Activated Fibroblast Program Orchestrates Tumor Initiation and Progression; Molecular Mechanisms and the Associated Therapeutic Strategies


Abstract
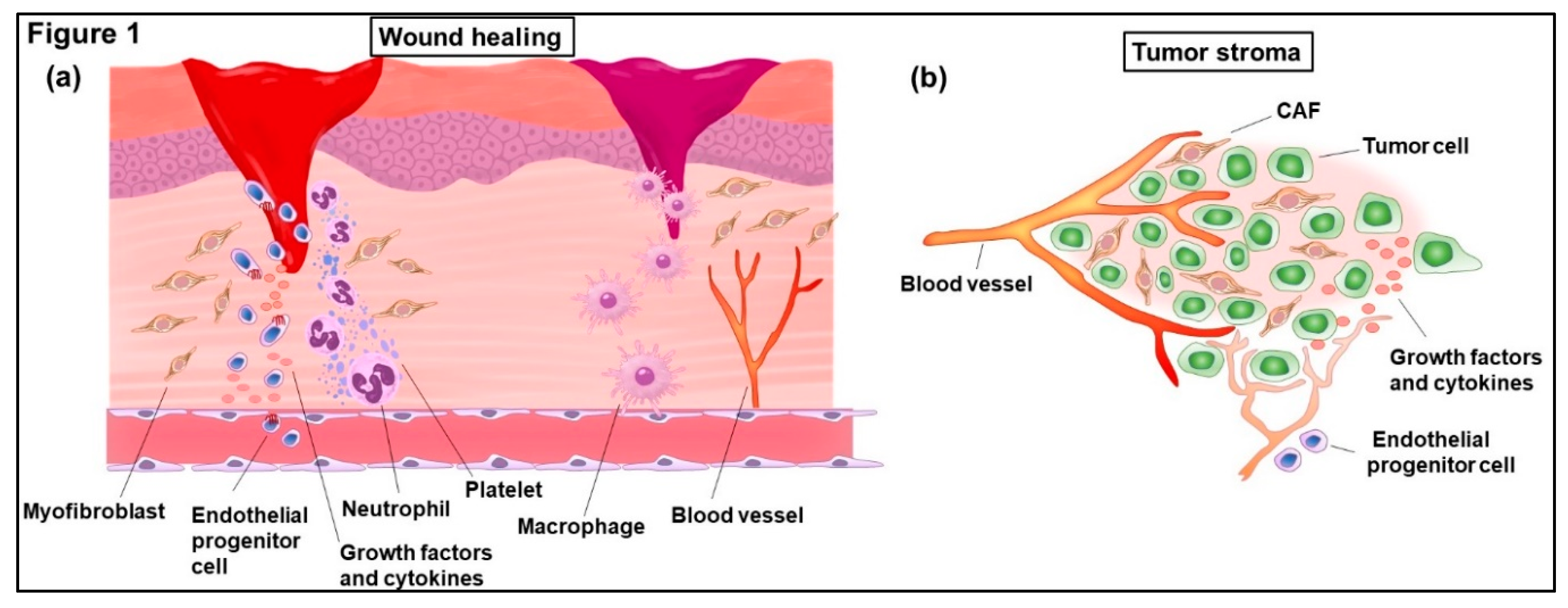
1. Significant Roles of Fibrosis in Cancer Development
1.1. Contributions of Fibrosis to Cancer Development
1.2. Epithelial-Mesenchymal Transition (EMT) and Endothelial-Mesenchymal Transition (EndoMT) in Fibrosis and Tumor Stroma
2. Fibrosis-Induced Tumor Progression
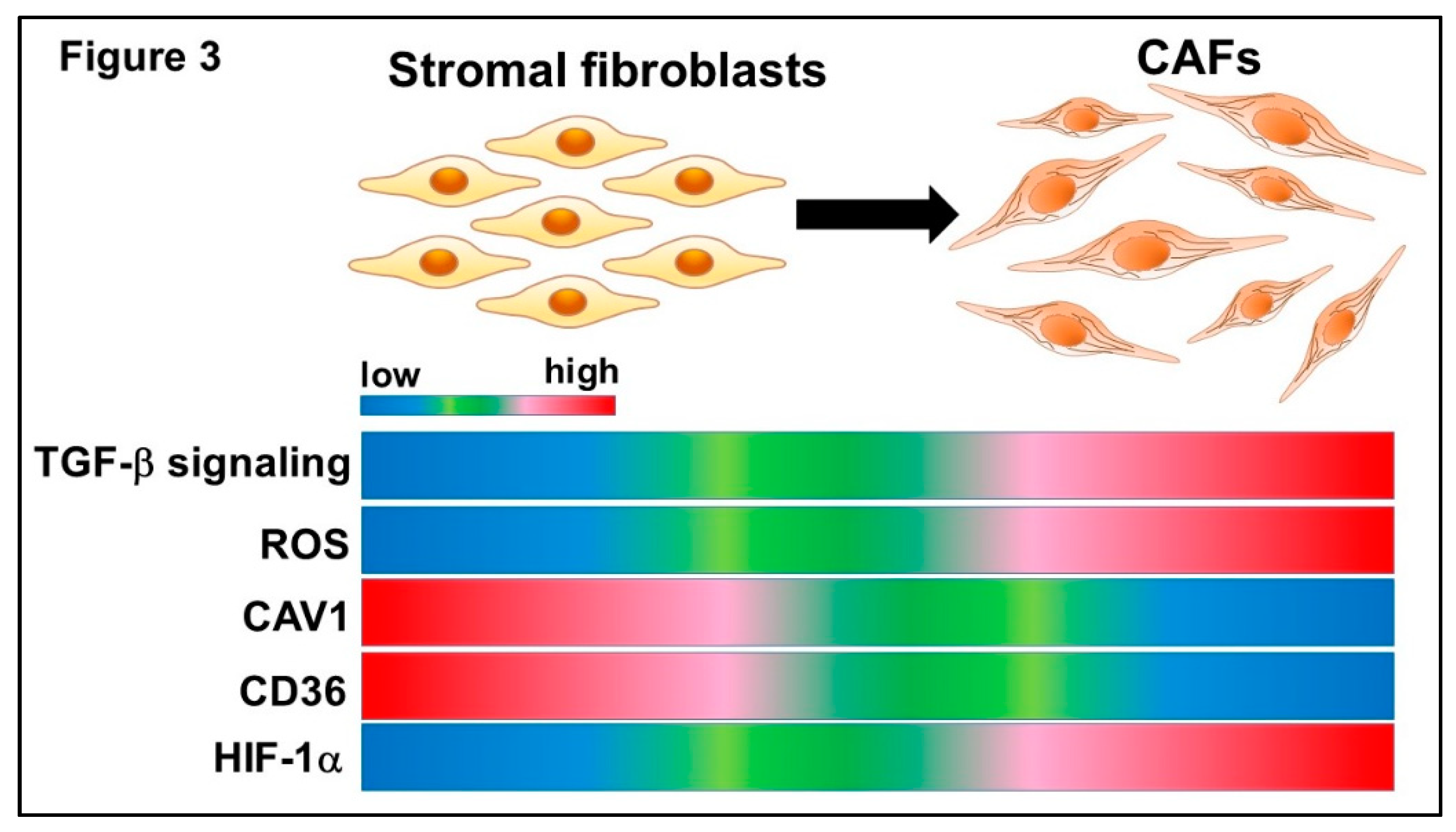
2.1. Origin and Differentiation into CAFs
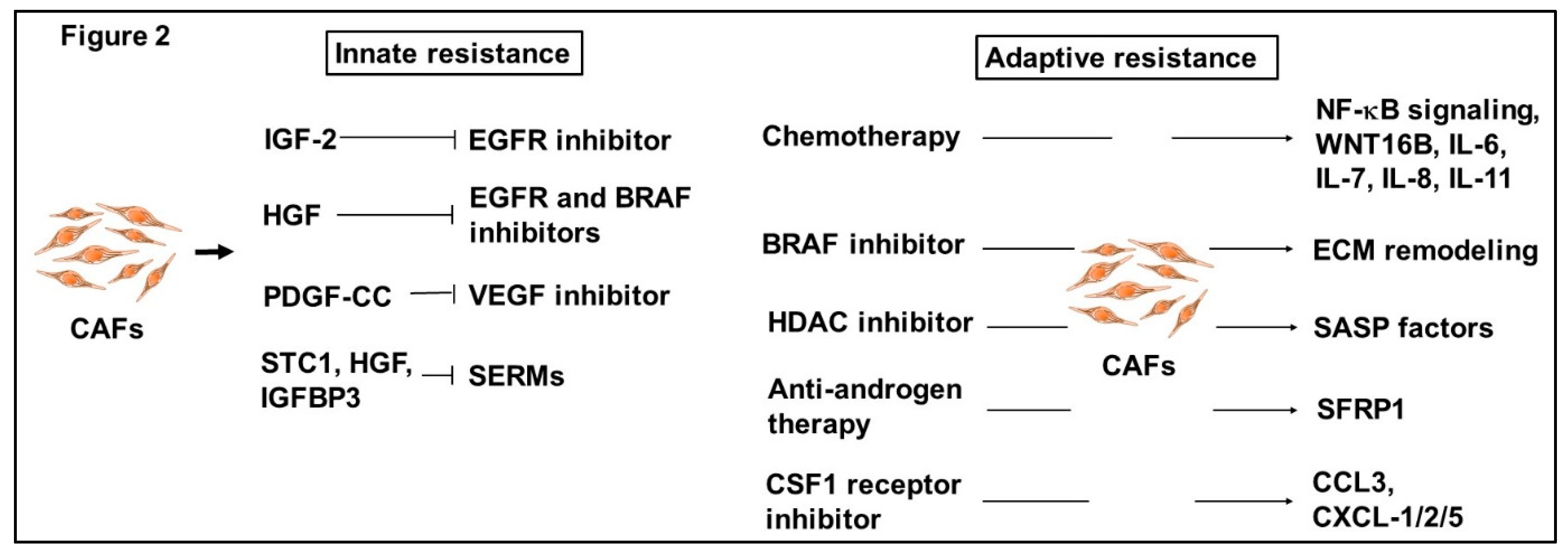
2.2. Emerging Roles of CAFs for Therapeutic Resistance
2.2.1. Innate Resistance of CAFs to Anti-Cancer Drugs
2.2.2. Adaptive Resistance of CAFs to Anti-Cancer Drugs
2.3. Cross-Talk between CAFs and Tumor Microenvironment
3. Metabolic Reprogramming of CAFs During Cancer Progression
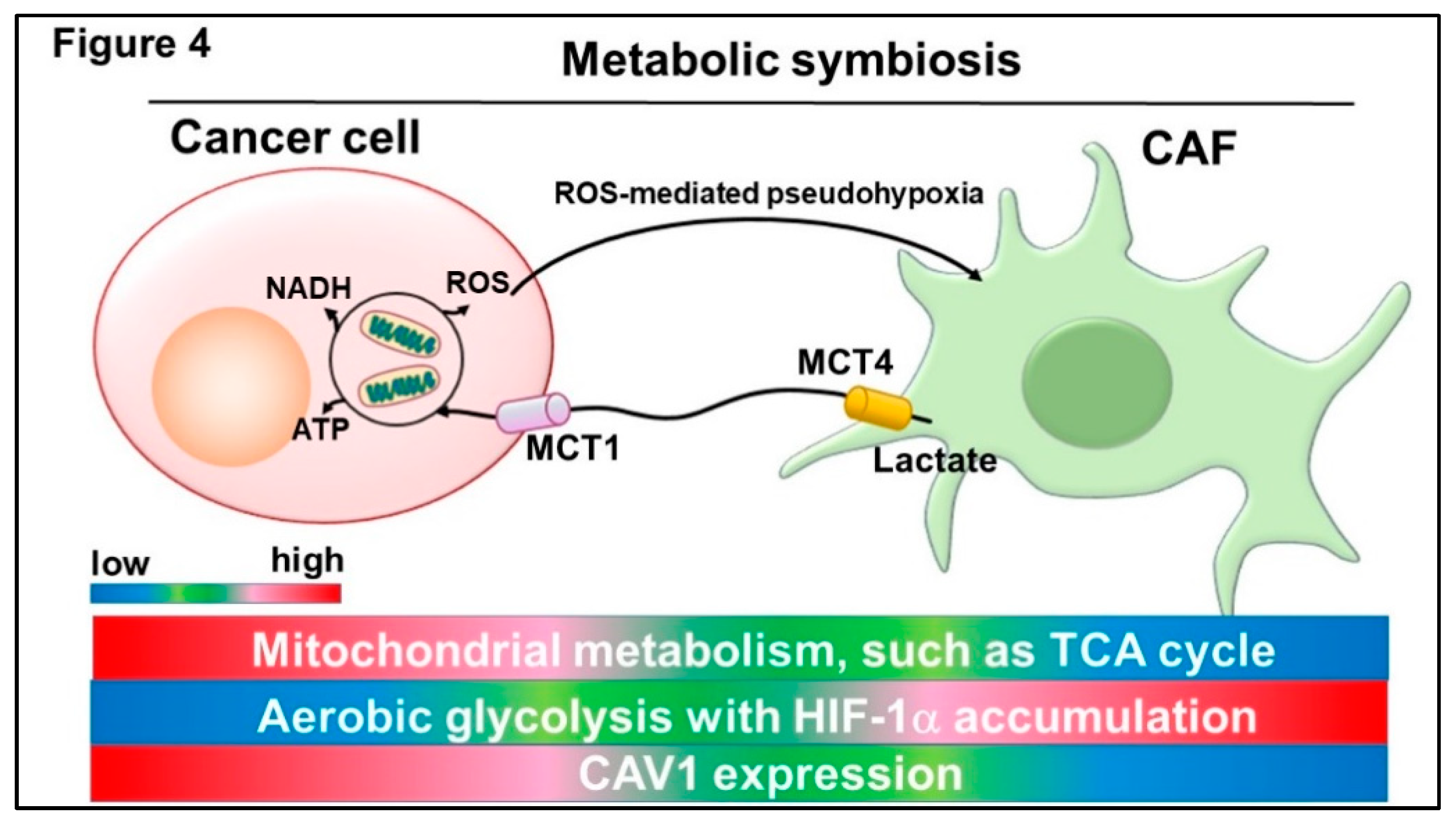
3.1. Metabolic Symbiosis Between Cancer Cells and CAFs
3.2. Signal Pathways Involved in Metabolic Reprogramming of CAFs
4. Targeting Tumor Stroma Fibroblasts to Attenuate Tumor Progression
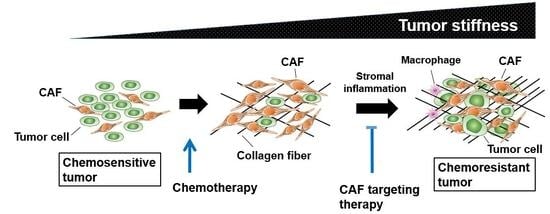
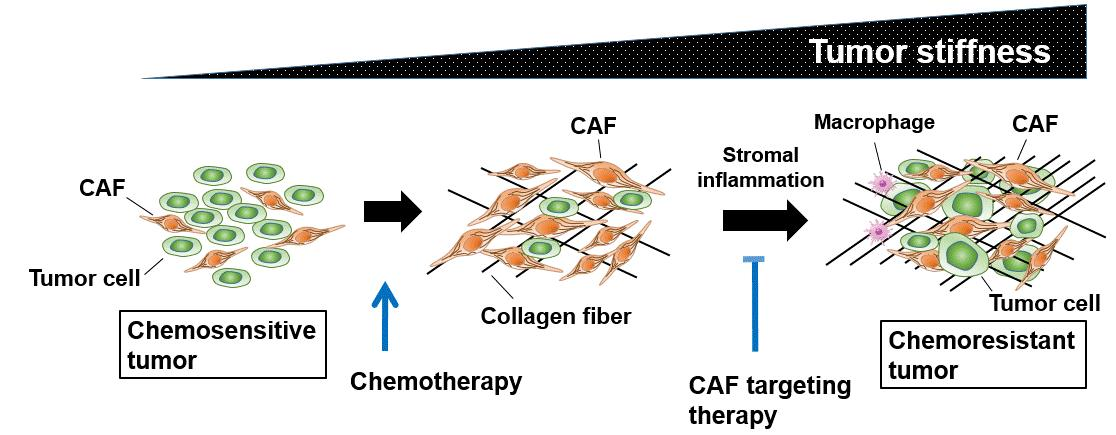
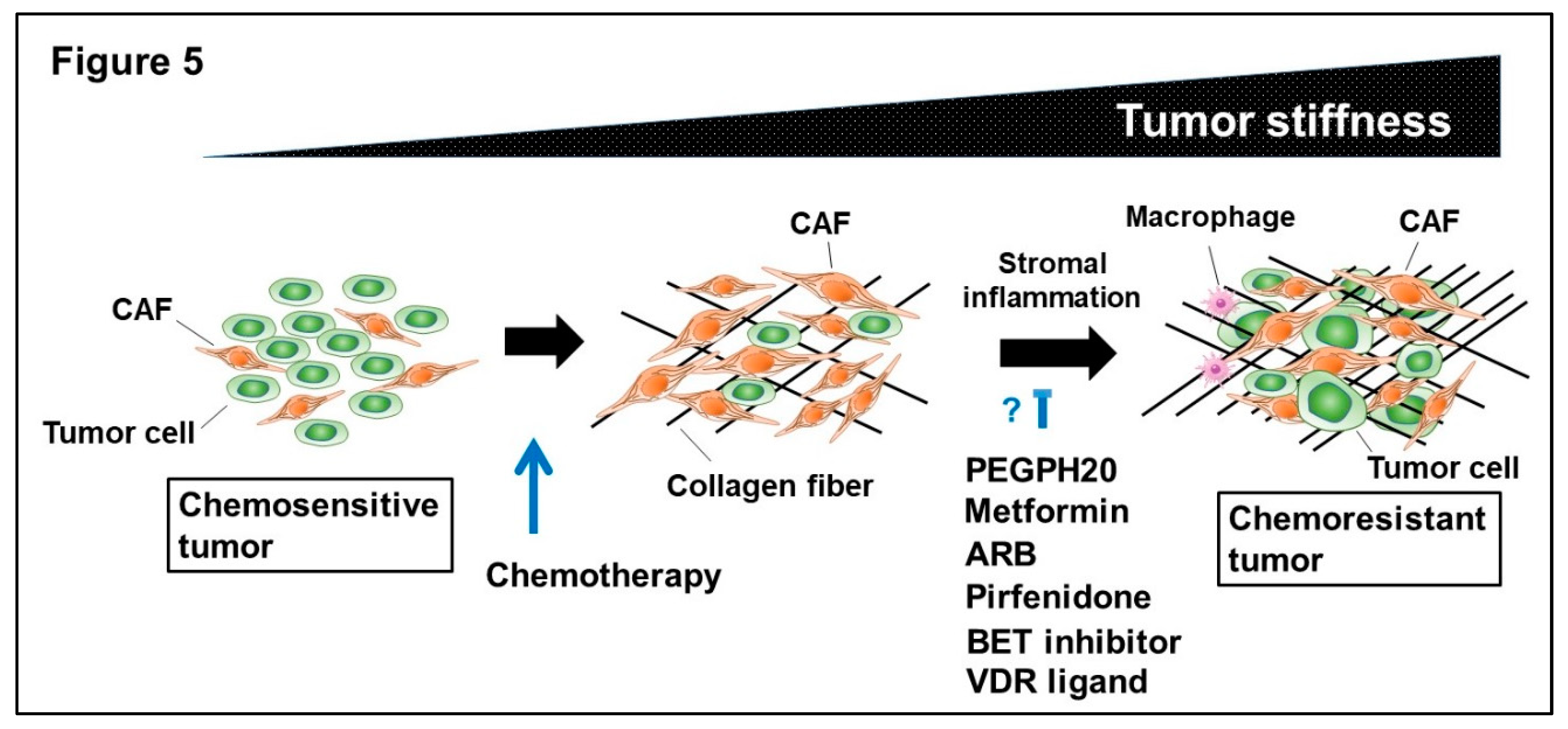
4.1. Tumor Stiffness and Enhanced Interstitial Fluid Pressure
4.2. Therapeutic Strategy Against Activated Tumor Stroma
5. Closing Remarks
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AngII | angiotensin II |
| ARB | angiotensin II type I receptor blocker |
| AT1R | angII type I receptor |
| α-SMA | α-smooth muscle actin |
| BRD4 | Bromodomain-containing protein 4 |
| CAFs | carcinoma-associated fibroblasts |
| CAV1 | caveolin 1 |
| c-Abl | c-Abl protein kinase |
| CSF1 | colony-stimulating factor 1 |
| DM | diabetes mellitus |
| ECM | extracellular matrix |
| EGF | epithelial growth factor |
| EGFR | epidermal growth factor receptor |
| EMT | epithelial-mesenchymal transition |
| EndoMT | endothelial-mesenchymal transition |
| EPCs | endothelial precursor cells |
| ERK | extracellular signal-regulated kinase |
| FAK | focal adhesion kinase |
| FGF | fibroblast growth factor |
| FGFRs | fibroblast growth factor receptors |
| GSK-3β | glycogen synthase kinase-3β |
| HA | hyaluronic acid |
| HCC | hepatocellular carcinoma |
| HGF | hepatocyte growth factor |
| Hh | Hedgehog |
| HIC1 | hypermethylated in cancer 1 |
| HIF-1α | hypoxia-induced factor-1α |
| HSCs | hepatic stellate cells |
| IFP | interstitial fluid pressures |
| IGF1R | insulin-like growth factor 1 receptor |
| IGF2 | insulin-like growth factor 2 |
| IL | interleukin |
| IPF | idiopathic pulmonary fibrosis |
| IR | insulin receptor |
| JAK1 | Janus kinase 1 |
| LIF | leukemia inhibitory factor |
| LOX | lysyl oxidase |
| MAPK | mitogen-activate protein kinase |
| MCT | monocarboxylate transporter |
| MLC | myosin light chain |
| MMP | matrix metalloproteinase |
| MSCs | mesenchymal stem cells |
| mTOR | mammalian target of rapamycin |
| NASH | non-alcoholic steatohepatitis |
| NSCLC | non-small cell lung cancer |
| PAI-1 | plasminogen activator inhibitor-1 |
| PDAC | pancreatic ductal adenocarcinoma |
| PDGF | platelet-derived growth factor |
| PDGFR | platelet-derived growth factor receptor |
| PGE2 | prostaglandin E2 |
| PI3K | phosphatidylinositol-3 kinase |
| PIP3 | phosphatidylinositol-3,4,5-triphosphate |
| PKC-δ | protein kinase C δ |
| KPC | Pdx1-Cre KrasG12D/+ Trp53R172H/+ |
| PMN-MDSCs | polymorpho-nuclear myeloid-derived suppressor cells |
| PTEN | phosphatase and tensin homologue deleted on chromosome 10 |
| ROCK | Rho-associated protein kinase |
| ROS | reactive oxygen species |
| SASP | senescence-associated secretory phenotype |
| SHP-1 | src homology region 2 domain-containing phosphatase-1 |
| SDF-1 | stromal cell-derived factor-1 |
| SMO | Smoothened |
| SPARC | secreted protein acidic and rich in cysteine |
| STAT3 | signal transducer and activator of transcription 3 |
| STC1 | stanniocalcin 1 |
| TAMs | tumor-associated macrophages |
| TCA | tricarboxylic acid |
| TGF-β | transforming growth factor-β |
| TKI | tyrosine kinase inhibitor |
| TNBC | triple-negative breast carcinoma |
| TIC | tumor-initiating cells |
| uPA | urokinase-type plasminogen activator |
| VDR | vitamin D receptor |
| VEGF | vascular endothelial growth factor |
| WNT16B | Wnt family member wingless-type MMTV integration site family member 16B |
| YAP | yes-associated protein |
References
- Hinz, B. Formation and function of the myofibroblast during tissue repair. J. Invest. Dermatol. 2007, 127, 526–537. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Lee, D.Y.; White, E.S.; Cui, Z.; Larios, J.M.; Chacon, R.; Horowitz, J.C.; Day, R.M.; Thomas, P.E. Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J. Biol. Chem. 2003, 278, 12384–12389. [Google Scholar] [CrossRef] [PubMed]
- Scharenberg, M.A.; Pippenger, B.E.; Sack, R.; Zingg, D.; Ferralli, J.; Schenk, S.; Martin, I.; Chiquet-Ehrismann, R. TGF-beta-induced differentiation into myofibroblasts involves specific regulation of two MKL1 isoforms. J. Cell Sci. 2014, 127, 1079–1091. [Google Scholar] [CrossRef]
- Foster, D.S.; Jones, R.E.; Ransom, R.C.; Longaker, M.T.; Norton, J.A. The evolving relationship of wound healing and tumor stroma. JCI Insight 2018, 3, e99911. [Google Scholar] [CrossRef]
- Carthy, J.M. TGFbeta signaling and the control of myofibroblast differentiation: Implications for chronic inflammatory disorders. J. Cell Physiol. 2018, 233, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Wipff, P.J.; Rifkin, D.B.; Meister, J.J.; Hinz, B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Uehara, T.; Ainslie, G.R.; Kutanzi, K.; Pogribny, I.P.; Muskhelishvili, L.; Izawa, T.; Yamate, J.; Kosyk, O.; Shymonyak, S.; Bradford, B.U.; et al. Molecular mechanisms of fibrosis-associated promotion of liver carcinogenesis. Toxicol. Sci. 2013, 132, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Dumur, C.I.; Holt, S.E.; Beckman, M.J.; Elmore, L.W. Multipotent adipose stromal cells and breast cancer development: Think globally, act locally. Mol. Carcinog 2010, 49, 923–927. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.R.; Erler, J.T. Molecular pathways: Connecting fibrosis and solid tumor metastasis. Clin. Cancer Res. 2014, 20, 3637–3643. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Nouraie, M.; Chen, X.; Zou, R.H.; Sellares, J.; Veraldi, K.L.; Chiarchiaro, J.; Lindell, K.; Wilson, D.O.; Kaminski, N.; et al. Characteristics of lung cancer among patients with idiopathic pulmonary fibrosis and interstitial lung disease—Analysis of institutional and population data. Respir. Res. 2018, 19, 195. [Google Scholar] [CrossRef] [PubMed]
- Tomassetti, S.; Gurioli, C.; Ryu, J.H.; Decker, P.A.; Ravaglia, C.; Tantalocco, P.; Buccioli, M.; Piciucchi, S.; Sverzellati, N.; Dubini, A.; et al. The impact of lung cancer on survival of idiopathic pulmonary fibrosis. Chest 2015, 147, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.L.; Sukkar, M.B. The SPARC protein: An overview of its role in lung cancer and pulmonary fibrosis and its potential role in chronic airways disease. Br. J. Pharmacol. 2017, 174, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Trombetta-Esilva, J.; Bradshaw, A.D. The Function of SPARC as a Mediator of Fibrosis. Open Rheumatol. J. 2012, 6, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Hohenester, E.; Sasaki, T.; Giudici, C.; Farndale, R.W.; Bachinger, H.P. Structural basis of sequence-specific collagen recognition by SPARC. Proc. Natl. Acad. Sci. USA 2008, 105, 18273–18277. [Google Scholar] [CrossRef]
- Chang, W.; Wei, K.; Jacobs, S.S.; Upadhyay, D.; Weill, D.; Rosen, G.D. SPARC suppresses apoptosis of idiopathic pulmonary fibrosis fibroblasts through constitutive activation of beta-catenin. J. Biol. Chem. 2010, 285, 8196–8206. [Google Scholar] [CrossRef]
- Francki, A.; McClure, T.D.; Brekken, R.A.; Motamed, K.; Murri, C.; Wang, T.; Sage, E.H. SPARC regulates TGF-beta1-dependent signaling in primary glomerular mesangial cells. J. Cell Biochem. 2004, 91, 915–925. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Giatromanolaki, A.; Brekken, R.A.; Sivridis, E.; Gatter, K.C.; Harris, A.L.; Sage, E.H. Enhanced expression of SPARC/osteonectin in the tumor-associated stroma of non-small cell lung cancer is correlated with markers of hypoxia/acidity and with poor prognosis of patients. Cancer Res. 2003, 63, 5376–5380. [Google Scholar]
- Infante, J.R.; Matsubayashi, H.; Sato, N.; Tonascia, J.; Klein, A.P.; Riall, T.A.; Yeo, C.; Iacobuzio-Donahue, C.; Goggins, M. Peritumoral fibroblast SPARC expression and patient outcome with resectable pancreatic adenocarcinoma. J. Clin. Oncol. 2007, 25, 319–325. [Google Scholar] [CrossRef]
- Said, N.; Frierson, H.F.; Sanchez-Carbayo, M.; Brekken, R.A.; Theodorescu, D. Loss of SPARC in bladder cancer enhances carcinogenesis and progression. J. Clin. Investig. 2013, 123, 751–766. [Google Scholar] [CrossRef]
- Baffy, G.; Brunt, E.M.; Caldwell, S.H. Hepatocellular carcinoma in non-alcoholic fatty liver disease: An emerging menace. J. Hepatol. 2012, 56, 1384–1391. [Google Scholar] [CrossRef]
- Lade, A.; Noon, L.A.; Friedman, S.L. Contributions of metabolic dysregulation and inflammation to nonalcoholic steatohepatitis, hepatic fibrosis, and cancer. Curr. Opin. Oncol. 2014, 26, 100–107. [Google Scholar] [CrossRef]
- Monga, S.P. beta-Catenin Signaling and Roles in Liver Homeostasis, Injury, and Tumorigenesis. Gastroenterology 2015, 148, 1294–1310. [Google Scholar] [CrossRef]
- Bakiri, L.; Hamacher, R.; Grana, O.; Guio-Carrion, A.; Campos-Olivas, R.; Martinez, L.; Dienes, H.P.; Thomsen, M.K.; Hasenfuss, S.C.; Wagner, E.F. Liver carcinogenesis by FOS-dependent inflammation and cholesterol dysregulation. J. Exp. Med. 2017, 214, 1387–1409. [Google Scholar] [CrossRef]
- Chun, Y.S.; Huang, M.; Rink, L.; Von Mehren, M. Expression levels of insulin-like growth factors and receptors in hepatocellular carcinoma: A retrospective study. World J. Surg. Oncol. 2014, 12, 231. [Google Scholar] [CrossRef]
- Enguita-German, M.; Fortes, P. Targeting the insulin-like growth factor pathway in hepatocellular carcinoma. World J. Hepatol. 2014, 6, 716–737. [Google Scholar] [CrossRef]
- Xu, Z.; Hu, J.; Cao, H.; Pilo, M.G.; Cigliano, A.; Shao, Z.; Xu, M.; Ribback, S.; Dombrowski, F.; Calvisi, D.F.; et al. Loss of Pten synergizes with c-Met to promote hepatocellular carcinoma development via mTORC2 pathway. Exp. Mol. Med. 2018, 50, e417. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar]
- Schafer, M.; Werner, S. Cancer as an overhealing wound: An old hypothesis revisited. Nat. Rev. Mol. Cell Biol. 2008, 9, 628–638. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Li, Z.; Jimenez, S.A. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 2011, 179, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Piera-Velazquez, S.; Mendoza, F.A.; Jimenez, S.A. Endothelial to Mesenchymal Transition (EndoMT) in the Pathogenesis of Human Fibrotic Diseases. J. Clin. Med. 2016, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis Model. Mech 2018, 11, dmm029447. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Kokudo, T.; Suzuki, Y.; Yoshimatsu, Y.; Yamazaki, T.; Watabe, T.; Miyazono, K. Snail is required for TGFbeta-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J. Cell Sci. 2008, 121, 3317–3324. [Google Scholar] [CrossRef]
- Medici, D.; Potenta, S.; Kalluri, R. Transforming growth factor-beta2 promotes Snail-mediated endothelial-mesenchymal transition through convergence of Smad-dependent and Smad-independent signalling. Biochem. J. 2011, 437, 515–520. [Google Scholar] [CrossRef]
- Batlle, E.; Sancho, E.; Franci, C.; Dominguez, D.; Monfar, M.; Baulida, J.; Garcia De Herreros, A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef]
- Li, Z.; Jimenez, S.A. Protein kinase Cdelta and c-Abl kinase are required for transforming growth factor beta induction of endothelial-mesenchymal transition in vitro. Arthritis Rheumatol. 2011, 63, 2473–2483. [Google Scholar] [CrossRef]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Prunotto, M.; Desmouliere, A.; Varga, J.; De Wever, O.; Mareel, M.; Gabbiani, G. Recent developments in myofibroblast biology: Paradigms for connective tissue remodeling. Am. J. Pathol. 2012, 180, 1340–1355. [Google Scholar] [CrossRef]
- Hudson, L.G.; Newkirk, K.M.; Chandler, H.L.; Choi, C.; Fossey, S.L.; Parent, A.E.; Kusewitt, D.F. Cutaneous wound reepithelialization is compromised in mice lacking functional Slug (Snai2). J. Dermatol. Sci. 2009, 56, 19–26. [Google Scholar] [CrossRef]
- Savagner, P.; Kusewitt, D.F.; Carver, E.A.; Magnino, F.; Choi, C.; Gridley, T.; Hudson, L.G. Developmental transcription factor slug is required for effective re-epithelialization by adult keratinocytes. J. Cell Physiol. 2005, 202, 858–866. [Google Scholar] [CrossRef]
- Arpino, V.; Brock, M.; Gill, S.E. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. 2015, 44–46, 247–254. [Google Scholar] [CrossRef]
- Vered, M.; Dayan, D.; Yahalom, R.; Dobriyan, A.; Barshack, I.; Bello, I.O.; Kantola, S.; Salo, T. Cancer-associated fibroblasts and epithelial-mesenchymal transition in metastatic oral tongue squamous cell carcinoma. Int. J. Cancer 2010, 127, 1356–1362. [Google Scholar] [CrossRef]
- Hassona, Y.; Cirillo, N.; Heesom, K.; Parkinson, E.K.; Prime, S.S. Senescent cancer-associated fibroblasts secrete active MMP-2 that promotes keratinocyte dis-cohesion and invasion. Br. J. Cancer 2014, 111, 1230–1237. [Google Scholar] [CrossRef]
- Tian, B.; Chen, X.; Zhang, H.; Li, X.; Wang, J.; Han, W.; Zhang, L.Y.; Fu, L.; Li, Y.; Nie, C.; et al. Urokinase plasminogen activator secreted by cancer-associated fibroblasts induces tumor progression via PI3K/AKT and ERK signaling in esophageal squamous cell carcinoma. Oncotarget 2017, 8, 42300–42313. [Google Scholar] [CrossRef][Green Version]
- Yamaguchi, Y.; Mann, D.M.; Ruoslahti, E. Negative regulation of transforming growth factor-beta by the proteoglycan decorin. Nature 1990, 346, 281–284. [Google Scholar] [CrossRef]
- Imai, K.; Hiramatsu, A.; Fukushima, D.; Pierschbacher, M.D.; Okada, Y. Degradation of decorin by matrix metalloproteinases: Identification of the cleavage sites, kinetic analyses and transforming growth factor-beta1 release. Biochem. J. 1997, 322 Pt 3, 809–814. [Google Scholar] [CrossRef]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef]
- Mezawa, Y.; Orimo, A. The roles of tumor- and metastasis-promoting carcinoma-associated fibroblasts in human carcinomas. Cell Tissue Res. 2016, 365, 675–689. [Google Scholar] [CrossRef]
- Pietras, K.; Ostman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef]
- Heneberg, P. Paracrine tumor signaling induces transdifferentiation of surrounding fibroblasts. Crit. Rev. Oncol. Hematol. 2016, 97, 303–311. [Google Scholar] [CrossRef]
- Tao, L.; Huang, G.; Song, H.; Chen, Y.; Chen, L. Cancer associated fibroblasts: An essential role in the tumor microenvironment. Oncol. Lett. 2017, 14, 2611–2620. [Google Scholar] [CrossRef]
- Polanska, U.M.; Orimo, A. Carcinoma-associated fibroblasts: Non-neoplastic tumour-promoting mesenchymal cells. J. Cell Physiol. 2013, 228, 1651–1657. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Ohlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef]
- Mishra, P.J.; Mishra, P.J.; Humeniuk, R.; Medina, D.J.; Alexe, G.; Mesirov, J.P.; Ganesan, S.; Glod, J.W.; Banerjee, D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008, 68, 4331–4339. [Google Scholar] [CrossRef]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef]
- Raz, Y.; Cohen, N.; Shani, O.; Bell, R.E.; Novitskiy, S.V.; Abramovitz, L.; Levy, C.; Milyavsky, M.; Leider-Trejo, L.; Moses, H.L.; et al. Bone marrow-derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J. Exp. Med. 2018, 215, 3075–3093. [Google Scholar] [CrossRef]
- Midgley, A.C.; Rogers, M.; Hallett, M.B.; Clayton, A.; Bowen, T.; Phillips, A.O.; Steadman, R. Transforming growth factor-beta1 (TGF-beta1)-stimulated fibroblast to myofibroblast differentiation is mediated by hyaluronan (HA)-facilitated epidermal growth factor receptor (EGFR) and CD44 co-localization in lipid rafts. J. Biol. Chem. 2013, 288, 14824–14838. [Google Scholar] [CrossRef]
- Bonner, J.C. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev. 2004, 15, 255–273. [Google Scholar] [CrossRef]
- Avgustinova, A.; Iravani, M.; Robertson, D.; Fearns, A.; Gao, Q.; Klingbeil, P.; Hanby, A.M.; Speirs, V.; Sahai, E.; Calvo, F.; et al. Tumour cell-derived Wnt7a recruits and activates fibroblasts to promote tumour aggressiveness. Nat. Commun. 2016, 7, 10305. [Google Scholar] [CrossRef]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef]
- Sharon, Y.; Raz, Y.; Cohen, N.; Ben-Shmuel, A.; Schwartz, H.; Geiger, T.; Erez, N. Tumor-derived osteopontin reprograms normal mammary fibroblasts to promote inflammation and tumor growth in breast cancer. Cancer Res. 2015, 75, 963–973. [Google Scholar] [CrossRef]
- Sanz-Moreno, V.; Gaggioli, C.; Yeo, M.; Albrengues, J.; Wallberg, F.; Viros, A.; Hooper, S.; Mitter, R.; Feral, C.C.; Cook, M.; et al. ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell 2011, 20, 229–245. [Google Scholar] [CrossRef]
- Albrengues, J.; Bertero, T.; Grasset, E.; Bonan, S.; Maiel, M.; Bourget, I.; Philippe, C.; Herraiz Serrano, C.; Benamar, S.; Croce, O.; et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat. Commun. 2015, 6, 10204. [Google Scholar] [CrossRef]
- Shien, K.; Papadimitrakopoulou, V.A.; Ruder, D.; Behrens, C.; Shen, L.; Kalhor, N.; Song, J.; Lee, J.J.; Wang, J.; Tang, X.; et al. JAK1/STAT3 Activation through a Proinflammatory Cytokine Pathway Leads to Resistance to Molecularly Targeted Therapy in Non-Small Cell Lung Cancer. Mol. Cancer Ther. 2017, 16, 2234–2245. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, H.Y.; Marzec, M.; Raghunath, P.N.; Nagasawa, T.; Wasik, M.A. STAT3- and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc. Natl. Acad. Sci. USA 2005, 102, 6948–6953. [Google Scholar] [CrossRef]
- Lee, H.; Zhang, P.; Herrmann, A.; Yang, C.; Xin, H.; Wang, Z.; Hoon, D.S.; Forman, S.J.; Jove, R.; Riggs, A.D.; et al. Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc. Natl. Acad. Sci. USA 2012, 109, 7765–7769. [Google Scholar] [CrossRef]
- Iwabu, A.; Smith, K.; Allen, F.D.; Lauffenburger, D.A.; Wells, A. Epidermal growth factor induces fibroblast contractility and motility via a protein kinase C delta-dependent pathway. J. Biol. Chem. 2004, 279, 14551–14560. [Google Scholar] [CrossRef]
- Mink, S.R.; Vashistha, S.; Zhang, W.; Hodge, A.; Agus, D.B.; Jain, A. Cancer-associated fibroblasts derived from EGFR-TKI-resistant tumors reverse EGFR pathway inhibition by EGFR-TKIs. Mol. Cancer Res. 2010, 8, 809–820. [Google Scholar] [CrossRef]
- Yun, Y.R.; Won, J.E.; Jeon, E.; Lee, S.; Kang, W.; Jo, H.; Jang, J.H.; Shin, U.S.; Kim, H.W. Fibroblast growth factors: Biology, function, and application for tissue regeneration. J. Tissue Eng. 2010, 2010, 218142. [Google Scholar] [CrossRef]
- Bai, Y.P.; Shang, K.; Chen, H.; Ding, F.; Wang, Z.; Liang, C.; Xu, Y.; Sun, M.H.; Li, Y.Y. FGF-1/-3/FGFR4 signaling in cancer-associated fibroblasts promotes tumor progression in colon cancer through Erk and MMP-7. Cancer Sci. 2015, 106, 1278–1287. [Google Scholar] [CrossRef]
- Mori, T.; Miyamoto, T.; Yoshida, H.; Asakawa, M.; Kawasumi, M.; Kobayashi, T.; Morioka, H.; Chiba, K.; Toyama, Y.; Yoshimura, A. IL-1beta and TNFalpha-initiated IL-6-STAT3 pathway is critical in mediating inflammatory cytokines and RANKL expression in inflammatory arthritis. Int. Immunol. 2011, 23, 701–712. [Google Scholar] [CrossRef]
- Tsuyada, A.; Chow, A.; Wu, J.; Somlo, G.; Chu, P.; Loera, S.; Luu, T.; Li, A.X.; Wu, X.; Ye, W.; et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. 2012, 72, 2768–2779. [Google Scholar] [CrossRef]
- Albrengues, J.; Bourget, I.; Pons, C.; Butet, V.; Hofman, P.; Tartare-Deckert, S.; Feral, C.C.; Meneguzzi, G.; Gaggioli, C. LIF mediates proinvasive activation of stromal fibroblasts in cancer. Cell Rep. 2014, 7, 1664–1678. [Google Scholar] [CrossRef]
- Li, W.; Fan, J.; Chen, M.; Guan, S.; Sawcer, D.; Bokoch, G.M.; Woodley, D.T. Mechanism of human dermal fibroblast migration driven by type I collagen and platelet-derived growth factor-BB. Mol. Biol. Cell 2004, 15, 294–309. [Google Scholar] [CrossRef]
- Roswall, P.; Bocci, M.; Bartoschek, M.; Li, H.; Kristiansen, G.; Jansson, S.; Lehn, S.; Sjolund, J.; Reid, S.; Larsson, C.; et al. Microenvironmental control of breast cancer subtype elicited through paracrine platelet-derived growth factor-CC signaling. Nat. Med. 2018, 24, 463–473. [Google Scholar] [CrossRef]
- Kabashima, K.; Sakabe, J.; Yoshiki, R.; Tabata, Y.; Kohno, K.; Tokura, Y. Involvement of Wnt signaling in dermal fibroblasts. Am. J. Pathol. 2010, 176, 721–732. [Google Scholar] [CrossRef][Green Version]
- Inada, M.; Takita, M.; Yokoyama, S.; Watanabe, K.; Tominari, T.; Matsumoto, C.; Hirata, M.; Maru, Y.; Maruyama, T.; Sugimoto, Y.; et al. Direct Melanoma Cell Contact Induces Stromal Cell Autocrine Prostaglandin E2-EP4 Receptor Signaling That Drives Tumor Growth, Angiogenesis, and Metastasis. J. Biol. Chem. 2015, 290, 29781–29793. [Google Scholar] [CrossRef]
- Guo, X.; Oshima, H.; Kitmura, T.; Taketo, M.M.; Oshima, M. Stromal fibroblasts activated by tumor cells promote angiogenesis in mouse gastric cancer. J. Biol. Chem. 2008, 283, 19864–19871. [Google Scholar] [CrossRef]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-beta signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef]
- Lohr, M.; Schmidt, C.; Ringel, J.; Kluth, M.; Muller, P.; Nizze, H.; Jesnowski, R. Transforming growth factor-beta1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. 2001, 61, 550–555. [Google Scholar]
- Berking, C.; Takemoto, R.; Schaider, H.; Showe, L.; Satyamoorthy, K.; Robbins, P.; Herlyn, M. Transforming growth factor-beta1 increases survival of human melanoma through stroma remodeling. Cancer Res. 2001, 61, 8306–8316. [Google Scholar]
- Guido, C.; Whitaker-Menezes, D.; Capparelli, C.; Balliet, R.; Lin, Z.; Pestell, R.G.; Howell, A.; Aquila, S.; Ando, S.; Martinez-Outschoorn, U.; et al. Metabolic reprogramming of cancer-associated fibroblasts by TGF-beta drives tumor growth: Connecting TGF-beta signaling with “Warburg-like” cancer metabolism and L-lactate production. Cell Cycle 2012, 11, 3019–3035. [Google Scholar] [CrossRef]
- Vaquero, J.; Lobe, C.; Tahraoui, S.; Claperon, A.; Mergey, M.; Merabtene, F.; Wendum, D.; Coulouarn, C.; Housset, C.; Desbois-Mouthon, C.; et al. The IGF2/IR/IGF1R Pathway in Tumor Cells and Myofibroblasts Mediates Resistance to EGFR Inhibition in Cholangiocarcinoma. Clin. Cancer Res. 2018, 24, 4282–4296. [Google Scholar] [CrossRef]
- Ireland, L.; Santos, A.; Ahmed, M.S.; Rainer, C.; Nielsen, S.R.; Quaranta, V.; Weyer-Czernilofsky, U.; Engle, D.D.; Perez-Mancera, P.A.; Coupland, S.E.; et al. Chemoresistance in Pancreatic Cancer Is Driven by Stroma-Derived Insulin-Like Growth Factors. Cancer Res. 2016, 76, 6851–6863. [Google Scholar] [CrossRef]
- Unger, C.; Kramer, N.; Unterleuthner, D.; Scherzer, M.; Burian, A.; Rudisch, A.; Stadler, M.; Schlederer, M.; Lenhardt, D.; Riedl, A.; et al. Stromal-derived IGF2 promotes colon cancer progression via paracrine and autocrine mechanisms. Oncogene 2017, 36, 5341–5355. [Google Scholar] [CrossRef]
- Chen, W.J.; Ho, C.C.; Chang, Y.L.; Chen, H.Y.; Lin, C.A.; Ling, T.Y.; Yu, S.L.; Yuan, S.S.; Chen, Y.J.; Lin, C.Y.; et al. Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat. Commun. 2014, 5, 3472. [Google Scholar] [CrossRef]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012, 487, 505–509. [Google Scholar] [CrossRef]
- Wang, W.; Li, Q.; Yamada, T.; Matsumoto, K.; Matsumoto, I.; Oda, M.; Watanabe, G.; Kayano, Y.; Nishioka, Y.; Sone, S.; et al. Crosstalk to stromal fibroblasts induces resistance of lung cancer to epidermal growth factor receptor tyrosine kinase inhibitors. Clin. Cancer Res. 2009, 15, 6630–6638. [Google Scholar] [CrossRef]
- Ferrara, N.; Kerbel, R.S. Angiogenesis as a therapeutic target. Nature 2005, 438, 967–974. [Google Scholar] [CrossRef]
- Crawford, Y.; Kasman, I.; Yu, L.; Zhong, C.; Wu, X.; Modrusan, Z.; Kaminker, J.; Ferrara, N. PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell 2009, 15, 21–34. [Google Scholar] [CrossRef]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10(+)GPR77(+) Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856. [Google Scholar] [CrossRef]
- Sun, Y.; Campisi, J.; Higano, C.; Beer, T.M.; Porter, P.; Coleman, I.; True, L.; Nelson, P.S. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat. Med. 2012, 18, 1359–1368. [Google Scholar] [CrossRef]
- Tao, L.; Huang, G.; Wang, R.; Pan, Y.; He, Z.; Chu, X.; Song, H.; Chen, L. Cancer-associated fibroblasts treated with cisplatin facilitates chemoresistance of lung adenocarcinoma through IL-11/IL-11R/STAT3 signaling pathway. Sci. Rep. 2016, 6, 38408. [Google Scholar] [CrossRef]
- Lotti, F.; Jarrar, A.M.; Pai, R.K.; Hitomi, M.; Lathia, J.; Mace, A.; Gantt, G.A., Jr.; Sukhdeo, K.; DeVecchio, J.; Vasanji, A.; et al. Chemotherapy activates cancer-associated fibroblasts to maintain colorectal cancer-initiating cells by IL-17A. J. Exp. Med. 2013, 210, 2851–2872. [Google Scholar] [CrossRef]
- Erdogan, B.; Webb, D.J. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem. Soc. Trans. 2017, 45, 229–236. [Google Scholar] [CrossRef]
- Karagiannis, G.S.; Poutahidis, T.; Erdman, S.E.; Kirsch, R.; Riddell, R.H.; Diamandis, E.P. Cancer-associated fibroblasts drive the progression of metastasis through both paracrine and mechanical pressure on cancer tissue. Mol. Cancer Res. 2012, 10, 1403–1418. [Google Scholar] [CrossRef]
- Hirata, E.; Girotti, M.R.; Viros, A.; Hooper, S.; Spencer-Dene, B.; Matsuda, M.; Larkin, J.; Marais, R.; Sahai, E. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin beta1/FAK signaling. Cancer Cell 2015, 27, 574–588. [Google Scholar] [CrossRef]
- Qayyum, M.A.; Insana, M.F. Stromal responses to fractionated radiotherapy. Int. J. Radiat. Biol. 2012, 88, 383–392. [Google Scholar] [CrossRef]
- Pazolli, E.; Alspach, E.; Milczarek, A.; Prior, J.; Piwnica-Worms, D.; Stewart, S.A. Chromatin remodeling underlies the senescence-associated secretory phenotype of tumor stromal fibroblasts that supports cancer progression. Cancer Res. 2012, 72, 2251–2261. [Google Scholar] [CrossRef]
- Nguyen, A.H.; Elliott, I.A.; Wu, N.; Matsumura, C.; Vogelauer, M.; Attar, N.; Dann, A.; Ghukasyan, R.; Toste, P.A.; Patel, S.G.; et al. Histone deacetylase inhibitors provoke a tumor supportive phenotype in pancreatic cancer associated fibroblasts. Oncotarget 2017, 8, 19074–19088. [Google Scholar] [CrossRef]
- Kato, M.; Placencio-Hickok, V.R.; Madhav, A.; Haldar, S.; Tripathi, M.; Billet, S.; Mishra, R.; Smith, B.; Rohena-Rivera, K.; Agarwal, P.; et al. Heterogeneous cancer-associated fibroblast population potentiates neuroendocrine differentiation and castrate resistance in a CD105-dependent manner. Oncogene 2018. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Molecular Pathways: Deciphering Mechanisms of Resistance to Macrophage-Targeted Therapies. Clin. Cancer Res. 2017, 23, 876–884. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018. [Google Scholar] [CrossRef]
- Kumar, V.; Donthireddy, L.; Marvel, D.; Condamine, T.; Wang, F.; Lavilla-Alonso, S.; Hashimoto, A.; Vonteddu, P.; Behera, R.; Goins, M.A.; et al. Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell 2017, 32, 654–668. [Google Scholar] [CrossRef]
- Darby, I.A.; Hewitson, T.D. Fibroblast differentiation in wound healing and fibrosis. Int. Rev. Cytol. 2007, 257, 143–179. [Google Scholar]
- Darby, I.A.; Laverdet, B.; Bonte, F.; Desmouliere, A. Fibroblasts and myofibroblasts in wound healing. Clin. Cosmet. Investig. Dermatol. 2014, 7, 301–311. [Google Scholar]
- Penn, M.S. SDF-1:CXCR4 axis is fundamental for tissue preservation and repair. Am. J. Pathol. 2010, 177, 2166–2168. [Google Scholar] [CrossRef]
- Schmidt, A.; Brixius, K.; Bloch, W. Endothelial precursor cell migration during vasculogenesis. Circ. Res. 2007, 101, 125–136. [Google Scholar] [CrossRef]
- Sewell-Loftin, M.K.; Bayer, S.V.H.; Crist, E.; Hughes, T.; Joison, S.M.; Longmore, G.D.; George, S.C. Cancer-associated fibroblasts support vascular growth through mechanical force. Sci. Rep. 2017, 7, 12574. [Google Scholar] [CrossRef]
- Fukumura, D.; Xavier, R.; Sugiura, T.; Chen, Y.; Park, E.C.; Lu, N.; Selig, M.; Nielsen, G.; Taksir, T.; Jain, R.K.; et al. Tumor induction of VEGF promoter activity in stromal cells. Cell 1998, 94, 715–725. [Google Scholar] [CrossRef]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.; Iglesias, M.; Cespedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.; et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef]
- De Wever, O.; Westbroek, W.; Verloes, A.; Bloemen, N.; Bracke, M.; Gespach, C.; Bruyneel, E.; Mareel, M. Critical role of N-cadherin in myofibroblast invasion and migration in vitro stimulated by colon-cancer-cell-derived TGF-beta or wounding. J. Cell Sci. 2004, 117, 4691–4703. [Google Scholar] [CrossRef]
- Ng, C.F.; Frieboes, H.B. Model of vascular desmoplastic multispecies tumor growth. J. Theor. Biol. 2017, 430, 245–282. [Google Scholar] [CrossRef]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef]
- Izumi, D.; Ishimoto, T.; Miyake, K.; Sugihara, H.; Eto, K.; Sawayama, H.; Yasuda, T.; Kiyozumi, Y.; Kaida, T.; Kurashige, J.; et al. CXCL12/CXCR4 activation by cancer-associated fibroblasts promotes integrin beta1 clustering and invasiveness in gastric cancer. Int. J. Cancer 2016, 138, 1207–1219. [Google Scholar] [CrossRef]
- Sun, X.; Cheng, G.; Hao, M.; Zheng, J.; Zhou, X.; Zhang, J.; Taichman, R.S.; Pienta, K.J.; Wang, J. CXCL12 / CXCR4 / CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010, 29, 709–722. [Google Scholar] [CrossRef]
- Teng, F.; Tian, W.Y.; Wang, Y.M.; Zhang, Y.F.; Guo, F.; Zhao, J.; Gao, C.; Xue, F.X. Cancer-associated fibroblasts promote the progression of endometrial cancer via the SDF-1/CXCR4 axis. J. Hematol. Oncol. 2016, 9, 8. [Google Scholar] [CrossRef]
- Jin, F.; Brockmeier, U.; Otterbach, F.; Metzen, E. New insight into the SDF-1/CXCR4 axis in a breast carcinoma model: Hypoxia-induced endothelial SDF-1 and tumor cell CXCR4 are required for tumor cell intravasation. Mol. Cancer Res. 2012, 10, 1021–1031. [Google Scholar] [CrossRef]
- Mego, M.; Cholujova, D.; Minarik, G.; Sedlackova, T.; Gronesova, P.; Karaba, M.; Benca, J.; Cingelova, S.; Cierna, Z.; Manasova, D.; et al. CXCR4-SDF-1 interaction potentially mediates trafficking of circulating tumor cells in primary breast cancer. BMC Cancer 2016, 16, 127. [Google Scholar] [CrossRef]
- Fujii, H.; Biel, M.A.; Zhou, W.; Weitzman, S.A.; Baylin, S.B.; Gabrielson, E. Methylation of the HIC-1 candidate tumor suppressor gene in human breast cancer. Oncogene 1998, 16, 2159–2164. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Xiao, G.; Wang, J.; Zu, L.; Hao, M.; Sun, X.; Fu, Y.; Hu, G.; Wang, J. Hypermethylated in cancer 1(HIC1) suppresses non-small cell lung cancer progression by targeting interleukin-6/Stat3 pathway. Oncotarget 2016, 7, 30350–30364. [Google Scholar] [CrossRef]
- Wang, Y.; Weng, X.; Wang, L.; Hao, M.; Li, Y.; Hou, L.; Liang, Y.; Wu, T.; Yao, M.; Lin, G.; et al. HIC1 deletion promotes breast cancer progression by activating tumor cell/fibroblast crosstalk. J. Clin. Investig. 2018. [Google Scholar] [CrossRef]
- Zheng, J.; Wang, J.; Sun, X.; Hao, M.; Ding, T.; Xiong, D.; Wang, X.; Zhu, Y.; Xiao, G.; Cheng, G.; et al. HIC1 modulates prostate cancer progression by epigenetic modification. Clin. Cancer Res. 2013, 19, 1400–1410. [Google Scholar] [CrossRef]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef]
- Grasset, E.M.; Bertero, T.; Bozec, A.; Friard, J.; Bourget, I.; Pisano, S.; Lecacheur, M.; Maiel, M.; Bailleux, C.; Emelyanov, A.; et al. Matrix Stiffening and EGFR Cooperate to Promote the Collective Invasion of Cancer Cells. Cancer Res. 2018, 78, 5229–5242. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Saitoh, M. Involvement of partial EMT in cancer progression. J. Biochem. 2018, 164, 257–264. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef]
- Byrne, C.; Schairer, C.; Wolfe, J.; Parekh, N.; Salane, M.; Brinton, L.A.; Hoover, R.; Haile, R. Mammographic features and breast cancer risk: Effects with time, age, and menopause status. J. Natl. Cancer Inst. 1995, 87, 1622–1629. [Google Scholar] [CrossRef]
- DeFilippis, R.A.; Chang, H.; Dumont, N.; Rabban, J.T.; Chen, Y.Y.; Fontenay, G.V.; Berman, H.K.; Gauthier, M.L.; Zhao, J.; Hu, D.; et al. CD36 repression activates a multicellular stromal program shared by high mammographic density and tumor tissues. Cancer Discov. 2012, 2, 826–839. [Google Scholar] [CrossRef]
- Plikus, M.V.; Guerrero-Juarez, C.F.; Ito, M.; Li, Y.R.; Dedhia, P.H.; Zheng, Y.; Shao, M.; Gay, D.L.; Ramos, R.; Hsi, T.C.; et al. Regeneration of fat cells from myofibroblasts during wound healing. Science 2017, 355, 748–752. [Google Scholar] [CrossRef]
- Thomson, D.M.; Winder, W.W. AMP-activated protein kinase control of fat metabolism in skeletal muscle. Acta Physiol. 2009, 196, 147–154. [Google Scholar] [CrossRef]
- Ladanyi, A.; Mukherjee, A.; Kenny, H.A.; Johnson, A.; Mitra, A.K.; Sundaresan, S.; Nieman, K.M.; Pascual, G.; Benitah, S.A.; Montag, A.; et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 2018, 37, 2285–2301. [Google Scholar] [CrossRef]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martin, M.; Castellanos, A.; Attolini, C.S.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Caveolae and signalling in cancer. Nat. Rev. Cancer 2015, 15, 225–237. [Google Scholar] [CrossRef]
- Santi, A.; Kugeratski, F.G.; Zanivan, S. Cancer Associated Fibroblasts: The Architects of Stroma Remodeling. Proteomics 2018, 18, e1700167. [Google Scholar] [CrossRef]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Lin, Z.; Ertel, A.; Flomenberg, N.; Witkiewicz, A.K.; Birbe, R.C.; Howell, A.; Pavlides, S.; Gandara, R.; et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle 2011, 10, 1772–1783. [Google Scholar] [CrossRef]
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Schwartz, L.; Supuran, C.T.; Alfarouk, K.O. The Warburg Effect and the Hallmarks of Cancer. Anticancer Agents Med. Chem. 2017, 17, 164–170. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, S.; Yin, S.; Niu, W.; Xiong, W.; Tan, M.; Li, G.; Zhou, M. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget 2017, 8, 57813–57825. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef]
- Jezierska-Drutel, A.; Rosenzweig, S.A.; Neumann, C.A. Role of oxidative stress and the microenvironment in breast cancer development and progression. Adv. Cancer Res. 2013, 119, 107–125. [Google Scholar]
- Mougeolle, A.; Poussard, S.; Decossas, M.; Lamaze, C.; Lambert, O.; Dargelos, E. Oxidative stress induces caveolin 1 degradation and impairs caveolae functions in skeletal muscle cells. PLoS ONE 2015, 10, e0122654. [Google Scholar] [CrossRef]
- Wang, S.; Wang, N.; Zheng, Y.; Zhang, J.; Zhang, F.; Wang, Z. Caveolin-1: An Oxidative Stress-Related Target for Cancer Prevention. Oxid. Med. Cell Longev. 2017, 2017, 7454031. [Google Scholar] [CrossRef]
- Avagliano, A.; Granato, G.; Ruocco, M.R.; Romano, V.; Belviso, I.; Carfora, A.; Montagnani, S.; Arcucci, A. Metabolic Reprogramming of Cancer Associated Fibroblasts: The Slavery of Stromal Fibroblasts. Biomed. Res. Int. 2018, 2018, 6075403. [Google Scholar] [CrossRef]
- Pavlides, S.; Vera, I.; Gandara, R.; Sneddon, S.; Pestell, R.G.; Mercier, I.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; et al. Warburg meets autophagy: Cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid. Redox Signal. 2012, 16, 1264–1284. [Google Scholar] [CrossRef]
- Xing, Y.; Zhao, S.; Zhou, B.P.; Mi, J. Metabolic reprogramming of the tumour microenvironment. FEBS J. 2015, 282, 3892–3898. [Google Scholar] [CrossRef]
- Capparelli, C.; Guido, C.; Whitaker-Menezes, D.; Bonuccelli, G.; Balliet, R.; Pestell, T.G.; Goldberg, A.F.; Pestell, R.G.; Howell, A.; Sneddon, S.; et al. Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis via glycolysis and ketone production. Cell Cycle 2012, 11, 2285–2302. [Google Scholar] [CrossRef]
- Allen, E.; Mieville, P.; Warren, C.M.; Saghafinia, S.; Li, L.; Peng, M.W.; Hanahan, D. Metabolic Symbiosis Enables Adaptive Resistance to Anti-angiogenic Therapy that Is Dependent on mTOR Signaling. Cell Rep. 2016, 15, 1144–1160. [Google Scholar] [CrossRef]
- Porporato, P.E.; Dhup, S.; Dadhich, R.K.; Copetti, T.; Sonveaux, P. Anticancer targets in the glycolytic metabolism of tumors: A comprehensive review. Front. Pharmacol. 2011, 2, 49. [Google Scholar] [CrossRef]
- Semenza, G.L. Tumor metabolism: Cancer cells give and take lactate. J. Clin. Investig. 2008, 118, 3835–3837. [Google Scholar] [CrossRef]
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef]
- Linares, J.F.; Cordes, T.; Duran, A.; Reina-Campos, M.; Valencia, T.; Ahn, C.S.; Castilla, E.A.; Moscat, J.; Metallo, C.M.; Diaz-Meco, M.T. ATF4-Induced Metabolic Reprograming Is a Synthetic Vulnerability of the p62-Deficient Tumor Stroma. Cell Metab. 2017, 26, 817–829. [Google Scholar] [CrossRef]
- Duran, A.; Hernandez, E.D.; Reina-Campos, M.; Castilla, E.A.; Subramaniam, S.; Raghunandan, S.; Roberts, L.R.; Kisseleva, T.; Karin, M.; Diaz-Meco, M.T.; et al. p62/SQSTM1 by Binding to Vitamin D Receptor Inhibits Hepatic Stellate Cell Activity, Fibrosis, and Liver Cancer. Cancer Cell 2016, 30, 595–609. [Google Scholar] [CrossRef]
- Reina-Campos, M.; Shelton, P.M.; Diaz-Meco, M.T.; Moscat, J. Metabolic reprogramming of the tumor microenvironment by p62 and its partners. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 88–95. [Google Scholar] [CrossRef]
- Goruppi, S.; Jo, S.H.; Laszlo, C.; Clocchiatti, A.; Neel, V.; Dotto, G.P. Autophagy Controls CSL/RBPJkappa Stability through a p62/SQSTM1-Dependent Mechanism. Cell Rep. 2018, 24, 3108–3114. [Google Scholar] [CrossRef]
- Valencia, T.; Kim, J.Y.; Abu-Baker, S.; Moscat-Pardos, J.; Ahn, C.S.; Reina-Campos, M.; Duran, A.; Castilla, E.A.; Metallo, C.M.; Diaz-Meco, M.T.; et al. Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell 2014, 26, 121–135. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Moscat, J.; Diaz-Meco, M.T. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009, 137, 1001–1004. [Google Scholar] [CrossRef]
- Huang, J.; Diaz-Meco, M.T.; Moscat, J. The macroenviromental control of cancer metabolism by p62. Cell Cycle 2018, 17, 2110–2121. [Google Scholar] [CrossRef]
- Jain, R.K.; Tong, R.T.; Munn, L.L. Effect of vascular normalization by antiangiogenic therapy on interstitial hypertension, peritumor edema, and lymphatic metastasis: Insights from a mathematical model. Cancer Res. 2007, 67, 2729–2735. [Google Scholar] [CrossRef]
- Munson, J.M.; Shieh, A.C. Interstitial fluid flow in cancer: Implications for disease progression and treatment. Cancer Manag. Res. 2014, 6, 317–328. [Google Scholar] [CrossRef]
- Omidi, Y.; Barar, J. Targeting tumor microenvironment: Crossing tumor interstitial fluid by multifunctional nanomedicines. Bioimpacts 2014, 4, 55–67. [Google Scholar]
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef]
- Christian, S.; Winkler, R.; Helfrich, I.; Boos, A.M.; Besemfelder, E.; Schadendorf, D.; Augustin, H.G. Endosialin (Tem1) is a marker of tumor-associated myofibroblasts and tumor vessel-associated mural cells. Am. J. Pathol. 2008, 172, 486–494. [Google Scholar] [CrossRef]
- Otranto, M.; Sarrazy, V.; Bonte, F.; Hinz, B.; Gabbiani, G.; Desmouliere, A. The role of the myofibroblast in tumor stroma remodeling. Cell Adhes. Migr. 2012, 6, 203–219. [Google Scholar] [CrossRef]
- Ribeiro, A.L.; Okamoto, O.K. Combined effects of pericytes in the tumor microenvironment. Stem Cells Int. 2015, 2015, 868475. [Google Scholar] [CrossRef]
- Liao, Z.; Tan, Z.W.; Zhu, P.; Tan, N.S. Cancer-associated fibroblasts in tumor microenvironment—Accomplices in tumor malignancy. Cell Immunol. 2018. [Google Scholar] [CrossRef]
- Boucher, Y.; Salehi, H.; Witwer, B.; Harsh, G.R.T.; Jain, R.K. Interstitial fluid pressure in intracranial tumours in patients and in rodents. Br. J. Cancer 1997, 75, 829–836. [Google Scholar] [CrossRef]
- Nathanson, S.D.; Nelson, L. Interstitial fluid pressure in breast cancer, benign breast conditions, and breast parenchyma. Ann. Surg. Oncol. 1994, 1, 333–338. [Google Scholar] [CrossRef]
- Simonsen, T.G.; Gaustad, J.V.; Leinaas, M.N.; Rofstad, E.K. High interstitial fluid pressure is associated with tumor-line specific vascular abnormalities in human melanoma xenografts. PLoS ONE 2012, 7, e40006. [Google Scholar] [CrossRef]
- Bhome, R.; Al Saihati, H.A.; Goh, R.W.; Bullock, M.D.; Primrose, J.N.; Thomas, G.J.; Sayan, A.E.; Mirnezami, A.H. Translational aspects in targeting the stromal tumour microenvironment: From bench to bedside. New Horiz Transl. Med. 2016, 3, 9–21. [Google Scholar] [CrossRef]
- Cazet, A.S.; Hui, M.N.; Elsworth, B.L.; Wu, S.Z.; Roden, D.; Chan, C.L.; Skhinas, J.N.; Collot, R.; Yang, J.; Harvey, K.; et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat. Commun. 2018, 9, 2897. [Google Scholar] [CrossRef]
- Giussani, M.; Merlino, G.; Cappelletti, V.; Tagliabue, E.; Daidone, M.G. Tumor-extracellular matrix interactions: Identification of tools associated with breast cancer progression. Semin. Cancer Biol. 2015, 35, 3–10. [Google Scholar] [CrossRef]
- Son, B.; Lee, S.; Youn, H.; Kim, E.; Kim, W.; Youn, B. The role of tumor microenvironment in therapeutic resistance. Oncotarget 2017, 8, 3933–3945. [Google Scholar] [CrossRef]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef]
- Peng, C.; Liu, J.; Yang, G.; Li, Y. Lysyl oxidase activates cancer stromal cells and promotes gastric cancer progression: Quantum dot-based identification of biomarkers in cancer stromal cells. Int. J. Nanomed. 2018, 13, 161–174. [Google Scholar] [CrossRef]
- Wang, T.H.; Hsia, S.M.; Shieh, T.M. Lysyl Oxidase and the Tumor Microenvironment. Int. J. Mol. Sci. 2016, 18, 62. [Google Scholar] [CrossRef]
- Reynaud, C.; Ferreras, L.; Di Mauro, P.; Kan, C.; Croset, M.; Bonnelye, E.; Pez, F.; Thomas, C.; Aimond, G.; Karnoub, A.E.; et al. Lysyl Oxidase Is a Strong Determinant of Tumor Cell Colonization in Bone. Cancer Res. 2017, 77, 268–278. [Google Scholar] [CrossRef]
- Cox, T.R.; Gartland, A.; Erler, J.T. Lysyl Oxidase, a Targetable Secreted Molecule Involved in Cancer Metastasis. Cancer Res. 2016, 76, 188–192. [Google Scholar] [CrossRef]
- Miller, B.W.; Morton, J.P.; Pinese, M.; Saturno, G.; Jamieson, N.B.; McGhee, E.; Timpson, P.; Leach, J.; McGarry, L.; Shanks, E.; et al. Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: Inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol. Med. 2015, 7, 1063–1076. [Google Scholar] [CrossRef]
- Kelleher, F.C. Hedgehog signaling and therapeutics in pancreatic cancer. Carcinogenesis 2011, 32, 445–451. [Google Scholar] [CrossRef]
- Erkan, M.; Hausmann, S.; Michalski, C.W.; Fingerle, A.A.; Dobritz, M.; Kleeff, J.; Friess, H. The role of stroma in pancreatic cancer: Diagnostic and therapeutic implications. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 454–467. [Google Scholar] [CrossRef]
- Mathew, E.; Zhang, Y.; Holtz, A.M.; Kane, K.T.; Song, J.Y.; Allen, B.L.; Pasca di Magliano, M. Dosage-dependent regulation of pancreatic cancer growth and angiogenesis by hedgehog signaling. Cell Rep. 2014, 9, 484–494. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhou, C.; Cheng, L.; Yan, B.; Chen, K.; Chen, X.; Zong, L.; Lei, J.; Duan, W.; Xu, Q.; et al. Inhibiting YAP expression suppresses pancreatic cancer progression by disrupting tumor-stromal interactions. J. Exp. Clin. Cancer Res. 2018, 37, 69. [Google Scholar] [CrossRef]
- Kharaishvili, G.; Simkova, D.; Bouchalova, K.; Gachechiladze, M.; Narsia, N.; Bouchal, J. The role of cancer-associated fibroblasts, solid stress and other microenvironmental factors in tumor progression and therapy resistance. Cancer Cell Int. 2014, 14, 41. [Google Scholar] [CrossRef]
- Warren, J.S.A.; Xiao, Y.; Lamar, J.M. YAP/TAZ Activation as a Target for Treating Metastatic Cancer. Cancers 2018, 10, 115. [Google Scholar] [CrossRef]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef]
- Campbell, I.; Polyak, K.; Haviv, I. Clonal mutations in the cancer-associated fibroblasts: The case against genetic coevolution. Cancer Res. 2009, 69, 6765–6768, discussion 6769. [Google Scholar] [CrossRef]
- Togo, S.; Polanska, U.M.; Horimoto, Y.; Orimo, A. Carcinoma-associated fibroblasts are a promising therapeutic target. Cancers 2013, 5, 149–169. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef]
- Tredan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef]
- Wu, M.; Frieboes, H.B.; Chaplain, M.A.; McDougall, S.R.; Cristini, V.; Lowengrub, J.S. The effect of interstitial pressure on therapeutic agent transport: Coupling with the tumor blood and lymphatic vascular systems. J. Theor. Biol. 2014, 355, 194–207. [Google Scholar] [CrossRef]
- Gourd, E. PEGPH20 for metastatic pancreatic ductal adenocarcinoma. Lancet Oncol. 2018, 19, e81. [Google Scholar] [CrossRef]
- Michl, P.; Gress, T.M. Improving drug delivery to pancreatic cancer: Breaching the stromal fortress by targeting hyaluronic acid. Gut 2012, 61, 1377–1379. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Zheng, L.; Bullock, A.J.; Seery, T.E.; Harris, W.P.; Sigal, D.S.; Braiteh, F.; Ritch, P.S.; Zalupski, M.M.; Bahary, N.; et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients With Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J. Clin. Oncol. 2018, 36, 359–366. [Google Scholar] [CrossRef]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef]
- Kourelis, T.V.; Siegel, R.D. Metformin and cancer: New applications for an old drug. Med. Oncol. 2012, 29, 1314–1327. [Google Scholar] [CrossRef]
- Leone, A.; Di Gennaro, E.; Bruzzese, F.; Avallone, A.; Budillon, A. New perspective for an old antidiabetic drug: Metformin as anticancer agent. Cancer Treat. Res. 2014, 159, 355–376. [Google Scholar]
- Saini, N.; Yang, X. Metformin as an anti-cancer agent: Actions and mechanisms targeting cancer stem cells. Acta Biochim. Biophys. Sin. 2018, 50, 133–143. [Google Scholar] [CrossRef]
- Yoshida, G.J. Therapeutic strategies of drug repositioning targeting autophagy to induce cancer cell death: From pathophysiology to treatment. J. Hematol. Oncol. 2017, 10, 67. [Google Scholar] [CrossRef]
- Nesti, L.; Natali, A. Metformin effects on the heart and the cardiovascular system: A review of experimental and clinical data. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 657–669. [Google Scholar] [CrossRef]
- Rangarajan, S.; Bone, N.B.; Zmijewska, A.A.; Jiang, S.; Park, D.W.; Bernard, K.; Locy, M.L.; Ravi, S.; Deshane, J.; Mannon, R.B.; et al. Metformin reverses established lung fibrosis in a bleomycin model. Nat. Med. 2018, 24, 1121–1127. [Google Scholar] [CrossRef]
- Shao, R.; Li, X.; Feng, Y.; Lin, J.F.; Billig, H. Direct effects of metformin in the endometrium: A hypothetical mechanism for the treatment of women with PCOS and endometrial carcinoma. J. Exp. Clin. Cancer Res. 2014, 33, 41. [Google Scholar] [CrossRef]
- Yi, H.; Huang, C.; Shi, Y.; Cao, Q.; Zhao, Y.; Zhang, L.; Chen, J.; Pollock, C.A.; Chen, X.M. Metformin attenuates folic-acid induced renal fibrosis in mice. J. Cell Physiol. 2018, 233, 7045–7054. [Google Scholar] [CrossRef]
- Li, L.; Huang, W.; Li, K.; Zhang, K.; Lin, C.; Han, R.; Lu, C.; Wang, Y.; Chen, H.; Sun, F.; et al. Metformin attenuates gefitinib-induced exacerbation of pulmonary fibrosis by inhibition of TGF-beta signaling pathway. Oncotarget 2015, 6, 43605–43619. [Google Scholar] [CrossRef]
- Xu, S.; Yang, Z.; Jin, P.; Yang, X.; Li, X.; Wei, X.; Wang, Y.; Long, S.; Zhang, T.; Chen, G.; et al. Metformin Suppresses Tumor Progression by Inactivating Stromal Fibroblasts in Ovarian Cancer. Mol. Cancer Ther. 2018, 17, 1291–1302. [Google Scholar] [CrossRef]
- Pinter, M.; Jain, R.K. Targeting the renin-angiotensin system to improve cancer treatment: Implications for immunotherapy. Sci. Transl. Med. 2017, 9, eaan5616. [Google Scholar] [CrossRef]
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013, 4, 2516. [Google Scholar] [CrossRef]
- Vennin, C.; Murphy, K.J.; Morton, J.P.; Cox, T.R.; Pajic, M.; Timpson, P. Reshaping the Tumor Stroma for Treatment of Pancreatic Cancer. Gastroenterology 2018, 154, 820–838. [Google Scholar] [CrossRef]
- Azuma, A. Pirfenidone treatment of idiopathic pulmonary fibrosis. Ther. Adv. Respir. Dis. 2012, 6, 107–114. [Google Scholar] [CrossRef]
- Didiasova, M.; Singh, R.; Wilhelm, J.; Kwapiszewska, G.; Wujak, L.; Zakrzewicz, D.; Schaefer, L.; Markart, P.; Seeger, W.; Lauth, M.; et al. Pirfenidone exerts antifibrotic effects through inhibition of GLI transcription factors. FASEB J. 2017, 31, 1916–1928. [Google Scholar] [CrossRef]
- Miura, Y.; Saito, T.; Tanaka, T.; Takoi, H.; Yatagai, Y.; Inomata, M.; Nei, T.; Saito, Y.; Gemma, A.; Azuma, A. Reduced incidence of lung cancer in patients with idiopathic pulmonary fibrosis treated with pirfenidone. Respir. Investig. 2018, 56, 72–79. [Google Scholar] [CrossRef]
- Mediavilla-Varela, M.; Boateng, K.; Noyes, D.; Antonia, S.J. The anti-fibrotic agent pirfenidone synergizes with cisplatin in killing tumor cells and cancer-associated fibroblasts. BMC Cancer 2016, 16, 176. [Google Scholar] [CrossRef]
- Ke, Z.; He, W.; Lai, Y.; Guo, X.; Chen, S.; Li, S.; Wang, Y.; Wang, L. Overexpression of collagen triple helix repeat containing 1 (CTHRC1) is associated with tumour aggressiveness and poor prognosis in human non-small cell lung cancer. Oncotarget 2014, 5, 9410–9424. [Google Scholar] [CrossRef]
- Takai, K.; Le, A.; Weaver, V.M.; Werb, Z. Targeting the cancer-associated fibroblasts as a treatment in triple-negative breast cancer. Oncotarget 2016, 7, 82889–82901. [Google Scholar] [CrossRef]
- Kozono, S.; Ohuchida, K.; Eguchi, D.; Ikenaga, N.; Fujiwara, K.; Cui, L.; Mizumoto, K.; Tanaka, M. Pirfenidone inhibits pancreatic cancer desmoplasia by regulating stellate cells. Cancer Res. 2013, 73, 2345–2356. [Google Scholar] [CrossRef]
- Polydorou, C.; Mpekris, F.; Papageorgis, P.; Voutouri, C.; Stylianopoulos, T. Pirfenidone normalizes the tumor microenvironment to improve chemotherapy. Oncotarget 2017, 8, 24506–24517. [Google Scholar] [CrossRef]
- Ding, N.; Hah, N.; Yu, R.T.; Sherman, M.H.; Benner, C.; Leblanc, M.; He, M.; Liddle, C.; Downes, M.; Evans, R.M. BRD4 is a novel therapeutic target for liver fibrosis. Proc. Natl. Acad. Sci. USA 2015, 112, 15713–15718. [Google Scholar] [CrossRef]
- Yamamoto, K.; Tateishi, K.; Kudo, Y.; Hoshikawa, M.; Tanaka, M.; Nakatsuka, T.; Fujiwara, H.; Miyabayashi, K.; Takahashi, R.; Tanaka, Y.; et al. Stromal remodeling by the BET bromodomain inhibitor JQ1 suppresses the progression of human pancreatic cancer. Oncotarget 2016, 7, 61469–61484. [Google Scholar] [CrossRef]
- Middleton, S.A.; Rajpal, N.; Cutler, L.; Mander, P.; Rioja, I.; Prinjha, R.K.; Rajpal, D.; Agarwal, P.; Kumar, V. BET Inhibition Improves NASH and Liver Fibrosis. Sci. Rep. 2018, 8, 17257. [Google Scholar] [CrossRef]
- Kim, D.E.; Procopio, M.G.; Ghosh, S.; Jo, S.H.; Goruppi, S.; Magliozzi, F.; Bordignon, P.; Neel, V.; Angelino, P.; Dotto, G.P. Convergent roles of ATF3 and CSL in chromatin control of cancer-associated fibroblast activation. J. Exp. Med. 2017, 214, 2349–2368. [Google Scholar] [CrossRef]
- Hah, N.; Sherman, M.H.; Yu, R.T.; Downes, M.; Evans, R.M. Targeting Transcriptional and Epigenetic Reprogramming in Stromal Cells in Fibrosis and Cancer. Cold Spring Harb. Symp. Quant. Biol. 2015, 80, 249–255. [Google Scholar] [CrossRef]
- Ding, N.; Yu, R.T.; Subramaniam, N.; Sherman, M.H.; Wilson, C.; Rao, R.; Leblanc, M.; Coulter, S.; He, M.; Scott, C.; et al. A vitamin D receptor/SMAD genomic circuit gates hepatic fibrotic response. Cell 2013, 153, 601–613. [Google Scholar] [CrossRef]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef]
- Shin, K.; Lim, A.; Zhao, C.; Sahoo, D.; Pan, Y.; Spiekerkoetter, E.; Liao, J.C.; Beachy, P.A. Hedgehog signaling restrains bladder cancer progression by eliciting stromal production of urothelial differentiation factors. Cancer Cell 2014, 26, 521–533. [Google Scholar] [CrossRef]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef]
- Gerling, M.; Buller, N.V.; Kirn, L.M.; Joost, S.; Frings, O.; Englert, B.; Bergstrom, A.; Kuiper, R.V.; Blaas, L.; Wielenga, M.C.; et al. Stromal Hedgehog signalling is downregulated in colon cancer and its restoration restrains tumour growth. Nat. Commun. 2016, 7, 12321. [Google Scholar] [CrossRef]
- Madar, S.; Goldstein, I.; Rotter, V. ‘Cancer associated fibroblasts’—More than meets the eye. Trends Mol. Med. 2013, 19, 447–453. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Signal Pathway | Wound-Induced Fibroblasts | CAFs |
|---|---|---|
| Epithelial growth factor (EGF) | EGF stimulation increases the phosphorylation of myosin light chain (MLC) subunit of myosin that promotes cell contractility in various different cell types. Activation of PKC with the PKC-δ isoform mediates the cell contraction by EGF-stimulated MLC phosphorylation in murine fibroblast cells [69]. | Resistance to the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) is partially medicated by CAFs in tumors through paracrine factors secreted from these fibroblasts [70]. |
| Fibroblast growth factor (FGF) | FGFs have the biological activity of stimulating the proliferation of fibroblasts and angiogenesis [71]. FGFs exert multiple functions through binding to and activation of fibroblast growth factor receptors (FGFRs), and the main signaling through the stimulation of FGFRs is the RAS/MAPK signal pathway. | CAFs secrete increased levels of FGF-1/-3 and promote cancer cell growth and angiogenesis through the activation of FGFR4, which is followed by the activation of extracellular signal-regulated kinase (ERK) and the modulation of MMP-7 expression [72]. In addition, FGF-1 and FGF-3 act as primary autocrine mediators of epithelial-stromal interactions in the tumor progression. |
| JAK/STAT | Synovial fibroblasts mediate chronic inflammation and joint destruction in patients suffered from rheumatoid arthritis (RA). Increased levels of IL-6, TNF-α and IL-1β production activate STAT3 signaling that in turn boosts expression levels of these cytokines in an autocrine fashion in synovial fibroblasts, promoting chronic inflammation [73]. STAT3 activation also induces receptor activator of nuclear factor kappa B ligand (RANKL) expression that stimulates osteoclastogenesis and thus promotes the joint destruction [73]. | CAFs release high levels of IL-6 and CCL2 upon STAT3 activation in co-culture system with cancer cells, promoting the self-renewal and spheroid forming potentials of cancer stem cells [74]. Furthermore, the leukemia inhibitory factor (LIF)-induced JAK1/STAT3 signaling pathway mediates expression of the invasive CAF phenotype [75]. |
| PDGF | PDGFs induce fibroblast activation and fibrosis. PDGF-BB stimulates polarization and provides enhancement and directionality for collagen-driven human dermal fibroblast migration. Akt processes both migratory and proliferative signals from PDGF receptors [76]. | Breast tumor cells produce PDGF-CC to activate stromal fibroblasts that in turn confer the basal and estrogen receptor α- negative phenotypes into cancer cells, rendering them unresponsive to endocrine treatment [77]. |
| PGE2-Wnt | Dermal fibroblasts expressing a low level of Dickkopf 1, a Wnt signaling antagonist, exhibit enhancement of the canonical Wnt/β-catenin signal pathway with accumulation of prostaglandin E2 (PGE2) [78]. The PGE2 signaling also increases nuclear β-catenin signaling in fibroblasts. | Autocrine activity of PGE2 regulates the production of angiogenic factors by fibroblasts, which are key to the vascularization of both primary and metastatic tumor growth [79]. Simultaneous activation of PGE2 and Wnt signals in transgenic mice causes gastric cancer with an abundance of vascular endothelial growth factor-A (VEGF-A) expressing CAFs, derived from bone marrow [80]. |
| TGF-β | Upon TGF-β stimulation, fibroblasts are activated and undergo phenotypic transition into myofibroblasts, the key effector cells under fibrotic conditions. The myofibroblast phenotype is characterized by the formation of gap junctions and by the acquisition of a contractile apparatus with associated contractile proteins. In healing wounds, myofibroblasts are required for tissue repair prior to their elimination due to the induction of apoptosis, but constitutively activated myofibroblasts promote fibrosis [81]. | Increased TGF-β production by tumor cells gives rise to the desmoplastic stroma in murine tumor models [82,83]. TGF-β potently suppresses immunity, induces angiogenesis and promotes cancer cell migration and invasion by stimulating EMT. Moreover, cancer cell-derived TGF-β activates TGF-β signaling in CAFs, inducing the up-regulation of monocarboxylate transporter 4 (MCT4) (a marker of glycolysis) and BNIP3 (a marker of autophagy) and the loss of caveolin-1 (CAV1) [84]. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshida, G.J.; Azuma, A.; Miura, Y.; Orimo, A. Activated Fibroblast Program Orchestrates Tumor Initiation and Progression; Molecular Mechanisms and the Associated Therapeutic Strategies. Int. J. Mol. Sci. 2019, 20, 2256. https://doi.org/10.3390/ijms20092256
Yoshida GJ, Azuma A, Miura Y, Orimo A. Activated Fibroblast Program Orchestrates Tumor Initiation and Progression; Molecular Mechanisms and the Associated Therapeutic Strategies. International Journal of Molecular Sciences. 2019; 20(9):2256. https://doi.org/10.3390/ijms20092256
Chicago/Turabian StyleYoshida, Go J., Arata Azuma, Yukiko Miura, and Akira Orimo. 2019. "Activated Fibroblast Program Orchestrates Tumor Initiation and Progression; Molecular Mechanisms and the Associated Therapeutic Strategies" International Journal of Molecular Sciences 20, no. 9: 2256. https://doi.org/10.3390/ijms20092256
APA StyleYoshida, G. J., Azuma, A., Miura, Y., & Orimo, A. (2019). Activated Fibroblast Program Orchestrates Tumor Initiation and Progression; Molecular Mechanisms and the Associated Therapeutic Strategies. International Journal of Molecular Sciences, 20(9), 2256. https://doi.org/10.3390/ijms20092256