Plasticity and Constraints of tRNA Aminoacylation Define Directed Evolution of Aminoacyl-tRNA Synthetases


Abstract
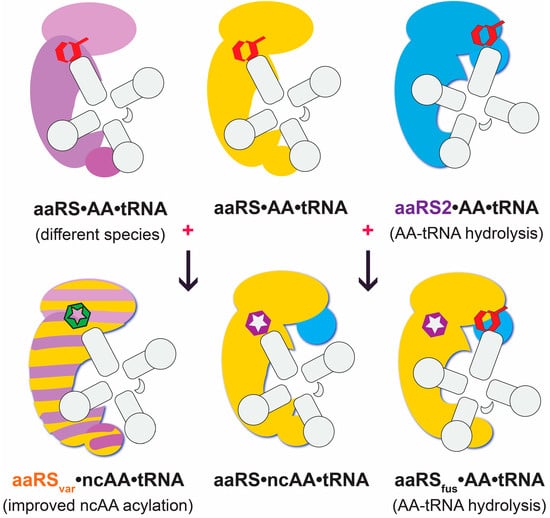
:
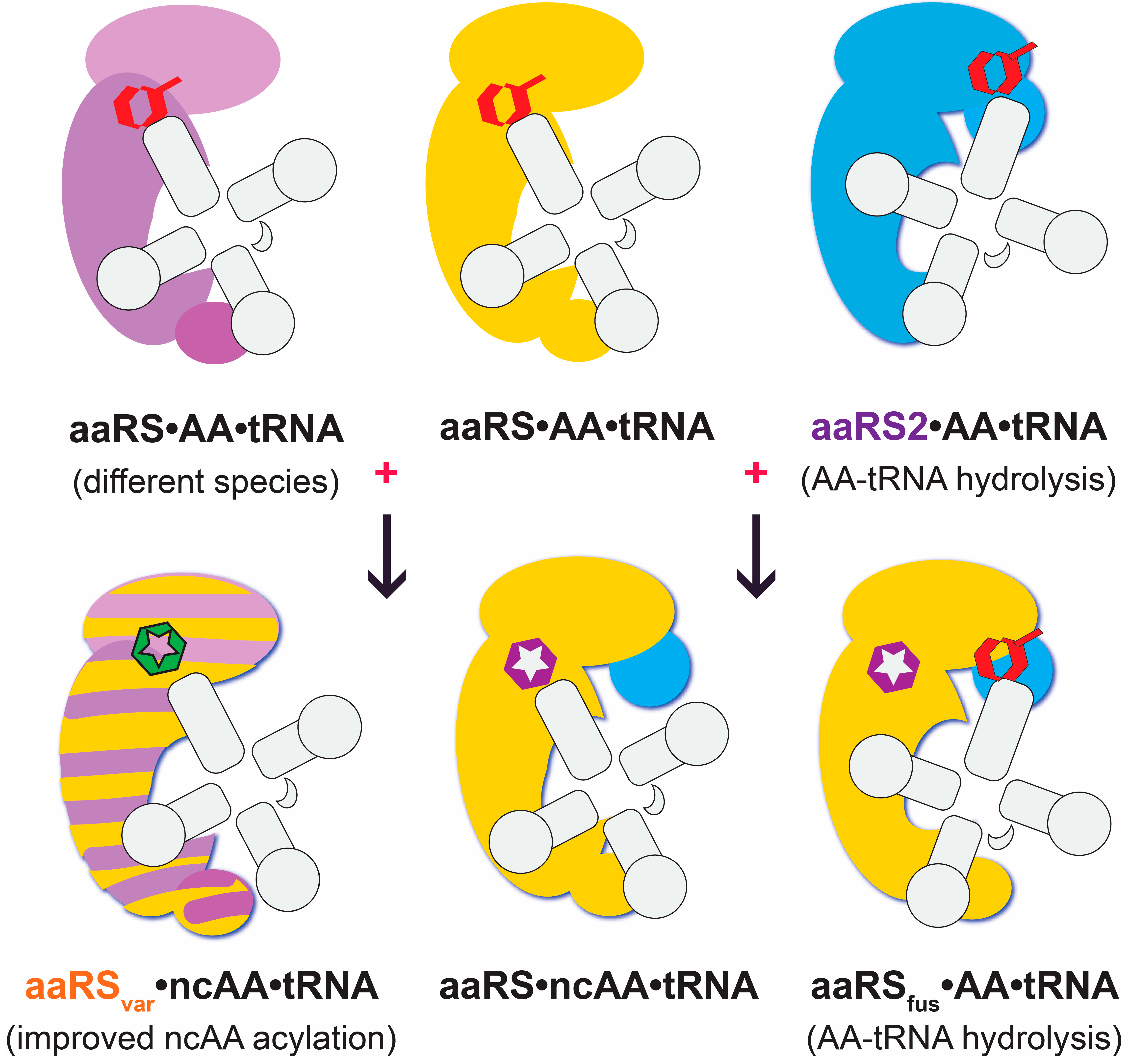{kind=link}
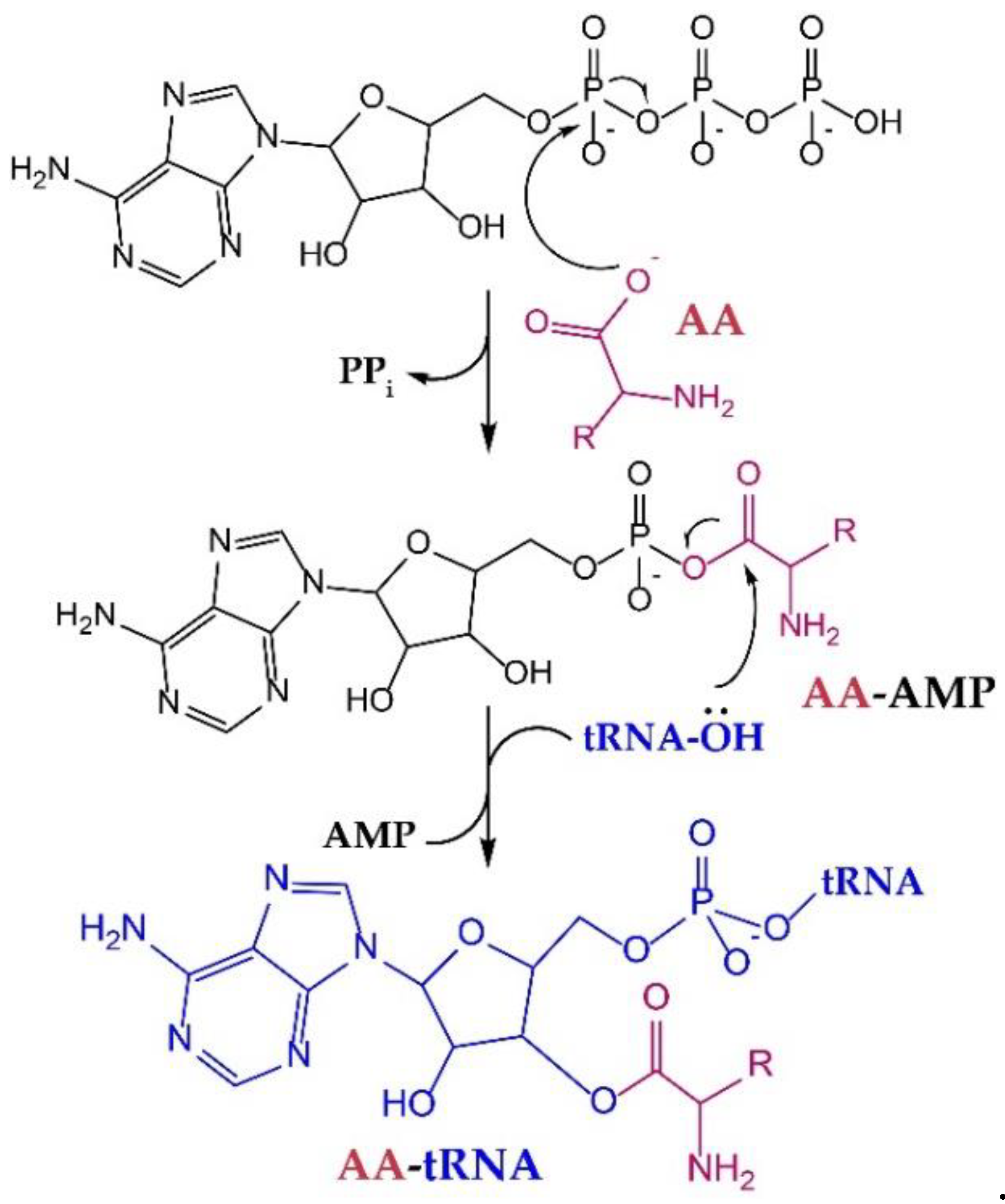{kind=link}
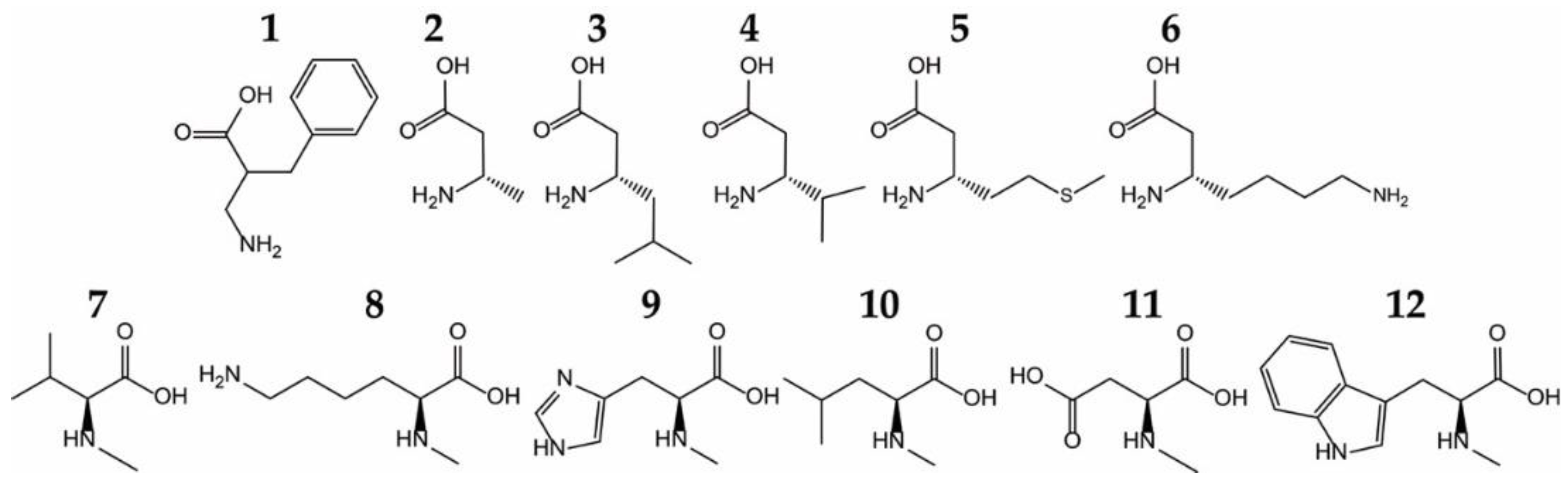{kind=link}
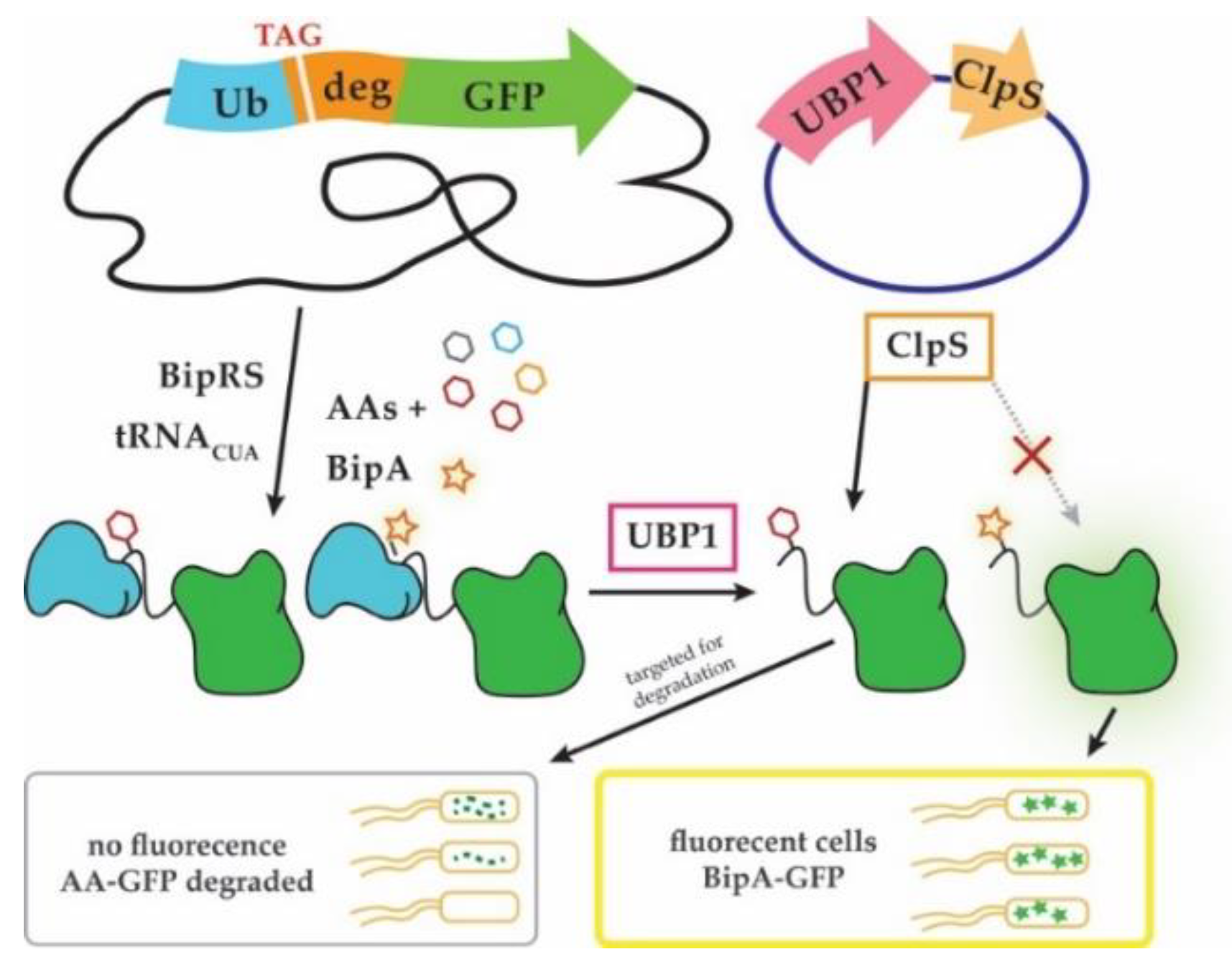{kind=link}
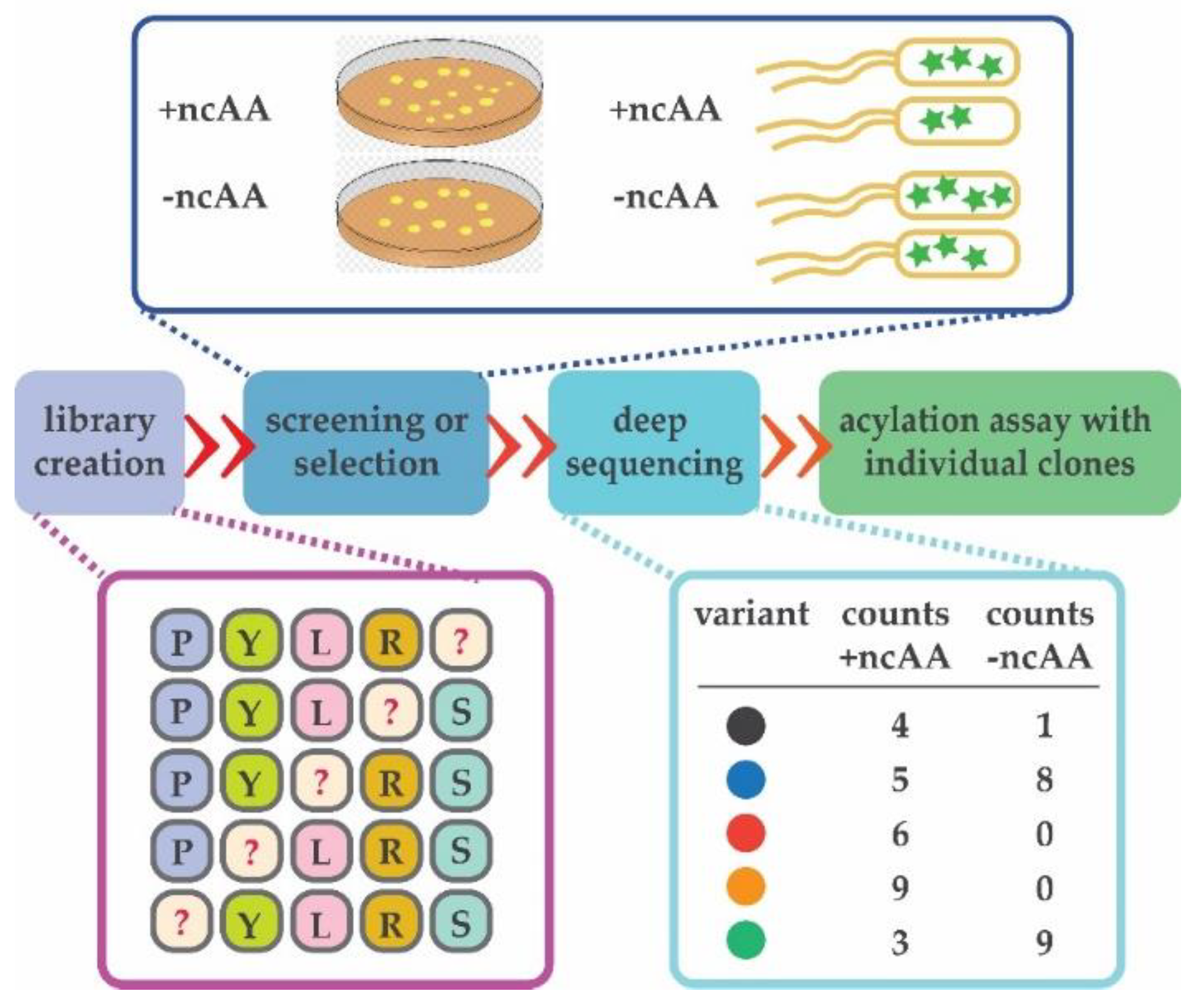
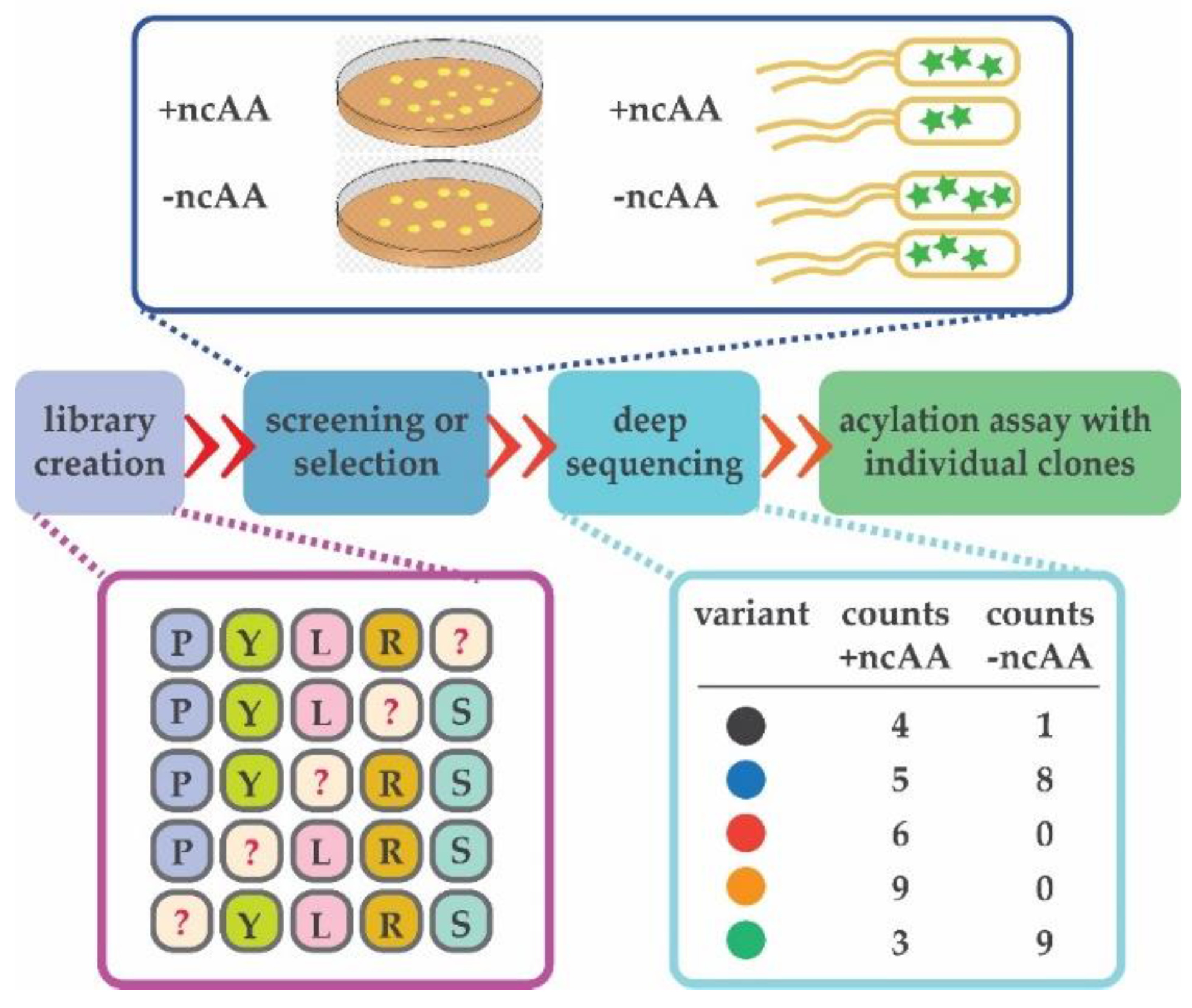{kind=link}
1. Introduction
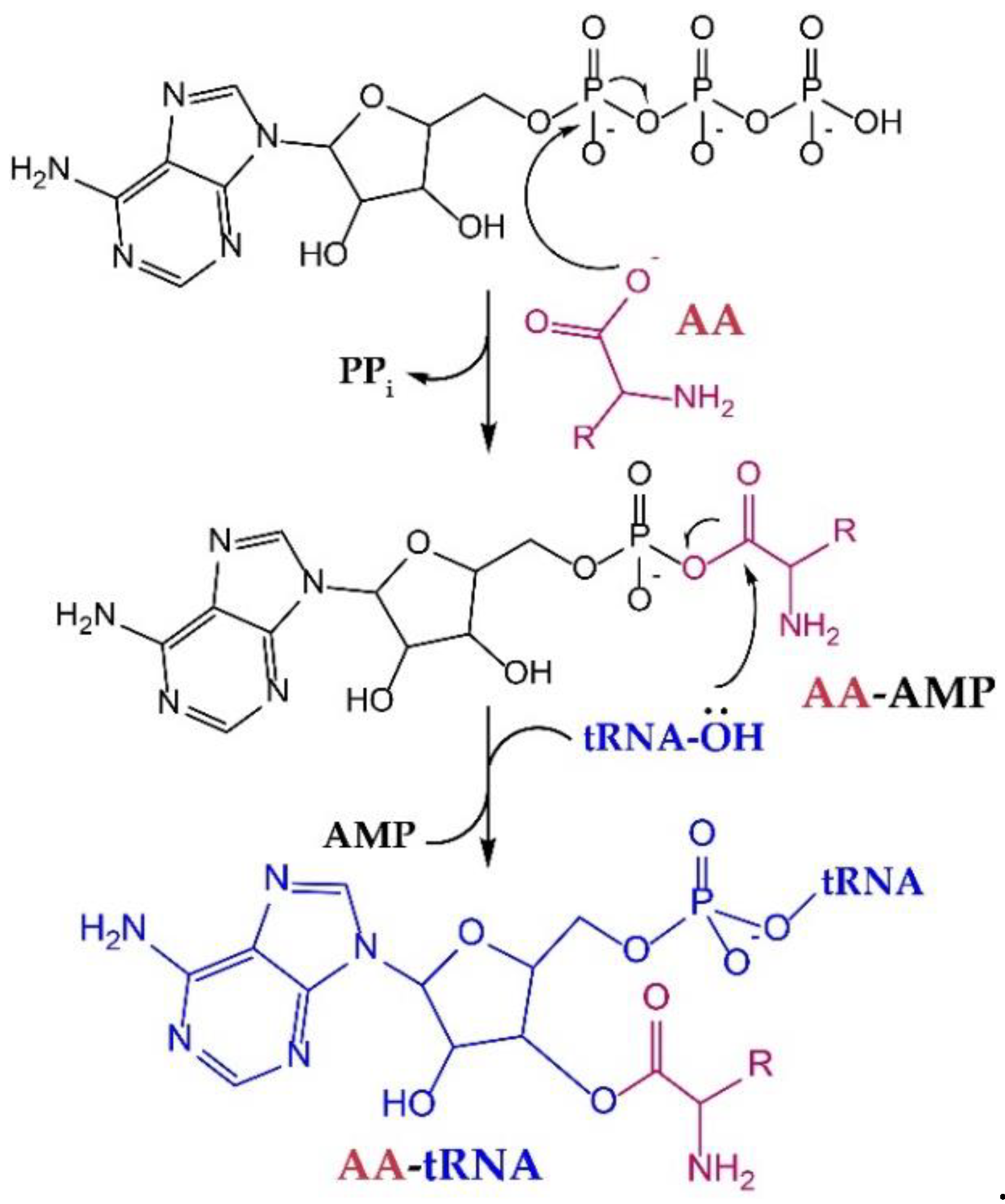
2. Flexibility of Wild-Type Synthetases
2.1. Implications of Substrate Recognition and Catalysis for Engineering of aaRSs
2.2. The Inherent AA Polyspecificity of aaRSs
2.3. Recognition of Fluorinated AA Analogs
2.4. Improving ncAA-tRNA Synthesis Using AA-tRNA Deacylases
2.5. Evolvability of 20 aaRSs
2.6. Availability of ncAA In Vivo May Dictate ncAA Incorporation
3. Directed Evolution of Orthogonal tRNA Synthetases
3.1. Sequence Randomization Methods
3.2. Selection Methods
3.3. Screening Methods
3.4. ncAA-Specific Reporters
3.5. Adaptation of Next-Generation Sequencing (NGS) to Sidestep a Negative Selection
4. Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| aaRS | Aminoacyl-tRNA synthetase |
| AA | Amino acid |
| ncAA | Noncanonical amino acid |
| PACE | Phage-assisted continuous evolution technique |
| PANCE | Phage-assisted noncontinuous evolution technique |
| MAGE | Multiplex automated genome engineering |
| NGS | Next-generation sequencing |
| GFP | Green fluorescent protein |
References
- Ramaswamy, K.; Saito, H.; Murakami, H.; Shiba, K.; Suga, H. Designer ribozymes: programming the tRNA specificity into flexizyme. J. Am. Chem. Soc. 2004, 126, 11454–11455. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Ji, X.; Gao, M.; Dedkova, L.M.; Hecht, S.M. In cellulo synthesis of proteins containing a fluorescent oxazole amino acid. J. Am. Chem. Soc. 2019. [Google Scholar] [CrossRef]
- Melo Czekster, C.; Robertson, W.E.; Walker, A.S.; Söll, D.; Schepartz, A. In vivo biosynthesis of a β-amino acid-containing protein. J. Am. Chem. Soc. 2016, 138, 5194–5197. [Google Scholar] [CrossRef]
- Katoh, T.; Iwane, Y.; Suga, H. Logical engineering of D-arm and T-stem of tRNA that enhances d-amino acid incorporation. Nucleic Acids Res. 2017, 45, 12601–12610. [Google Scholar] [CrossRef] [PubMed]
- Katoh, T.; Suga, H. Ribosomal incorporation of consecutive β-amino acids. J. Am. Chem. Soc. 2018, 140, 12159–12167. [Google Scholar] [CrossRef]
- Woese, C.R.; Olsen, G.J.; Ibba, M.; Söll, D. Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiol. Mol. Biol. Rev. 2000, 64, 202–236. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.C.; Josephson, K.; Szostak, J.W. Enzymatic aminoacylation of tRNA with unnatural amino acids. Proc. Natl. Acad. Sci. USA 2006, 103, 4356–4361. [Google Scholar] [CrossRef] [Green Version]
- Hoesl, M.G.; Budisa, N. Recent advances in genetic code engineering in Escherichia coli. Curr. Opin. Biotech. 2012, 23, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Senn, H.; Gsell, B.; Vetter, W.; Baron, C.; Böck, A. The formation of diselenide bridges in proteins by incorporation of selenocysteine residues: Biosynthesis and characterization of (Se)2-thioredoxin. Biochemistry 2002, 33, 3404–3412. [Google Scholar] [CrossRef]
- Campos-Olivas, R.; Aziz, R.; Helms, G.L.; Evans, J.N.; Gronenborn, A.M. Placement of 19F into the center of GB1: effects on structure and stability. FEBS Lett. 2002, 517, 55–60. [Google Scholar] [CrossRef]
- Reynolds, N.M.; Vargas-Rodriguez, O.; Söll, D.; Crnkovic, A. The central role of tRNA in genetic code expansion. BBA-Gen. Subjects 2017, 1861, 3001–3008. [Google Scholar] [CrossRef]
- Winter, G.; Fersht, A.R.; Wilkinson, A.J.; Zoller, M.; Smith, M. Redesigning enzyme structure by site-directed mutagenesis: Tyrosyl tRNA synthetase and ATP binding. Nature 1982, 299, 756–758. [Google Scholar] [CrossRef]
- Giegé, R.; Sissler, M.; Florentz, C. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 1998, 26, 5017–5035. [Google Scholar] [CrossRef] [Green Version]
- Rogerson, D.T.; Sachdeva, A.; Wang, K.; Haq, T.; Kazlauskaite, A.; Hancock, S.M.; Huguenin-Dezot, N.; Muqit, M.M.; Fry, A.M.; Bayliss, R.; et al. Efficient genetic encoding of phosphoserine and its nonhydrolyzable analog. Nat. Chem. Biol. 2015, 11, 496–503. [Google Scholar] [CrossRef]
- Rauch, B.J.; Porter, J.J.; Mehl, R.A.; Perona, J.J. Improved incorporation of noncanonical amino acids by an engineered tRNATyr suppressor. Biochemistry 2016, 55, 618–628. [Google Scholar] [CrossRef]
- Perona, J.J.; Gruic-Sovulj, I. Synthetic and editing mechanisms of aminoacyl-tRNA synthetases. Top. Curr. Chem. 2014, 344, 1–41. [Google Scholar] [CrossRef]
- Santoro, S.W.; Anderson, J.C.; Lakshman, V.; Schultz, P.G. An archaebacteria-derived glutamyl-tRNA synthetase and tRNA pair for unnatural amino acid mutagenesis of proteins in Escherichia coli. Nucleic Acids Res. 2003, 31, 6700–6709. [Google Scholar] [CrossRef]
- Liu, D.R.; Schultz, P.G. Progress toward the evolution of an organism with an expanded genetic code. Proc. Natl. Acad. Sci. USA 1999, 96, 4780–4785. [Google Scholar] [CrossRef] [Green Version]
- Avis, J.M.; Day, A.G.; Garcia, G.A.; Fersht, A.R. Reaction of modified and unmodified tRNATyr substrates with tyrosyl-tRNA synthetase (Bacillus stearothermophilus). Biochemistry 1993, 32, 5312–5320. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, L.; Schultz, P.G.; Wilson, I.A. Crystal structures of apo wild-type M. jannaschii tyrosyl-tRNA synthetase (TyrRS) and an engineered TyrRS specific for O-methyl-l-tyrosine. Protein Sci. 2005, 14, 1340–1349. [Google Scholar] [CrossRef] [PubMed]
- Hauf, M.; Richter, F.; Schneider, T.; Faidt, T.; Martins, B.M.; Baumann, T.; Durkin, P.; Dobbek, H.; Jacobs, K.; Möglich, A.; et al. Photoactivatable mussel-based underwater adhesive proteins by an expanded genetic code. ChemBioChem 2017, 18, 1819–1823. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Ho, J.M.L.; Chirathivat, N.; Söll, D.; Wang, Y.S. Exploring the substrate range of wild-type aminoacyl-tRNA synthetases. ChemBioChem 2014, 15, 1805–1809. [Google Scholar] [CrossRef]
- Dumas, A.; Lercher, L.; Spicer, C.D.; Davis, B.G. Designing logical codon reassignment—Expanding the chemistry in biology. Chem. Sci. 2015, 6, 50–69. [Google Scholar] [CrossRef]
- Wang, P.; Tang, Y.; Tirrell, D.A. Incorporation of trifluoroisoleucine into proteins in vivo. J. Am. Chem. Soc. 2003, 125, 6900–6906. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Tirrell, D.A. Biosynthesis of a highly stable coiled-coil protein containing hexafluoroleucine in an engineered bacterial host. J. Am. Chem. Soc. 2001, 123, 11089–11090. [Google Scholar] [CrossRef]
- Eichler, J.F.; Cramer, J.C.; Kirk, K.L.; Bann, J.G. Biosynthetic incorporation of fluorohistidine into proteins in E. coli: a new probe of macromolecular structure. ChemBioChem 2005, 6, 2170–2173. [Google Scholar] [CrossRef]
- Xu, Z.J.; Love, M.L.; Ma, L.Y.; Blum, M.; Bronskill, P.M.; Bernstein, J.; Grey, A.A.; Hofmann, T.; Camerman, N.; Wong, J.T. Tryptophanyl-tRNA synthetase from Bacillus subtilis. Characterization and role of hydrophobicity in substrate recognition. J. Biol. Chem. 1989, 264, 4304–4311. [Google Scholar] [PubMed]
- Zhang, Q.S.; Shen, L.; Wang, E.D.; Wang, Y.L. Biosynthesis and characterization of 4-fluorotryptophan-labeled Escherichia coli arginyl-tRNA synthetase. J. Protein Chem. 1999, 18, 187–192. [Google Scholar] [CrossRef]
- Gao, R.; Zhang, Y.; Dedkova, L.; Choudhury, A.K.; Rahier, N.J.; Hecht, S.M. Effects of modification of the active site tyrosine of human DNA topoisomerase I. Biochemistry 2006, 45, 8402–8410. [Google Scholar] [CrossRef]
- Hartman, M.C.; Josephson, K.; Lin, C.W.; Szostak, J.W. An expanded set of amino acid analogs for the ribosomal translation of unnatural peptides. PLoS ONE 2007, 2, e972. [Google Scholar] [CrossRef]
- Völler, J.S.; Dulic, M.; Gerling-Driessen, U.I.; Biava, H.; Baumann, T.; Budisa, N.; Gruic-Sovulj, I.; Koksch, B. Discovery and investigation of natural editing function against artificial amino acids in protein translation. ACS Cent. Sci. 2017, 3, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, E.S.; Dods, K.K.; Hartman, M.C.T. Ribosomal incorporation of backbone modified amino acids via an editing-deficient aminoacyl-tRNA synthetase. Org. Biomol. Chem. 2018, 16, 1073–1078. [Google Scholar] [CrossRef]
- Roy, H.; Ling, J.; Irnov, M.; Ibba, M. Post-transfer editing in vitro and in vivo by the β subunit of phenylalanyl-tRNA synthetase. EMBO J. 2004, 23, 4639–4648. [Google Scholar] [CrossRef] [Green Version]
- Oki, K.; Sakamoto, K.; Kobayashi, T.; Sasaki, H.M.; Yokoyama, S. Transplantation of a tyrosine editing domain into a tyrosyl-tRNA synthetase variant enhances its specificity for a tyrosine analog. Proc. Natl. Acad. Sci. USA 2008, 105, 13298–13303. [Google Scholar] [CrossRef] [Green Version]
- Richardson, C.J.; First, E.A. Altering the enantioselectivity of tyrosyl-tRNA synthetase by insertion of a stereospecific editing domain. Biochemistry 2016, 55, 1541–1553. [Google Scholar] [CrossRef]
- Kartvelishvili, E.; Peretz, M.; Tworowski, D.; Moor, N.; Safro, M. Chimeric human mitochondrial PheRS exhibits editing activity to discriminate nonprotein amino acids. Protein Sci. 2016, 25, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, K.; O’Donoghue, P.; Gundllapalli, S.; Araiso, Y.; Ishitani, R.; Umehara, T.; Söll, D.; Nureki, O. Pyrrolysyl-tRNA synthetase–tRNAPyl structure reveals the molecular basis of orthogonality. Nature 2008, 457, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Nureki, O.; Ishitani, R.; Yaremchuk, A.; Tukalo, M.; Cusack, S.; Sakamoto, K.; Yokoyama, S. Structural basis for orthogonal tRNA specificities of tyrosyl-tRNA synthetases for genetic code expansion. Nat. Struct. Mol. Biol. 2003, 10, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, T.; Umehara, T.; Sakamoto, K.; Yokoyama, S. Expanded genetic code technologies for incorporating modified lysine at multiple sites. ChemBioChem 2014, 15, 2181–2187. [Google Scholar] [CrossRef]
- Suzuki, T.; Miller, C.; Guo, L.T.; Ho, J.M.L.; Bryson, D.I.; Wang, Y.S.; Liu, D.R.; Söll, D. Crystal structures reveal an elusive functional domain of pyrrolysyl-tRNA synthetase. Nat. Chem. Biol. 2017, 13, 1261–1266. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Yanagisawa, T.; Sakamoto, K.; Yokoyama, S. Recognition of non-α-amino substrates by pyrrolysyl-tRNA synthetase. J. Mol. Biol. 2009, 385, 1352–1360. [Google Scholar] [CrossRef]
- Doublié, S.; Bricogne, G.; Gilmore, C.; Carter, C.W., Jr. Tryptophanyl-tRNA synthetase crystal structure reveals an unexpected homology to tyrosyl-tRNA synthetase. Structure 1995, 3, 17–31. [Google Scholar] [CrossRef] [Green Version]
- Romero, P.A.; Arnold, F.H. Exploring protein fitness landscapes by directed evolution. Nat. Rev. Mol. Cell Biol. 2009, 10, 866–876. [Google Scholar] [CrossRef] [Green Version]
- Bennett, B.D.; Kimball, E.H.; Gao, M.; Osterhout, R.; van Dien, S.J.; Rabinowitz, J.D. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 2009, 5, 593–599. [Google Scholar] [CrossRef]
- Venkat, S.; Gregory, C.; Gan, Q.; Fan, C. Biochemical characterization of the lysine acetylation of tyrosyl-tRNA synthetase in Escherichia coli. ChemBioChem 2017, 18, 1928–1934. [Google Scholar] [CrossRef]
- Hamano-Takaku, F.; Iwama, T.; Saito-Yano, S.; Takaku, K.; Monden, Y.; Kitabatake, M.; Söll, D.; Nishimura, S. A mutant Escherichia coli tyrosyl-tRNA synthetase utilizes the unnatural amino acid azatyrosine more efficiently than tyrosine. J. Biol. Chem. 2000, 275, 40324–40328. [Google Scholar] [CrossRef]
- Hauenstein, S.I.; Hou, Y.M.; Perona, J.J. The homotetrameric phosphoseryl-tRNA synthetase from Methanosarcina mazei exhibits half-of-the-sites activity. J. Biol. Chem. 2008, 283, 21997–22006. [Google Scholar] [CrossRef]
- Steinfeld, J.B.; Aerni, H.R.; Rogulina, S.; Liu, Y.; Rinehart, J. Expanded cellular amino acid pools containing phosphoserine, phosphothreonine, and phosphotyrosine. ACS Chem. Biol. 2014, 9, 1104–1112. [Google Scholar] [CrossRef]
- Burkovski, A.; Kramer, R. Bacterial amino acid transport proteins: occurrence, functions, and significance for biotechnological applications. Appl. Microbiol. Biot. 2002, 58, 265–274. [Google Scholar]
- Boehm, J.C.; Kingsbury, W.D.; Perry, D.; Gilvarg, C. The use of cysteinyl peptides to effect portage transport of sulfhydryl-containing compounds in Escherichia coli. J. Biol. Chem. 1983, 258, 14850–14855. [Google Scholar]
- Smith, M.W.; Tyreman, D.R.; Payne, G.M.; Marshall, N.J.; Payne, J.W. Substrate specificity of the periplasmic dipeptide-binding protein from Escherichia coli: Experimental basis for the design of peptide prodrugs. Microbiology 1999, 145, 2891–2901. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Fu, G.; Wang, R.E.; Zhu, X.; Zambaldo, C.; Liu, R.; Liu, T.; Lyu, X.; Du, J.; Xuan, W.; et al. Genetically encoding phosphotyrosine and its nonhydrolyzable analog in bacteria. Nat. Chem. Biol. 2017, 13, 845–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tame, J.R.; Dodson, E.J.; Murshudov, G.; Higgins, C.F.; Wilkinson, A.J. The crystal structures of the oligopeptide-binding protein OppA complexed with tripeptide and tetrapeptide ligands. Structure 1995, 3, 1395–1406. [Google Scholar] [CrossRef] [Green Version]
- Kuenzl, T.; Sroka, M.; Srivastava, P.; Herdewijn, P.; Marliere, P.; Panke, S. Overcoming the membrane barrier: Recruitment of γ-glutamyl transferase for intracellular release of metabolic cargo from peptide vectors. Metab. Eng. 2017, 39, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Hohn, M.J.; Umehara, T.; Guo, L.T.; Osborne, E.M.; Benner, J.; Noren, C.J.; Rinehart, J.; Söll, D. Expanding the genetic code of Escherichia coli with phosphoserine. Science 2011, 333, 1151–1154. [Google Scholar] [CrossRef] [PubMed]
- Beránek, V.; Reinkemeier, C.D.; Zhang, M.S.; Liang, A.D.; Kym, G.; Chin, J.W. Genetically encoded protein phosphorylation in mammalian cells. Cell Chem. Biol. 2018, 25, 1067–1074. [Google Scholar] [CrossRef]
- Longstaff, D.G.; Larue, R.C.; Faust, J.E.; Mahapatra, A.; Zhang, L.; Green-Church, K.B.; Krzycki, J.A. A natural genetic code expansion cassette enables transmissible biosynthesis and genetic encoding of pyrrolysine. Proc. Natl. Acad. Sci. USA 2007, 104, 1021–1026. [Google Scholar] [CrossRef] [Green Version]
- Fan, C.; Fromm, H.J.; Bobik, T.A. Kinetic and functional analysis of l-threonine kinase, the PduX enzyme of Salmonella enterica. J. Biol. Chem. 2009, 284, 20240–20248. [Google Scholar] [CrossRef]
- Zhang, M.S.; Brunner, S.F.; Huguenin-Dezot, N.; Liang, A.D.; Schmied, W.H.; Rogerson, D.T.; Chin, J.W. Biosynthesis and genetic encoding of phosphothreonine through parallel selection and deep sequencing. Nat. Methods 2017, 14, 729–736. [Google Scholar] [CrossRef] [Green Version]
- Niemann, C.; Rapport, M.M. The toxicity of 3-fluoro-d(+)- and l(-)-tyrosine. J. Am. Chem. Soc. 1946, 68, 1671. [Google Scholar] [CrossRef]
- Reichau, S.; Blackmore, N.J.; Jiao, W.; Parker, E.J. Probing the sophisticated synergistic allosteric regulation of aromatic amino acid biosynthesis in Mycobacterium tuberculosis using d-amino acids. PLoS ONE 2016, 11, e0152723. [Google Scholar] [CrossRef]
- Wanner, B.L.; Metcalf, W.W. Molecular genetic studies of a 10.9-kb operon in Escherichia coli for phosphonate uptake and biodegradation. FEMS Microbiol. Lett. 1992, 100, 133–139. [Google Scholar] [CrossRef]
- Ko, J.H.; Wang, Y.S.; Nakamura, A.; Guo, L.T.; Söll, D.; Umehara, T. Pyrrolysyl-tRNA synthetase variants reveal ancestral aminoacylation function. FEBS Lett. 2013, 587, 3243–3248. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.R.; Magliery, T.J.; Pasternak, M.; Schultz, P.G. Engineering a tRNA and aminoacyl-tRNA synthetase for the site-specific incorporation of unnatural amino acids into proteins in vivo. Proc. Natl. Acad. Sci. USA 1997, 94, 10092–10097. [Google Scholar] [CrossRef] [Green Version]
- Santoro, S.W.; Schultz, P.G. Directed evolution of the substrate specificities of a site-specific recombinase and an aminoacyl-tRNA synthetase using fluorescence-activated cell sorting (FACS). Methods Mol. Biol. 2003, 230, 291–312. [Google Scholar]
- Acevedo-Rocha, C.G.; Reetz, M.T.; Nov, Y. Economical analysis of saturation mutagenesis experiments. Sci. Rep. 2015, 5, 10654. [Google Scholar] [CrossRef] [Green Version]
- Richter, F.; Leaver-Fay, A.; Khare, S.D.; Bjelic, S.; Baker, D. De novo enzyme design using Rosetta3. PLoS ONE 2011, 6, e19230. [Google Scholar] [CrossRef]
- Amiram, M.; Haimovich, A.D.; Fan, C.; Wang, Y.S.; Aerni, H.R.; Ntai, I.; Moonan, D.W.; Ma, N.J.; Rovner, A.J.; Hong, S.H.; et al. Evolution of translation machinery in recoded bacteria enables multi-site incorporation of nonstandard amino acids. Nat. Biotechnol. 2015, 33, 1272–1279. [Google Scholar] [CrossRef] [Green Version]
- Bryson, D.I.; Fan, C.; Guo, L.T.; Miller, C.; Söll, D.; Liu, D.R. Continuous directed evolution of aminoacyl-tRNA synthetases. Nat. Chem. Biol. 2017, 13, 1253–1260. [Google Scholar] [CrossRef] [Green Version]
- Hoesl, M.G.; Oehm, S.; Durkin, P.; Darmon, E.; Peil, L.; Aerni, H.R.; Rappsilber, J.; Rinehart, J.; Leach, D.; Söll, D.; et al. Chemical evolution of a bacterial proteome. Angew. Chem. Int. Edit. 2015, 54, 10030–10034. [Google Scholar] [CrossRef]
- Wang, H.H.; Isaacs, F.J.; Carr, P.A.; Sun, Z.Z.; Xu, G.; Forest, C.R.; Church, G.M. Programming cells by multiplex genome engineering and accelerated evolution. Nature 2009, 460, 894–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, H.M.; Yu, D.; DiTizio, T.; Court, D.L. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc. Natl. Acad. Sci. USA 2001, 98, 6742–6746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, S.M.; Rubini, M.; Fuhrmann, M.; Theobald, I.; Skerra, A. Engineering of an orthogonal aminoacyl-tRNA synthetase for efficient incorporation of the non-natural amino acid O-methyl-l-tyrosine using fluorescence-based bacterial cell sorting. J. Mol. Biol. 2010, 404, 70–87. [Google Scholar] [CrossRef] [PubMed]
- Kwok, H.S.; Vargas-Rodriguez, O.; Melnikov, S.V.; Söll, D. Engineered aminoacyl-tRNA synthetases with improved selectivity towards non-canonical amino acids. ACS Chem. Biol. 2019, 14, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Badran, A.H.; Liu, D.R. Development of potent in vivo mutagenesis plasmids with broad mutational spectra. Nat. Commun. 2015, 6, 8425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esvelt, K.M.; Carlson, J.C.; Liu, D.R. A system for the continuous directed evolution of biomolecules. Nature 2011, 472, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Schultz, P.G. A general approach for the generation of orthogonal tRNAs. Chem. Biol. 2001, 8, 883–890. [Google Scholar] [CrossRef] [Green Version]
- Umehara, T.; Kim, J.; Lee, S.; Guo, L.T.; Söll, D.; Park, H.S. N-acetyl lysyl-tRNA synthetases evolved by a CcdB-based selection possess N-acetyl lysine specificity in vitro and in vivo. FEBS Lett. 2012, 586, 729–733. [Google Scholar] [CrossRef]
- Maranhao, A.C.; Ellington, A.D. Evolving orthogonal suppressor tRNAs to incorporate modified amino acids. ACS Synth. Biol. 2017, 6, 108–119. [Google Scholar] [CrossRef]
- Owens, A.E.; Grasso, K.T.; Ziegler, C.A.; Fasan, R. Two-tier screening platform for directed evolution of aminoacyl-tRNA synthetases with enhanced stop codon suppression efficiency. ChemBioChem 2017, 18, 1109–1116. [Google Scholar] [CrossRef]
- Thyer, R.; Shroff, R.; Klein, D.R.; d’Oelsnitz, S.; Cotham, V.C.; Byrom, M.; Brodbelt, J.S.; Ellington, A.D. Custom selenoprotein production enabled by laboratory evolution of recoded bacterial strains. Nat. Biotechnol. 2018, 36, 624–631. [Google Scholar] [CrossRef]
- Volkwein, W.; Maier, C.; Krafczyk, R.; Jung, K.; Lassak, J. A versatile toolbox for the control of protein levels using Ne-acetyl-L-lysine dependent amber suppression. ACS Synth. Biol. 2017, 6, 1892–1902. [Google Scholar] [CrossRef]
- McKinnon, K.M. Flow cytometry: An overview. Curr. Protoc. Immunol. 2018, 120, 5 1 1–5 1 11. [Google Scholar]
- Young, T.S.; Ahmad, I.; Yin, J.A.; Schultz, P.G. An enhanced system for unnatural amino acid mutagenesis in E. coli. J. Mol. Biol. 2010, 395, 361–374. [Google Scholar] [CrossRef]
- Guo, L.T.; Wang, Y.S.; Nakamura, A.; Eiler, D.; Kavran, J.M.; Wong, M.; Kiessling, L.L.; Steitz, T.A.; O’Donoghue, P.; Söll, D. Polyspecific pyrrolysyl-tRNA synthetases from directed evolution. Proc. Natl. Acad. Sci. USA 2014, 111, 16724–16729. [Google Scholar] [CrossRef]
- Young, D.D.; Jockush, S.; Turro, N.J.; Schultz, P.G. Synthetase polyspecificity as a tool to modulate protein function. Bioorg. Med. Chem. Lett. 2011, 21, 7502–7504. [Google Scholar] [CrossRef] [Green Version]
- Stokes, A.L.; Miyake-Stoner, S.J.; Peeler, J.C.; Nguyen, D.P.; Hammer, R.P.; Mehl, R.A. Enhancing the utility of unnatural amino acid synthetases by manipulating broad substrate specificity. Mol. Biosyst. 2009, 5, 1032–1038. [Google Scholar] [CrossRef]
- Aharoni, A.; Gaidukov, L.; Khersonsky, O.; Gould, S.M.; Roodveldt, C.; Tawfik, D.S. The “evolvability” of promiscuous protein functions. Nat. Genet. 2005, 37, 73–76. [Google Scholar] [CrossRef]
- Kunjapur, A.M.; Stork, D.A.; Kuru, E.; Vargas-Rodriguez, O.; Landon, M.; Söll, D.; Church, G.M. Engineering posttranslational proofreading to discriminate nonstandard amino acids. Proc. Natl. Acad. Sci. USA 2018, 115, 619–624. [Google Scholar] [CrossRef]
- Bajaj, K.; Chakrabarti, P.; Varadarajan, R. Mutagenesis-based definitions and probes of residue burial in proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 16221–16226. [Google Scholar] [CrossRef] [Green Version]
- Aldag, C.; Bröcker, M.J.; Hohn, M.J.; Prat, L.; Hammond, G.; Plummer, A.; Söll, D. Rewiring translation for elongation factor Tu-dependent selenocysteine incorporation. Angew. Chem. Int. Edit. 2013, 52, 1441–1445. [Google Scholar] [CrossRef]
- Yoshizawa, S.; Böck, A. The many levels of control on bacterial selenoprotein synthesis. Biochim. Biophys. Acta 2009, 1790, 1404–1414. [Google Scholar] [CrossRef]
- Haruna, K.; Alkazemi, M.H.; Liu, Y.; Söll, D.; Englert, M. Engineering the elongation factor Tu for efficient selenoprotein synthesis. Nucleic Acids Res. 2014, 42, 9976–9983. [Google Scholar] [CrossRef] [Green Version]
- Thyer, R.; Robotham, S.A.; Brodbelt, J.S.; Ellington, A.D. Evolving tRNASec for efficient canonical incorporation of selenocysteine. J. Am. Chem. Soc. 2015, 137, 46–49. [Google Scholar] [CrossRef]
- Mukai, T.; Sevostyanova, A.; Suzuki, T.; Fu, X.; Söll, D. A facile method for producing selenocysteine-containing proteins. Angew. Chem. Int. Edit. 2018, 57, 7215–7219. [Google Scholar] [CrossRef]
- Fu, X.; Crnkovic, A.; Sevostyanova, A.; Söll, D. Designing seryl-tRNA synthetase for improved serylation of selenocysteine tRNAs. FEBS Lett. 2018, 592, 3759–3768. [Google Scholar] [CrossRef]
- Humbard, M.A.; Surkov, S.; de Donatis, G.M.; Jenkins, L.M.; Maurizi, M.R. The N-degradome of Escherichia coli: Limited proteolysis in vivo generates a large pool of proteins bearing N-degrons. J. Biol. Chem. 2013, 288, 28913–28924. [Google Scholar] [CrossRef]
- Fowler, D.M.; Araya, C.L.; Fleishman, S.J.; Kellogg, E.H.; Stephany, J.J.; Baker, D.; Fields, S. High-resolution mapping of protein sequence-function relationships. Nat. Methods 2010, 7, 741–746. [Google Scholar] [CrossRef] [Green Version]
- Wrenbeck, E.E.; Faber, M.S.; Whitehead, T.A. Deep sequencing methods for protein engineering and design. Curr. Opin. Struct. Biol. 2017, 45, 36–44. [Google Scholar] [CrossRef]
- Uyeda, A.; Watanabe, T.; Kato, Y.; Watanabe, H.; Yomo, T.; Hohsaka, T.; Matsuura, T. Liposome-based in vitro evolution of aminoacyl-tRNA synthetase for enhanced pyrrolysine derivative incorporation. ChemBioChem 2015, 16, 1797–1802. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crnković, A.; Vargas-Rodriguez, O.; Söll, D. Plasticity and Constraints of tRNA Aminoacylation Define Directed Evolution of Aminoacyl-tRNA Synthetases. Int. J. Mol. Sci. 2019, 20, 2294. https://doi.org/10.3390/ijms20092294
Crnković A, Vargas-Rodriguez O, Söll D. Plasticity and Constraints of tRNA Aminoacylation Define Directed Evolution of Aminoacyl-tRNA Synthetases. International Journal of Molecular Sciences. 2019; 20(9):2294. https://doi.org/10.3390/ijms20092294
Chicago/Turabian StyleCrnković, Ana, Oscar Vargas-Rodriguez, and Dieter Söll. 2019. "Plasticity and Constraints of tRNA Aminoacylation Define Directed Evolution of Aminoacyl-tRNA Synthetases" International Journal of Molecular Sciences 20, no. 9: 2294. https://doi.org/10.3390/ijms20092294
APA StyleCrnković, A., Vargas-Rodriguez, O., & Söll, D. (2019). Plasticity and Constraints of tRNA Aminoacylation Define Directed Evolution of Aminoacyl-tRNA Synthetases. International Journal of Molecular Sciences, 20(9), 2294. https://doi.org/10.3390/ijms20092294