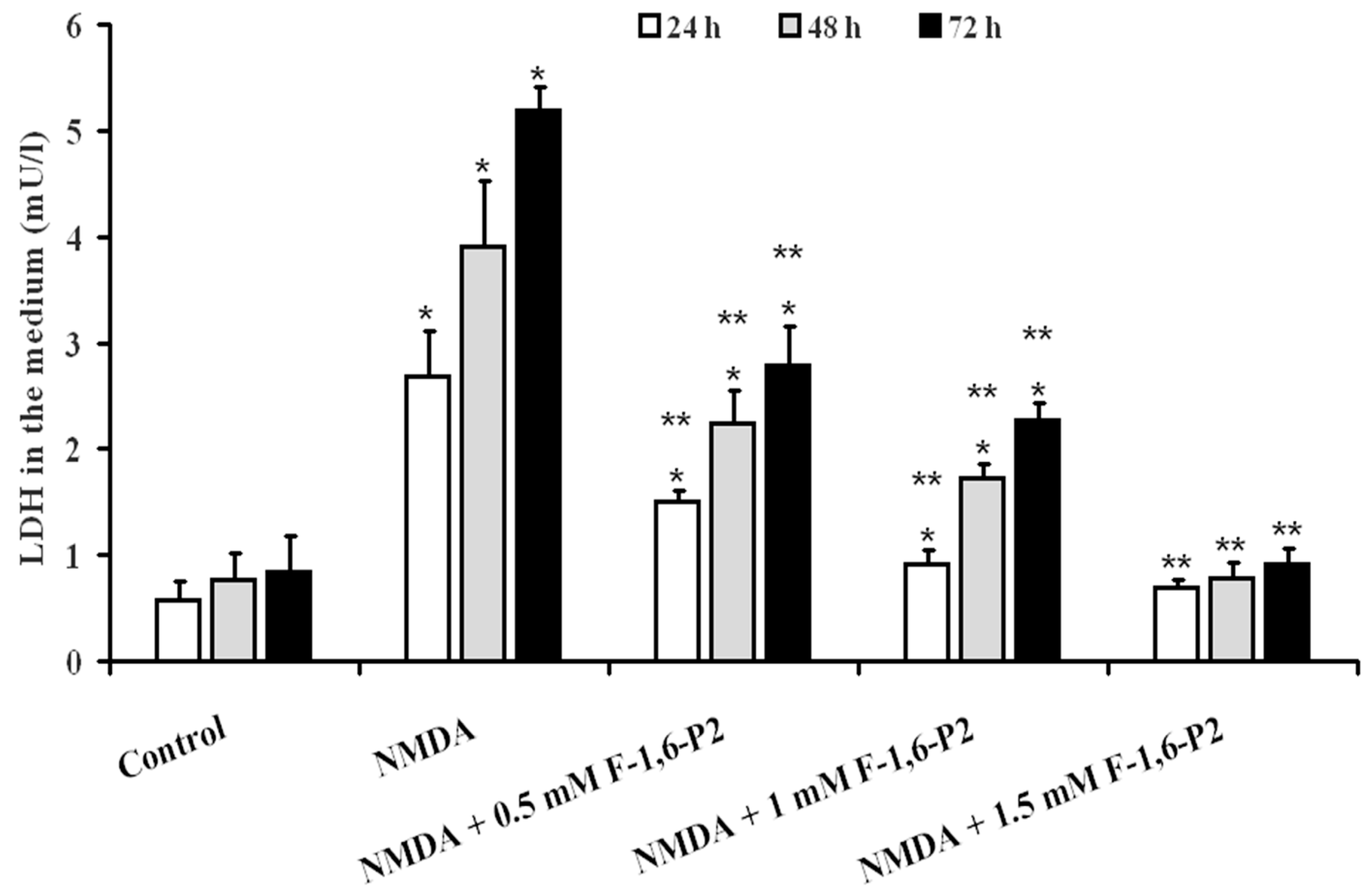
2.1. NMDA Cytotoxicity and Protection by F-1,6-P2
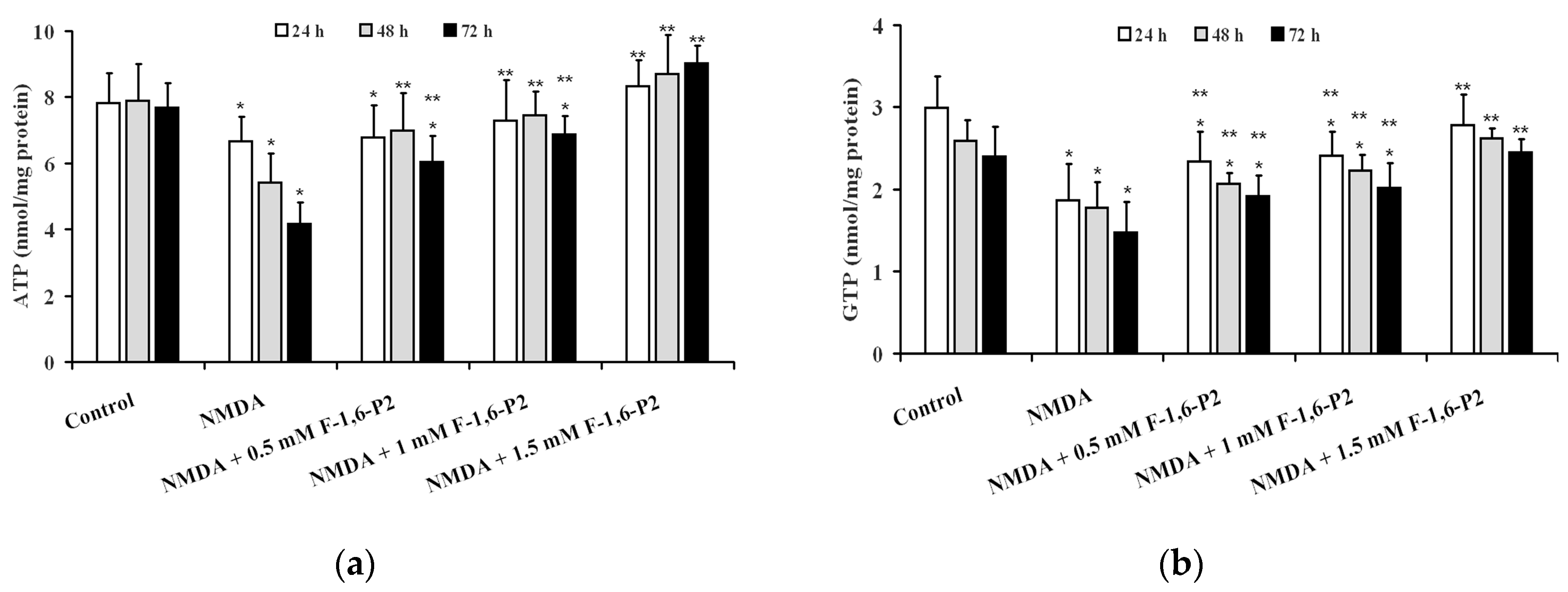
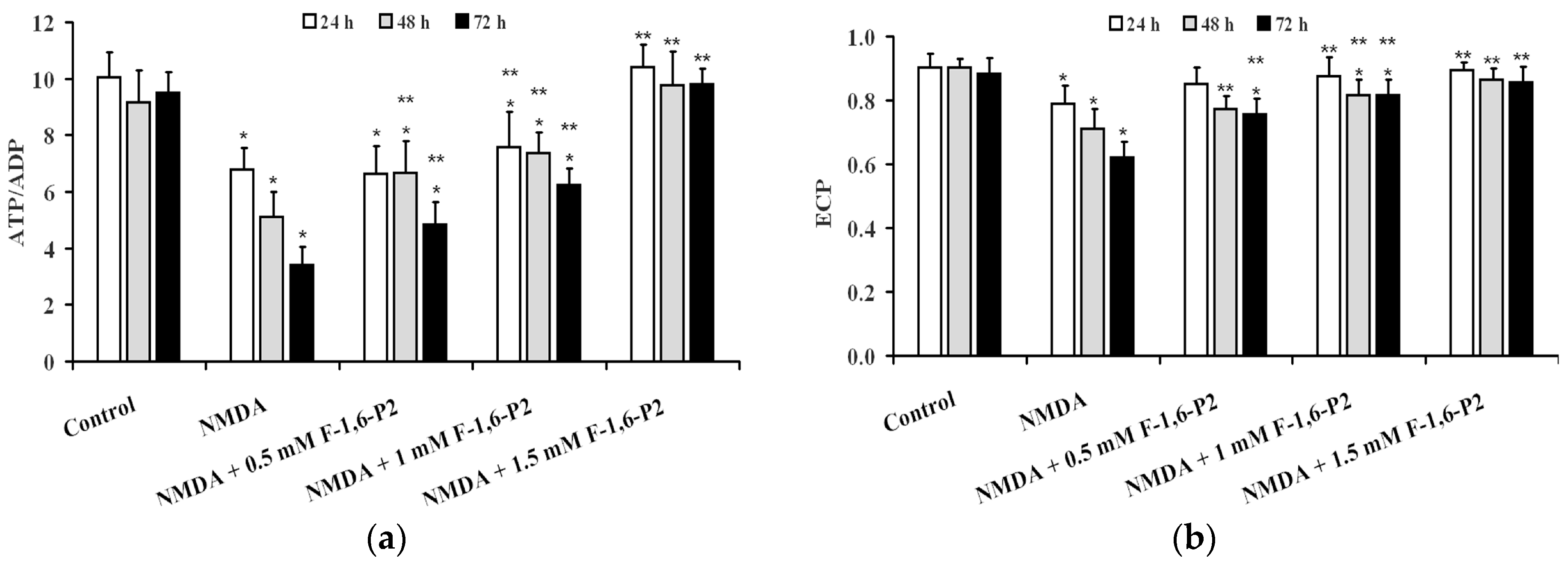
2.2. NMDA-Induced Imbalance of OHSC Energy Metabolism and Protection by F-1,6-P2
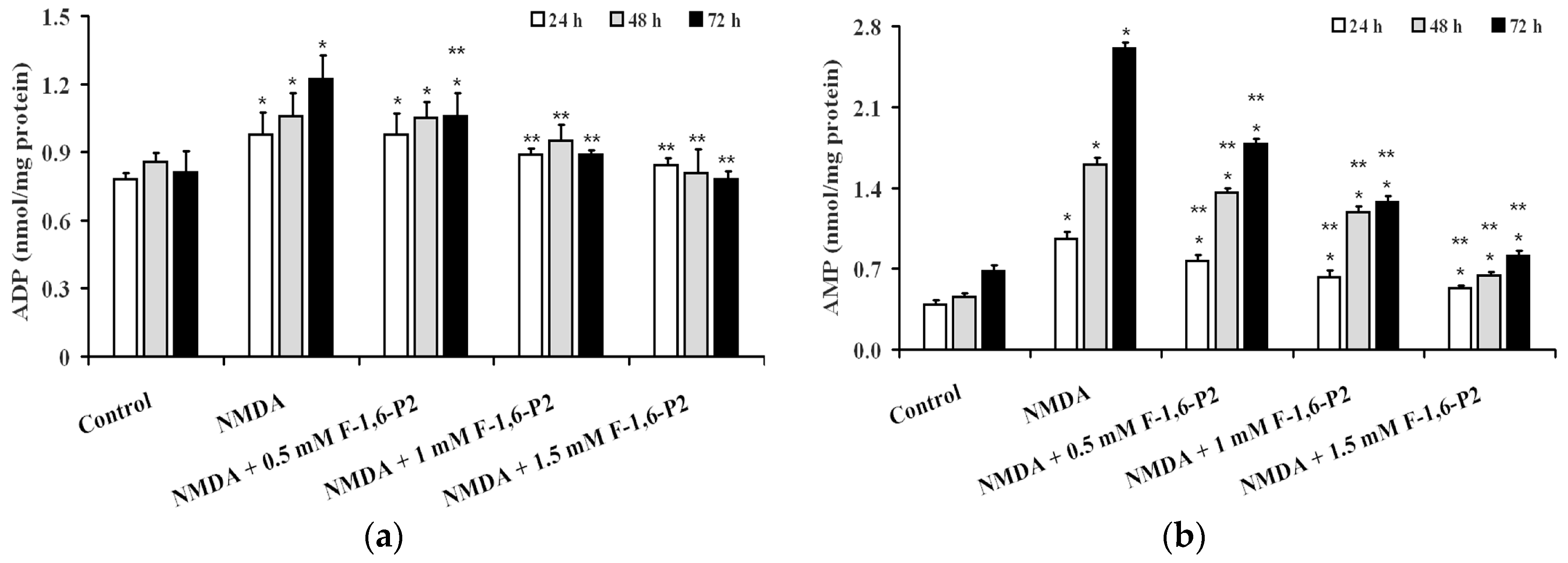
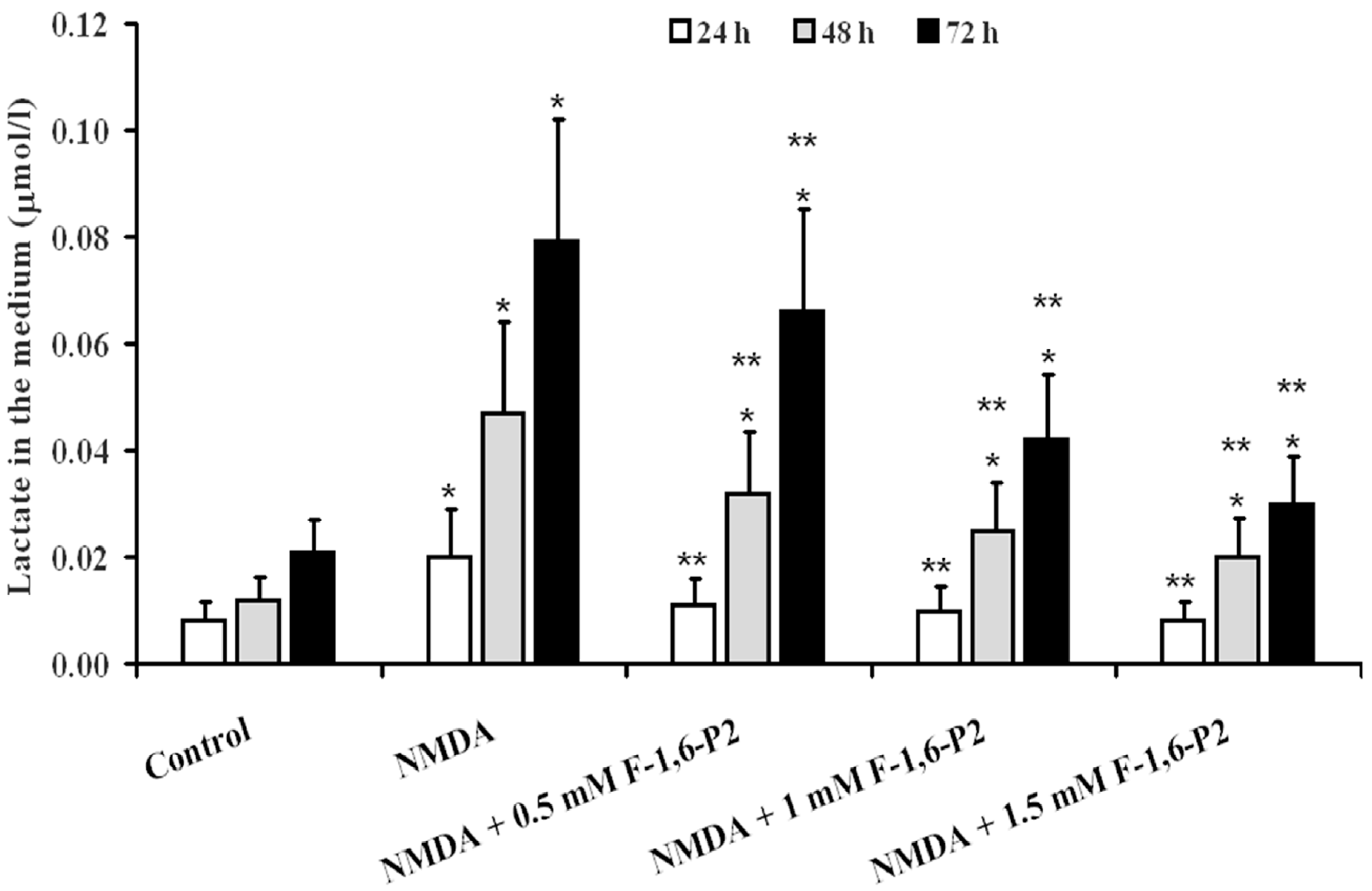
2.3. Effects of F-1,6-P2 on Adenine Nucleotide Catabolism and Lactate Production in OHSC Challenged with NMDA
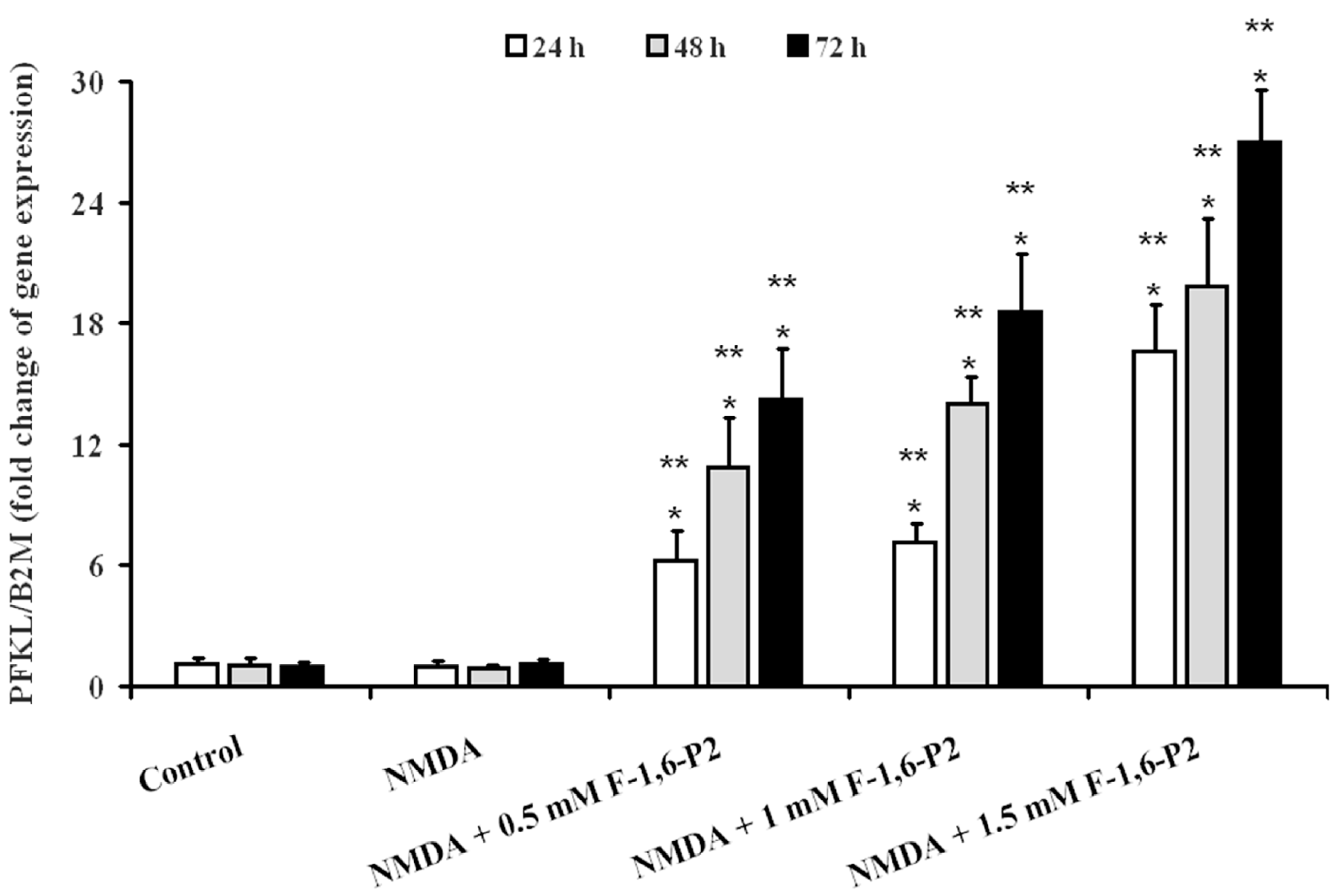
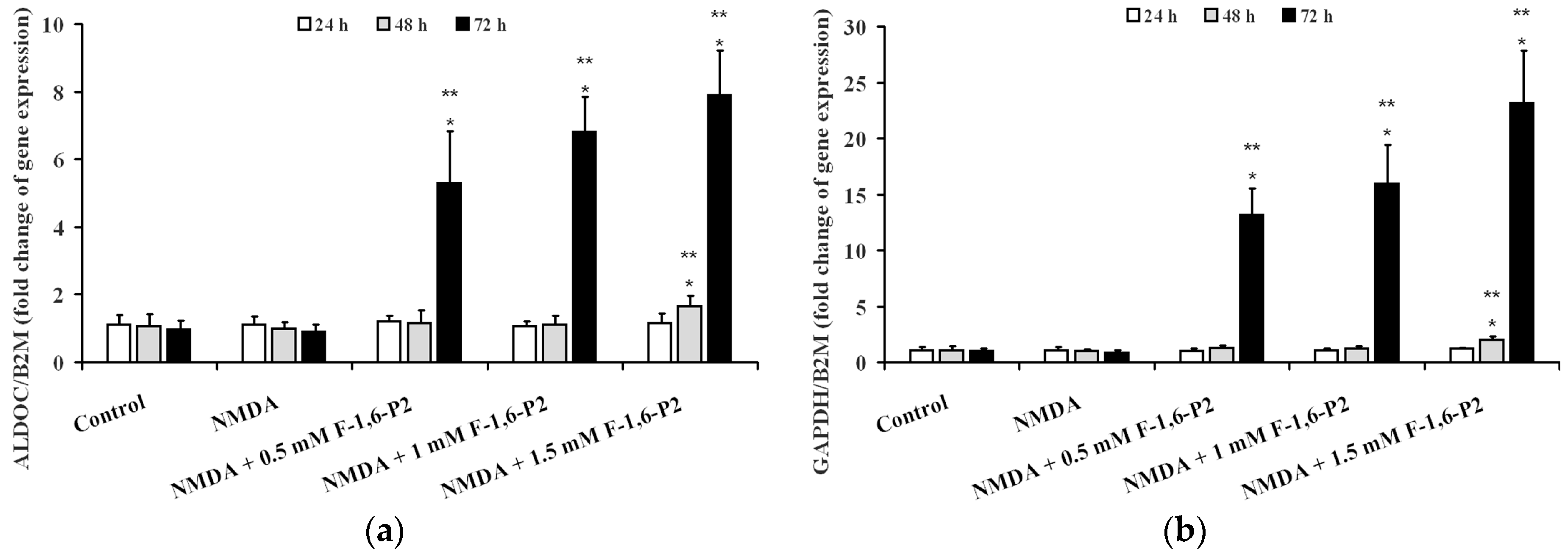
2.4. Expression of Genes Regulating the Synthesis of Glycolytic Enzymes Involved in F-1,6-P2 Metabolism
3. Discussion
4. Materials and Methods
4.1. Organotypic Hippocampal Slice Cultures
4.2. NMDA and F-1,6-P2 Treatments
4.3. LDH and Lactate Assays
4.4. HPLC Analysis of Metabolites
4.5. RNA Extraction and Real Time PCR Analysis
4.6. Statistics
Author Contributions
Funding
Conflicts of Interest
References
- de Lores Arnaiz, G.R.; Ordieres, M.G. Brain Na(+), K(+)-ATPase activity in aging and disease. Int. J. Biomed. Sci. 2014, 10, 85–102. [Google Scholar]
- Anandhan, A.; Jacome, M.S.; Lei, S.; Hernandez-Franco, P.; Pappa, A.; Panayiotidis, M.I.; Powers, R.; Franco, R. Metabolic dysfunction in Parkinson’s disease: Bioenergetics, redox homeostasis and central carbon metabolism. Brain Res. Bull. 2017, 133, 12–30. [Google Scholar] [CrossRef]
- Piacentini, R.; Li Puma, D.D.; Mainardi, M.; Lazzarino, G.; Tavazzi, B.; Arancio, O.; Grassi, C. Reduced gliotransmitter release from astrocytes mediates tau-induced synaptic dysfunction in cultured hippocampal neurons. Glia 2017, 65, 1302–1316. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, F.; Li, L.; Tang, F.; Siedlak, S.L.; Fujioka, H.; Liu, Y.; Su, B.; Pi, Y.; Wang, X. MFN2 couples glutamate excitotoxicity and mitochondrial dysfunction in motor neurons. J. Biol. Chem. 2015, 290, 168–182. [Google Scholar] [CrossRef]
- Kumagai, A.; Sasaki, T.; Matsuoka, K.; Abe, M.; Tabata, T.; Itoh, Y.; Fuchino, H.; Wugangerile, S.; Suga, M.; Yamaguchi, T.; et al. Monitoring of glutamate-induced excitotoxicity by mitochondrial oxygen consumption. Synapse 2018. [Google Scholar] [CrossRef]
- Serpa, A.; Pinto, I.; Bernardino, L.; Cascalheira, J.F. Combined neuroprotective action of adenosine A1 and cannabinoid CB1 receptors against NMDA-induced excitotoxicity in the hippocampus. Neurochem. Int. 2015, 87, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Amorini, A.M.; Lazzarino, G.; Di Pietro, V.; Signoretti, S.; Lazzarino, G.; Belli, A.; Tavazzi, B. Severity of experimental traumatic brain injury modulates changes in concentrations of cerebral free amino acids. J. Cell. Mol. Med. 2017, 21, 530–542. [Google Scholar] [CrossRef]
- Amorini, A.M.; Lazzarino, G.; Di Pietro, V.; Signoretti, S.; Lazzarino, G.; Belli, A.; Tavazzi, B. Metabolic, enzymatic and gene involvement in cerebral glucose dysmetabolism after traumatic brain injury. Biochim. Biophys. Acta 2016, 1862, 679–687. [Google Scholar] [CrossRef]
- Fresta, C.G.; Chakraborty, A.; Wijesinghe, M.B.; Amorini, A.M.; Lazzarino, G.; Lazzarino, G.; Tavazzi, B.; Lunte, S.M.; Caraci, F.; Dhar, P.; et al. Non-toxic engineered carbon nanodiamond concentrations induce oxidative/nitrosative stress, imbalance of energy metabolism, and mitochondrial dysfunction in microglial and alveolar basal epithelial cells. Cell Death Dis. 2018, 9, 245. [Google Scholar] [CrossRef]
- Gray, J.J.; Zommer, A.E.; Bouchard, R.J.; Duval, N.; Blackstone, C.; Linseman, D.A. N-terminal cleavage of the mitochondrial fusion GTPase OPA1 occurs via a caspase-independent mechanism in cerebellar granule neurons exposed to oxidative or nitrosative stress. Brain Res. 2013, 1494, 28–43. [Google Scholar] [CrossRef] [PubMed]
- Gerbatin, R.D.R.; Cassol, G.; Dobrachinski, F.; Ferreira, A.P.O.; Quines, C.B.; Pace, I.D.D.; Busanello, G.L.; Gutierres, J.M.; Nogueira, C.W.; Oliveira, M.S.; et al. Guanosine protects against traumatic brain injury-induced functional impairments and neuronal loss by modulating excitotoxicity, mitochondrial dysfunction, and inflammation. Mol. Neurobiol. 2017, 54, 7585–7596. [Google Scholar] [CrossRef]
- Shalaby, H.N.; El-Tanbouly, D.M.; Zaki, H.F. Topiramate mitigates 3-nitropropionic acid-induced striatal neurotoxicity via modulation of AMPA receptors. Food Chem. Toxicol. 2018, 118, 227–234. [Google Scholar] [CrossRef]
- Tai, S.H.; Lee, W.T.; Lee, A.C.; Lin, Y.W.; Hung, H.Y.; Huang, S.Y.; Wu, T.S.; Lee, E.J. Therapeutic window for YC-1 following glutamate-induced neuronal damage and transient focal cerebral ischemia. Mol. Med. Rep. 2018, 17, 6490–6496. [Google Scholar] [CrossRef]
- Lao, K.; Ji, N.; Zhang, X.; Qiao, W.; Tang, Z.; Gou, X. Drug development for Alzheimer’s disease: Review. J. Drug Target 2018, 20, 1–10. [Google Scholar] [CrossRef]
- Schumacher, M.; Denier, C.; Oudinet, J.P.; Adams, D.; Guennoun, R. Progesterone neuroprotection: The background of clinical trial failure. J. Steroid Biochem. Mol. Biol. 2016, 160, 53–66. [Google Scholar] [CrossRef]
- Oláh, J.; Orosz, F.; Puskás, L.G.; Hackler, L., Jr.; Horányi, M.; Polgár, L.; Hollán, S.; Ovádi, J. Triosephosphate isomerase deficiency: Consequences of an inherited mutation at mRNA, protein and metabolic levels. Biochem. J. 2005, 392 Pt 3, 675–683. [Google Scholar] [CrossRef]
- Lazzarino, G.; Cattani, L.; Costrini, R.; Mulieri, L.; Candiani, A.; Galzigna, L. Increase of intraerythrocytic fructose-1,6-diphosphate after incubation of whole human blood with fructose-1,6-diphosphate. Clin. Biochem. 1984, 17, 42–45. [Google Scholar] [CrossRef]
- Alva, N.; Alva, R.; Carbonell, T. Fructose 1,6-bisphosphate: A summary of its cytoprotective mechanism. Curr. Med. Chem. 2016, 23, 4396–4417. [Google Scholar] [CrossRef]
- Lazzarino, G.; Nuutinen, M.E.; Tavazzi, B.; Cerroni, L.; Di Pierro, D.; Giardina, B. Preserving effect of fructose-1,6-bisphosphate on high-energy phosphate compounds during anoxia and reperfusion in isolated langendorff-perfused rat hearts. J. Mol. Cell. Cardiol. 1991, 23, 13–23. [Google Scholar] [CrossRef]
- Tavazzi, B.; Cerroni, L.; Di Pierro, D.; Lazzarino, G.; Nuutinen, M.; Starnes, J.W.; Giardina, B. Oxygen radical injury and loss of high-energy compounds in anoxic and reperfused rat heart: Prevention by exogenous fructose-1,6-bisphosphate. Free Radic. Res. Commun. 1990, 10, 167–176. [Google Scholar] [CrossRef]
- Cohen, J.E.; Atluri, P.; Taylor, M.D.; Grand, T.J.; Liao, G.P.; Panlilio, C.M.; Suarez, E.E.; Zentko, S.E.; Hsu, V.M.; Berry, M.F.; et al. Fructose 1,6-diphosphate administration attenuates post-ischemic ventricular dysfunction. Heart Lung Circ. 2006, 15, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Romsi, P.; Kaakinen, T.; Kiviluoma, K.; Vainionpää, V.; Hirvonen, J.; Pokela, M.; Ohtonen, P.; Biancari, F.; Nuutinen, M.; Juvonen, T. Fructose-1,6-bisphosphate for improved outcome after hypothermic circulatory arrest in pigs. J. Thorac. Cardiovasc. Surg. 2003, 125, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Markov, A.K.; Brumley, M.A.; Figueroa, A.; Skelton, T.N.; Lehan, P.H. Hemodynamic effects of fructose 1,6-diphosphate in patients with normal and impaired left ventricular function. Am. Heart J. 1997, 133, 541–549. [Google Scholar] [CrossRef]
- Riedel, B.J.; Gal, J.; Ellis, G.; Marangos, P.J.; Fox, A.W.; Royston, D. Myocardial protection using fructose-1,6-diphosphate during coronary artery bypass graft surgery: A randomized, placebo-controlled clinical trial. Anesth. Analg. 2004, 98, 20–29. [Google Scholar] [CrossRef]
- Kaakinen, T.; Naukkarinen, A.; Tuominen, H.; Romsi, P.; Nuutinen, M.; Biancari, F.; Juvonen, T. Neuronal ultrastructure is preserved by fructose-1,6-bisphosphate after hypothermic circulatory arrest in pigs. J. Thorac. Cardiovasc. Surg. 2005, 130, 1475–1476. [Google Scholar] [CrossRef] [PubMed]
- Kaakinen, T.; Heikkinen, J.; Dahlbacka, S.; Alaoja, H.; Laurila, P.; Kiviluoma, K.; Salomäki, T.; Romsi, P.; Tuominen, H.; Biancari, F.; et al. Fructose-1,6-bisphosphate supports cerebral energy metabolism in pigs after ischemic brain injury caused by experimental particle embolization. Heart Surg. Forum 2006, 9, E828–E835. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, F.; Zhang, J.; Gao, H.; Yang, Y.; Fu, R. Repeated febrile convulsions impair hippocampal neurons and cause synaptic damage in immature rats: Neuroprotective effect of fructose-1,6-diphosphate. Neural Regen. Res. 2014, 9, 937–942. [Google Scholar] [CrossRef]
- Catarina, A.V.; Luft, C.; Greggio, S.; Venturin, G.T.; Ferreira, F.; Marques, E.P.; Rodrigues, L.; Wartchow, K.; Leite, M.C.; Gonçalves, C.A.; et al. Fructose-1,6-bisphosphate preserves glucose metabolism integrity and reduces reactive oxygen species in the brain during experimental sepsis. Brain Res. 2018, 1698, 54–61. [Google Scholar] [CrossRef]
- Liu, J.; Hirai, K.; Litt, L. Fructose-1,6-bisphosphate does not preserve ATP in hypoxic-ischemic neonatal cerebrocortical slices. Brain Res. 2008, 1238, 230–238. [Google Scholar] [CrossRef]
- Hofer, R.E.; Wagner, S.R., 4th; Pasternak, J.J.; Albrecht, R.F., 2nd; Gallagher, W.J.; Lanier, W.L. Fructose-1,6-bisphosphate and fructose-2,6-bisphosphate do not influence brain carbohydrate or high-energy phosphate metabolism in a rat model of forebrain ischemia. J. Neurosurg. Anesthesiol. 2009, 21, 31–39. [Google Scholar] [CrossRef]
- Lossi, L.; Alasia, S.; Salio, C.; Merighi, A. Cell death and proliferation in acute slices and organotypic cultures of mammalian CNS. Prog. Neurobiol. 2009, 88, 221–245. [Google Scholar] [CrossRef]
- Alasia, S.; Aimar, P.; Merighi, A.; Lossi, L. Context-dependent toxicity of amyloid-β peptides on mouse cerebellar cells. J. Alzheimers Dis. 2012, 30, 41–51. [Google Scholar] [CrossRef]
- Grabiec, U.; Hohmann, T.; Hammer, N.; Dehghani, F. Organotypic hippocampal slice cultures as a model to study neuroprotection and invasiveness of tumor cells. J. Vis. Exp. 2017. [Google Scholar] [CrossRef]
- Di Pietro, V.; Amin, D.; Pernagallo, S.; Lazzarino, G.; Tavazzi, B.; Vagnozzi, R.; Pringle, A.; Belli, A. Transcriptomics of traumatic brain injury: Gene expression and molecular pathways of different grades of insult in a rat organotypic hippocampal culture model. J. Neurotrauma 2010, 27, 349–359. [Google Scholar] [CrossRef]
- Krzyżanowska, W.; Pomierny, B.; Bystrowska, B.; Pomierny-Chamioło, L.; Filip, M.; Budziszewska, B.; Pera, J. Ceftriaxone- and N-acetylcysteine-induced brain tolerance to ischemia: Influence on glutamate levels in focal cerebral ischemia. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [CrossRef]
- Lin, C.J.; Chen, T.H.; Yang, L.Y.; Shih, C.M. Resveratrol protects astrocytes against traumatic brain injury through inhibiting apoptotic and autophagic cell death. Cell Death Dis. 2014. [Google Scholar] [CrossRef]
- Jang, S.; Kim, H.; Kim, H.J.; Lee, S.K.; Kim, E.W.; Namkoong, K.; Kim, E. Long-term culture of organotypic hippocampal slice from old 3xTg-AD mouse: An ex vivo model of Alzheimer’s disease. Psychiatry Investig. 2018, 15, 205–213. [Google Scholar] [CrossRef]
- Saliba, S.W.; Marcotegui, A.R.; Fortwängler, E.; Ditrich, J.; Perazzo, J.C.; Muñoz, E.; de Oliveira, A.C.P.; Fiebich, B.L. AM404, paracetamol metabolite, prevents prostaglandin synthesis in activated microglia by inhibiting COX activity. J. Neuroinflamm. 2017, 14, 246. [Google Scholar] [CrossRef]
- Maldonado, E.N.; Lemasters, J.J. ATP/ADP ratio, the missed connection between mitochondria and the Warburg effect. Mitochondrion 2014, 19, 78–84. [Google Scholar] [CrossRef]
- Lee, J.S.; Wang, R.X.; Alexeev, E.E.; Lanis, J.M.; Battista, K.D.; Glover, L.E.; Colgan, S.P. Hypoxanthine is a checkpoint stress metabolite in colonic epithelial energy modulation and barrier function. J. Biol. Chem. 2018, 293, 6039–6051. [Google Scholar] [CrossRef] [PubMed]
- Lama, S.; Auer, R.N.; Tyson, R.; Gallagher, C.N.; Tomanek, B.; Sutherland, G.R. Lactate storm marks cerebral metabolism following brain trauma. J. Biol. Chem. 2014, 289, 20200–20208. [Google Scholar] [CrossRef]
- Hill, D.E.; Hammes, G.G. An equilibrium binding study of the interaction of fructose 6-phosphate and fructose-1,6-bisphosphate with rabbit muscle phosphofructokinase. Biochemistry 1975, 14, 203–213. [Google Scholar] [CrossRef]
- Tavazzi, B.; Starnes, J.W.; Lazzarino, G.; Di Pierro, D.; Nuutinen, E.M.; Giardina, B. Exogenous fructose-1,6-bisphosphate is a metabolizable substrate for the isolated normoxic rat heart. Basic Res. Cardiol. 1992, 87, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Hardin, C.D.; Lazzarino, G.; Tavazzi, B.; Di Pierro, D.; Roberts, T.M.; Giardina, B.; Rovetto, M.J. Myocardial metabolism of exogenous FDP is consistent with transport by a dicarboxylate transporter. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H2654–H2660. [Google Scholar] [CrossRef] [PubMed]
- Pajor, A.M.; Gangula, R.; Yao, X. Cloning and functional characterization of a high-affinity Na(+)/dicarboxylate cotransporter from mouse brain. Am. J. Physiol. Cell Physiol. 2001, 280, C1215–C1223. [Google Scholar] [CrossRef] [PubMed]
- Yodoya, E.; Wada, M.; Shimada, A.; Katsukawa, H.; Okada, N.; Yamamoto, A.; Ganapathy, V.; Fujita, T. Functional and molecular identification of sodium-coupled dicarboxylate transporters in rat primary cultured cerebrocortical astrocytes and neurons. J. Neurochem. 2006, 97, 162–173. [Google Scholar] [CrossRef]
- Nuutinen, E.M.; Lazzarino, G.; Giardina, B.; Hassinen, I.E. Effect of exogenous fructose-1,6-bisphosphate on glycolysis in the isolated perfused rat heart. Am. Heart J. 1991, 122, 523–527. [Google Scholar] [CrossRef]
- Hassinen, I.E.; Nuutinen, E.M.; Ito, K.; Nioka, S.; Lazzarino, G.; Giardina, B.; Chance, B. Mechanism of the effect of exogenous fructose 1,6-bisphosphate on myocardial energy metabolism. Circulation 1991, 83, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Yu, Y.; Ma, Z.Y.; Sun, H.; Zhang, Y.L.; Wang, X.T.; Wang, C.; Fan, W.M.; Zheng, Q.Y.; Ma, C.L. NMDAR-Mediated hippocampal neuronal death is exacerbated by activities of ASIC1a. Neurotox. Res. 2015, 28, 122–137. [Google Scholar] [CrossRef]
- Di Pietro, V.; Lazzarino, G.; Amorini, A.M.; Signoretti, S.; Hill, L.J.; Porto, E.; Tavazzi, B.; Lazzarino, G.; Belli, A. Fusion or fission: The destiny of mitochondria in traumatic brain injury of different severities. Sci. Rep. 2017, 7, 9189. [Google Scholar] [CrossRef]
- Sandoval, M.; Luarte, A.; Herrera-Molina, R.; Varas-Godoy, M.; Santibáñez, M.; Rubio, F.J.; Smit, A.B.; Gundelfinger, E.D.; Li, K.W.; Smalla, K.H.; et al. The glycolytic enzyme aldolase C is up-regulated in rat forebrain microsomes and in the cerebrospinal fluid after repetitive fluoxetine treatment. Brain Res. 2013, 1520, 1–14. [Google Scholar] [CrossRef]
- Mamczur, P.; Borsuk, B.; Paszko, J.; Sas, Z.; Mozrzymas, J.; Wiśniewski, J.R.; Gizak, A.; Rakus, D. Astrocyte-neuron crosstalk regulates the expression and subcellular localization of carbohydrate metabolism enzymes. Glia 2015, 63, 328–340. [Google Scholar] [CrossRef]
- Tsutsumi, S.; Yamazaki, M.; Miyazaki, T.; Watanabe, M.; Sakimura, K.; Kano, M.; Kitamura, K. Structure-function relationships between aldolase C/zebrin II expression and complex spike synchrony in the cerebellum. J. Neurosci. 2015, 35, 843–852. [Google Scholar] [CrossRef]
- Schousboe, A.; Sickmann, H.M.; Bak, L.K.; Schousboe, I.; Jajo, F.S.; Faek, S.A.; Waagepetersen, H.S. Neuron-glia interactions in glutamatergic neurotransmission: Roles of oxidative and glycolytic adenosine triphosphate as energy source. J. Neurosci. Res. 2011, 89, 1926–1934. [Google Scholar] [CrossRef]
- Lazarev, V.F.; Dutysheva, E.A.; Komarova, E.Y.; Mikhaylova, E.R.; Guzhova, I.V.; Margulis, B.A. GAPDH-targeted therapy—A new approach for secondary damage after traumatic brain injury on rats. Biochem. Biophys. Res. Commun. 2018, 501, 1003–1008. [Google Scholar] [CrossRef]
- Sen, T.; Saha, P.; Sen, N. Nitrosylation of GAPDH augments pathological tau acetylation upon exposure to amyloid-β. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef]
- Di Pietro, V.; Amorini, A.M.; Lazzarino, G.; Yakoub, K.M.; D’Urso, S.; Lazzarino, G.; Belli, A. S100B and glial fibrillary acidic protein as indexes to monitor damage severity in an in vitro model of traumatic brain injury. Neurochem. Res. 2015, 40, 991–999. [Google Scholar] [CrossRef]
- Ikegaya, Y.; Kim, J.A.; Baba, M.; Iwatsubo, T.; Nishiyama, N.; Matsuki, N. Rapid and reversible changes in dendrite morphology and synaptic efficacy following NMDA receptor activation: Implication for a cellular defense against excitotoxicity. J. Cell Sci. 2001, 114, 4083–4093. [Google Scholar]
- Lazzarino, G.; Amorini, A.M.; Petzold, A.; Gasperini, C.; Ruggieri, S.; Quartuccio, M.E.; Lazzarino, G.; Di Stasio, E.; Tavazzi, B. Serum compounds of energy metabolism impairment are related to disability, disease course and neuroimaging in multiple sclerosis. Mol. Neurobiol. 2017, 54, 7520–7533. [Google Scholar] [CrossRef]
- Bergmeyer, H.U. Methods of Enzymatic Analysis, 2nd ed.; Academic Press: New York, NY, USA, 1974; Volume 1-2. [Google Scholar]
- Artiss, J.D.; Karcher, R.E.; Cavanagh, K.T.; Collins, S.L.; Peterson, V.J.; Varma, S.; Zak, B. A liquid-stable reagent for lactic acid levels. Application to the Hitachi 911 and Beckman CX7. Am. J. Clin. Pathol. 2000, 114, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Bracko, O.; Di Pietro, V.; Lazzarino, G.; Amorini, A.M.; Tavazzi, B.; Artmann, J.; Wong, E.C.; Buxton, R.B.; Weller, M.; Luft, A.R.; et al. 3-Nitropropionic acid-induced ischemia tolerance in the rat brain is mediated by reduced metabolic activity and cerebral blood flow. J. Cereb. Blood Flow Metab. 2014, 34, 1522–1530. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Time (Hours) | PFKL/BD2M (Fold Increase) | ALDOC/BD2M (Fold Increase) | GAPDH/BD2M (Fold Increase) |
|---|---|---|---|---|
| Control | 24 | 1.10 ± 0.30 | 1.02 ± 0.18 | 1.15 ± 0.13 |
| 48 | 1.07 ± 0.26 | 1.15 ± 0.21 | 1.07 ± 0.20 | |
| 72 | 0.98 ± 0.21 | 1.08 ± 0.23 | 0.99 ± 0.14 | |
| 0.5 mM F-1,6-P2 | 24 | 4.64 ± 0.62 a,b | 0.94 ± 0.11 | 1.14 ± 0.25 |
| 48 | 7.89 ± 0.88 a,b | 1.09 ± 0.18 | 1.09 ± 0.18 | |
| 72 | 11.26 ± 1.23 a,b | 3.67 ± 0.43 a,b | 8.38 ± 1.23 a,b | |
| 1 mM F-1,6-P2 | 24 | 5.16 ± 0.49 a,b | 1.11 ± 0.17 | 1.06 ± 0.16 |
| 48 | 10.54 ± 1.18 a,b | 0.97 ± 0.09 | 0.94 ± 0.10 | |
| 72 | 15.92 ± 1.66 a,b | 4.71 ± 0.75 a,b | 11.55 ± 2.03 a,b | |
| 1.5 mM F-1,6-P2 | 24 | 9.75 ± 0.95 a,b | 1.21 ± 0.26 | 1.19 ± 0.21 |
| 48 | 14.21 ± 1.58 a,b | 1.34 ± 0.31 | 1.32 ± 0.18 | |
| 72 | 21.09 ± 2.36 a,b | 6.92 ± 0.95 a,b | 17.46 ± 2.67 a,b |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yakoub, K.M.; Lazzarino, G.; Amorini, A.M.; Caruso, G.; Scazzone, C.; Ciaccio, M.; Tavazzi, B.; Lazzarino, G.; Belli, A.; Di Pietro, V. Fructose-1,6-Bisphosphate Protects Hippocampal Rat Slices from NMDA Excitotoxicity. Int. J. Mol. Sci. 2019, 20, 2239. https://doi.org/10.3390/ijms20092239
Yakoub KM, Lazzarino G, Amorini AM, Caruso G, Scazzone C, Ciaccio M, Tavazzi B, Lazzarino G, Belli A, Di Pietro V. Fructose-1,6-Bisphosphate Protects Hippocampal Rat Slices from NMDA Excitotoxicity. International Journal of Molecular Sciences. 2019; 20(9):2239. https://doi.org/10.3390/ijms20092239
Chicago/Turabian StyleYakoub, Kamal M., Giacomo Lazzarino, Angela M. Amorini, Giuseppe Caruso, Concetta Scazzone, Marcello Ciaccio, Barbara Tavazzi, Giuseppe Lazzarino, Antonio Belli, and Valentina Di Pietro. 2019. "Fructose-1,6-Bisphosphate Protects Hippocampal Rat Slices from NMDA Excitotoxicity" International Journal of Molecular Sciences 20, no. 9: 2239. https://doi.org/10.3390/ijms20092239
APA StyleYakoub, K. M., Lazzarino, G., Amorini, A. M., Caruso, G., Scazzone, C., Ciaccio, M., Tavazzi, B., Lazzarino, G., Belli, A., & Di Pietro, V. (2019). Fructose-1,6-Bisphosphate Protects Hippocampal Rat Slices from NMDA Excitotoxicity. International Journal of Molecular Sciences, 20(9), 2239. https://doi.org/10.3390/ijms20092239