Endoplasmic Reticulum Stress Pathway, the Unfolded Protein Response, Modulates Immune Function in the Tumor Microenvironment to Impact Tumor Progression and Therapeutic Response

Abstract
:1. Introduction
2. Body
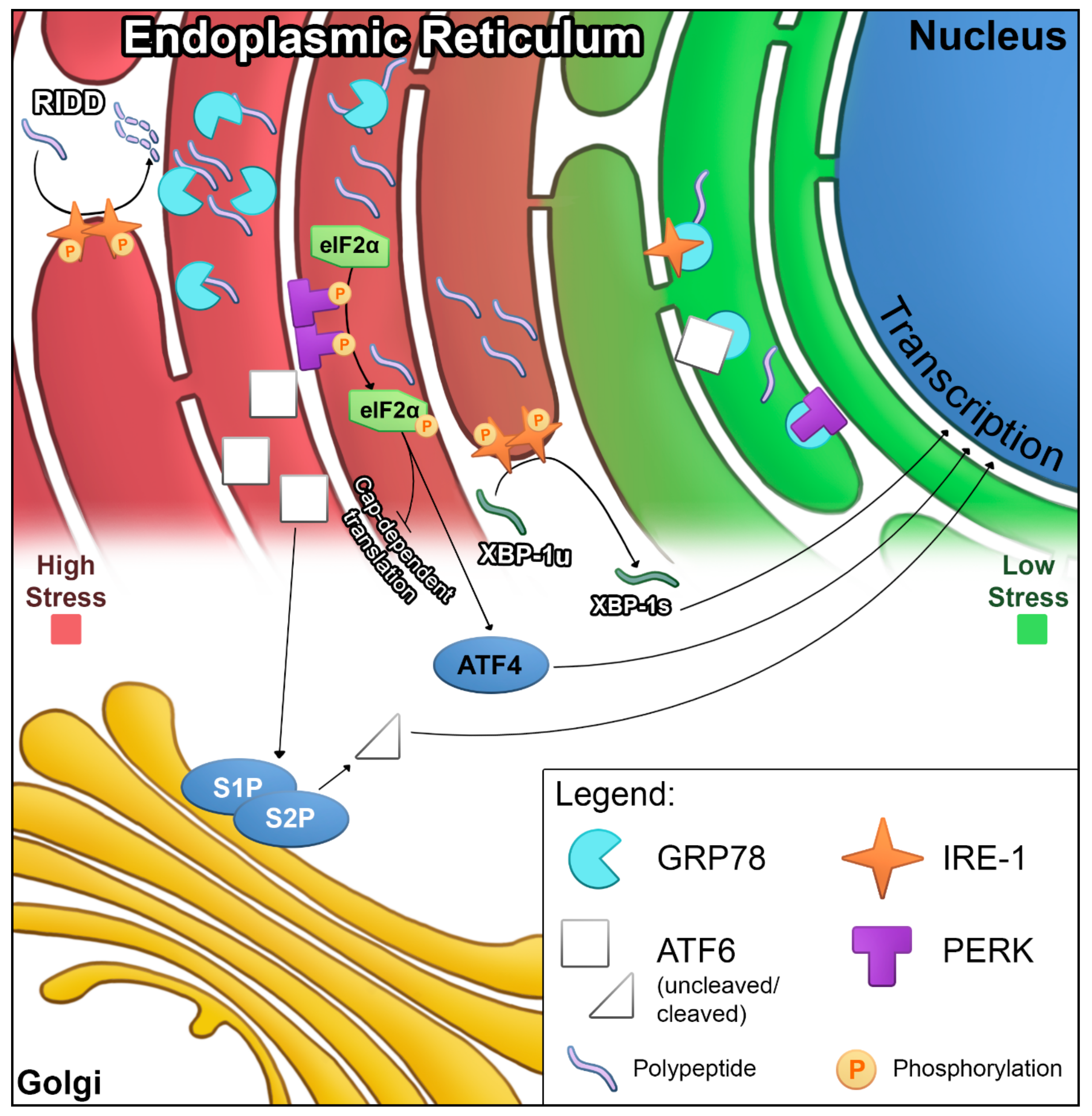
2.1. ER Stress and UPR Signaling
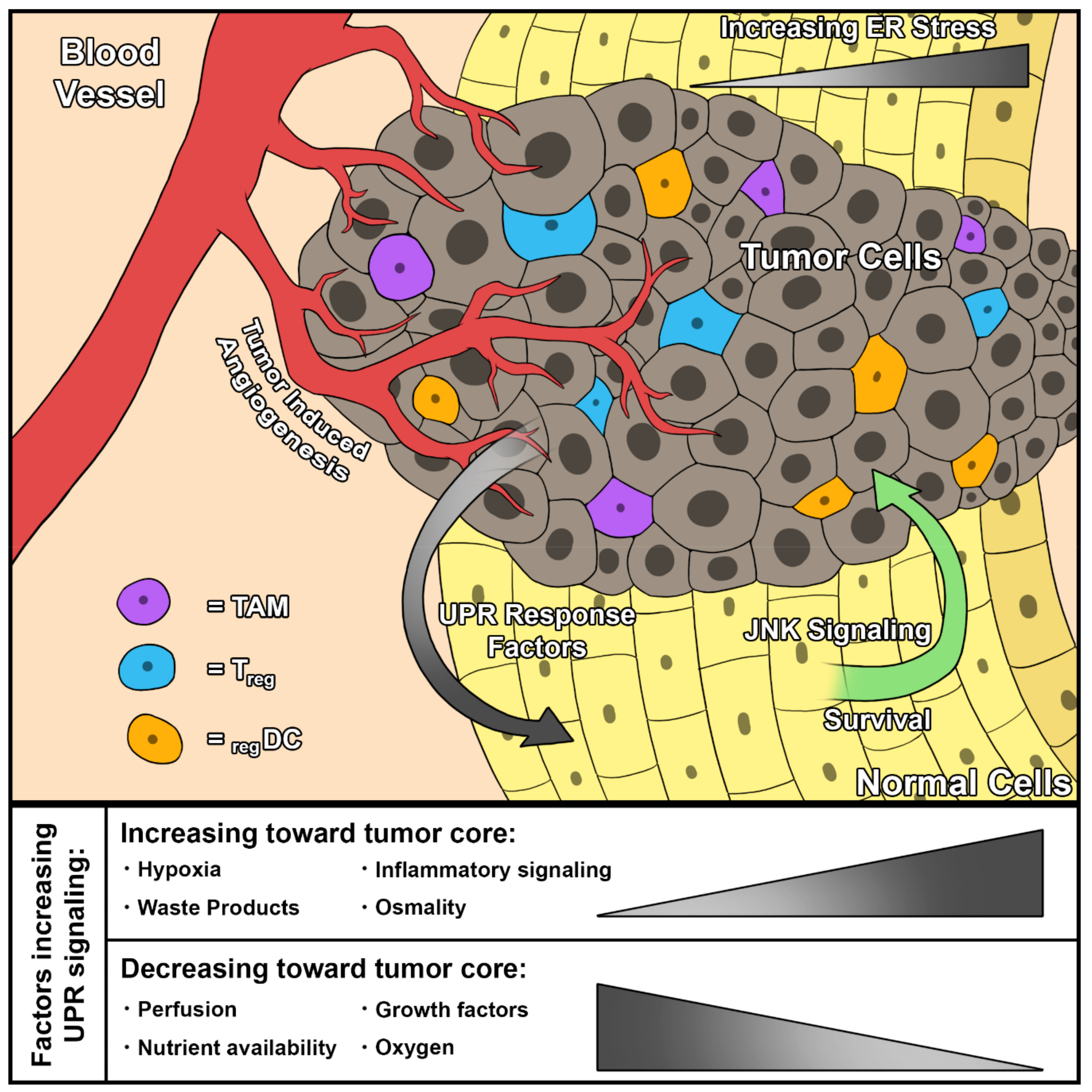
2.2. ER Stress and the UPR in the Tumor Microenvironment
2.3. Tumor Immunology
2.4. UPR Signaling in Dendritic (Myeloid-Derived Suppressor) Cells
2.5. UPR Signaling in Macrophages
2.6. UPR Signaling in T Cells
2.7. UPR Signaling and Cancer-Associated Fibroblasts
2.8. Implications for UPR-Targeting Drugs in Cancer Therapy
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feitelson, M.A.; Arzumanyan, A.; Kulathinal, R.J.; Blain, S.W.; Holcombe, R.F.; Mahajna, J.; Marino, M.; Martinez-Chantar, M.L.; Nawroth, R.; Sanchez-Garcia, I.; et al. Sustained proliferation in cancer: Mechanisms and novel therapeutic targets. Semin. Cancer Biol. 2015, 35, S25–S54. [Google Scholar] [CrossRef] [PubMed]
- Messerschmidt, J.L.; Prendergast, G.C.; Messerschmidt, G.L. How Cancers Escape Immune Destruction and Mechanisms of Action for the New Significantly Active Immune Therapies: Helping Nonimmunologists Decipher Recent Advances. Oncologist 2016, 21, 233–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- McGee, H.M.; Jiang, D.; Soto-Pantoja, D.R.; Nevler, A.; Giaccia, A.J.; Woodward, W.A. Targeting the Tumor Microenvironment in Radiation Oncology: Proceedings from the 2018 ASTRO-AACR Research Workshop. Clin. Cancer Res. 2019, 25, 2969–2974. [Google Scholar] [CrossRef] [Green Version]
- Dvorak, H.F. Tumors: Wounds that do not heal-redux. Cancer Immunol. Res. 2015, 3, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckl) 2015, 3, 83–92. [Google Scholar] [CrossRef] [Green Version]
- McCann, J.V.; Xiao, L.; Kim, D.J.; Khan, O.F.; Kowalski, P.S.; Anderson, D.G.; Pecot, C.V.; Azam, S.H.; Parker, J.S.; Tsai, Y.S.; et al. Endothelial miR-30c suppresses tumor growth via inhibition of TGF-beta-induced Serpine1. J. Clin. Investig. 2019, 130, 1654–1670. [Google Scholar] [CrossRef]
- Fulbright, L.E.; Ellermann, M.; Arthur, J.C. The microbiome and the hallmarks of cancer. PLoS Pathog. 2017, 13, e1006480. [Google Scholar] [CrossRef]
- Ma, Y.; Hendershot, L.M. The unfolding tale of the unfolded protein response. Cell 2001, 107, 827–830. [Google Scholar] [CrossRef] [Green Version]
- Haas, I.G.; Wabl, M. Immunoglobulin heavy chain binding protein. Nature 1983, 306, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. Coordinated regulation of a set of genes by glucose and calcium ionophores in mammalian cells. Trends Biochem. Sci. 1987, 12, 20–23. [Google Scholar] [CrossRef]
- Clarke, R.; Cook, K.L. Unfolding the Role of Stress Response Signaling in Endocrine Resistant Breast Cancers. Front. Oncol. 2015, 5, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, R.; Cook, K.L.; Hu, R.; Facey, C.O.; Tavassoly, I.; Schwartz, J.L.; Baumann, W.T.; Tyson, J.J.; Xuan, J.; Wang, Y.; et al. Endoplasmic reticulum stress, the unfolded protein response, autophagy, and the integrated regulation of breast cancer cell fate. Cancer Res. 2012, 72, 1321–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Tirasophon, W.; Shen, X.; Michalak, M.; Prywes, R.; Okada, T.; Yoshida, H.; Mori, K.; Kaufman, R.J. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002, 16, 452–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, B.M.; Walter, P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 2011, 333, 1891–1894. [Google Scholar] [CrossRef] [Green Version]
- Cullinan, S.B.; Diehl, J.A. Coordination of ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2006, 38, 317–332. [Google Scholar] [CrossRef]
- Rodvold, J.J.; Chiu, K.T.; Hiramatsu, N.; Nussbacher, J.K.; Galimberti, V.; Mahadevan, N.R.; Willert, K.; Lin, J.H.; Zanetti, M. Intercellular transmission of the unfolded protein response promotes survival and drug resistance in cancer cells. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Mahadevan, N.R.; Rodvold, J.; Sepulveda, H.; Rossi, S.; Drew, A.F.; Zanetti, M. Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells. Proc. Natl. Acad. Sci. USA 2011, 108, 6561–6566. [Google Scholar] [CrossRef] [Green Version]
- Jain, B.P. An Overview of Unfolded Protein Response Signaling and Its Role in Cancer. Cancer Biother. Radiopharm. 2017, 32, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Rufo, N.; Garg, A.D.; Agostinis, P. The Unfolded Protein Response in Immunogenic Cell Death and Cancer Immunotherapy. Trends Cancer 2017, 3, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Wouters, B.G.; Koritzinsky, M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat. Rev. Cancer 2008, 8, 851–864. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.C.; Weissman, A.M. The Unfolded Protein Response, Degradation from Endoplasmic Reticulum and Cancer. Genes Cancer 2010, 1, 764–778. [Google Scholar] [CrossRef] [PubMed]
- Tsachaki, M.; Mladenovic, N.; Stambergova, H.; Birk, J.; Odermatt, A. Hexose-6-phosphate dehydrogenase controls cancer cell proliferation and migration through pleiotropic effects on the unfolded-protein response, calcium homeostasis, and redox balance. FASEB J. 2018, 32, 2690–2705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shajahan-Haq, A.N.; Cook, K.L.; Schwartz-Roberts, J.L.; Eltayeb, A.E.; Demas, D.M.; Warri, A.M.; Facey, C.O.; Hilakivi-Clarke, L.A.; Clarke, R. MYC regulates the unfolded protein response and glucose and glutamine uptake in endocrine resistant breast cancer. Mol. Cancer 2014, 13, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, S.; Furuno, A.; Sakurai, J.; Sakamoto, A.; Park, H.R.; Shin-Ya, K.; Tsuruo, T.; Tomida, A. Chemical genomics identifies the unfolded protein response as a target for selective cancer cell killing during glucose deprivation. Cancer Res. 2009, 69, 4225–4234. [Google Scholar] [CrossRef] [Green Version]
- Nagelkerke, A.; Bussink, J.; Mujcic, H.; Wouters, B.G.; Lehmann, S.; Sweep, F.C.; Span, P.N. Hypoxia stimulates migration of breast cancer cells via the PERK/ATF4/LAMP3-arm of the unfolded protein response. Breast Cancer Res. 2013, 15. [Google Scholar] [CrossRef] [Green Version]
- Bartkowiak, K.; Effenberger, K.E.; Harder, S.; Andreas, A.; Buck, F.; Peter-Katalinic, J.; Pantel, K.; Brandt, B.H. Discovery of a novel unfolded protein response phenotype of cancer stem/progenitor cells from the bone marrow of breast cancer patients. J. Proteome Res. 2010, 9, 3158–3168. [Google Scholar] [CrossRef]
- Bartkowiak, K.; Kwiatkowski, M.; Buck, F.; Gorges, T.M.; Nilse, L.; Assmann, V.; Andreas, A.; Muller, V.; Wikman, H.; Riethdorf, S.; et al. Disseminated Tumor Cells Persist in the Bone Marrow of Breast Cancer Patients through Sustained Activation of the Unfolded Protein Response. Cancer Res. 2015, 75, 5367–5377. [Google Scholar] [CrossRef] [Green Version]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic Reticulum Stress, Unfolded Protein Response, and Cancer Cell Fate. Front. Oncol. 2017, 7, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, X.; Xue, Y.; Si, Y.; Wang, Q.; Wang, Z.; Yuan, J.; Zhang, X. The unfolded protein response potentiates epithelial-to-mesenchymal transition (EMT) of gastric cancer cells under severe hypoxic conditions. Med. Oncol. 2015, 32, 447. [Google Scholar] [CrossRef] [PubMed]
- Mujumdar, N.; Banerjee, S.; Chen, Z.; Sangwan, V.; Chugh, R.; Dudeja, V.; Yamamoto, M.; Vickers, S.M.; Saluja, A.K. Triptolide activates unfolded protein response leading to chronic ER stress in pancreatic cancer cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G1011–G1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, K.L.; Soto-Pantoja, D.R. “UPRegulation” of CD47 by the endoplasmic reticulum stress pathway controls anti-tumor immune responses. Biomark. Res. 2017, 5, 26. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.L.; Soto-Pantoja, D.R.; Clarke, P.A.; Cruz, M.I.; Zwart, A.; Warri, A.; Hilakivi-Clarke, L.; Roberts, D.D.; Clarke, R. Endoplasmic Reticulum Stress Protein GRP78 Modulates Lipid Metabolism to Control Drug Sensitivity and Antitumor Immunity in Breast Cancer. Cancer Res. 2016, 76, 5657–5670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto-Pantoja, D.R.; Wilson, A.S.; Clear, K.Y.; Westwood, B.; Triozzi, P.L.; Cook, K.L. Unfolded protein response signaling impacts macrophage polarity to modulate breast cancer cell clearance and melanoma immune checkpoint therapy responsiveness. Oncotarget 2017, 8, 80545–80559. [Google Scholar] [CrossRef]
- Misra, U.K.; Pizzo, S.V. Modulation of the unfolded protein response in prostate cancer cells by antibody-directed against the carboxyl-terminal domain of GRP78. Apoptosis 2010, 15, 173–182. [Google Scholar] [CrossRef]
- Thornton, M.; Aslam, M.A.; Tweedle, E.M.; Ang, C.; Campbell, F.; Jackson, R.; Costello, E.; Rooney, P.S.; Vlatkovic, N.; Boyd, M.T. The unfolded protein response regulator GRP78 is a novel predictive biomarker in colorectal cancer. Int. J. Cancer 2013, 133, 1408–1418. [Google Scholar] [CrossRef] [Green Version]
- So, A.Y.; de la Fuente, E.; Walter, P.; Shuman, M.; Bernales, S. The unfolded protein response during prostate cancer development. Cancer Metastasis Rev. 2009, 28, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Sheng, X.; Arnoldussen, Y.J.; Storm, M.; Tesikova, M.; Nenseth, H.Z.; Zhao, S.; Fazli, L.; Rennie, P.; Risberg, B.; Waehre, H.; et al. Divergent androgen regulation of unfolded protein response pathways drives prostate cancer. EMBO Mol. Med. 2015, 7, 788–801. [Google Scholar] [CrossRef]
- Storm, M.; Sheng, X.; Arnoldussen, Y.J.; Saatcioglu, F. Prostate cancer and the unfolded protein response. Oncotarget 2016, 7, 54051–54066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scriven, P.; Coulson, S.; Haines, R.; Balasubramanian, S.; Cross, S.; Wyld, L. Activation and clinical significance of the unfolded protein response in breast cancer. Br. J. Cancer 2009, 101, 1692–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Yin, Y.; Hua, H.; Li, M.; Luo, T.; Xu, L.; Wang, R.; Liu, D.; Zhang, Y.; Jiang, Y. Blockade of GRP78 sensitizes breast cancer cells to microtubules-interfering agents that induce the unfolded protein response. J. Cell Mol. Med. 2009, 13, 3888–3897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Notte, A.; Rebucci, M.; Fransolet, M.; Roegiers, E.; Genin, M.; Tellier, C.; Watillon, K.; Fattaccioli, A.; Arnould, T.; Michiels, C. Taxol-induced unfolded protein response activation in breast cancer cells exposed to hypoxia: ATF4 activation regulates autophagy and inhibits apoptosis. Int. J. Biochem. Cell Biol. 2015, 62, 1–14. [Google Scholar] [CrossRef]
- Cook, K.L.; Clarke, P.A.; Clarke, R. Targeting GRP78 and antiestrogen resistance in breast cancer. Future Med. Chem. 2013, 5, 1047–1057. [Google Scholar] [CrossRef]
- Rajapaksa, G.; Thomas, C.; Gustafsson, J.A. Estrogen signaling and unfolded protein response in breast cancer. J. Steroid Biochem. Mol. Biol. 2016, 163, 45–50. [Google Scholar] [CrossRef]
- Andruska, N.; Zheng, X.; Yang, X.; Helferich, W.G.; Shapiro, D.J. Anticipatory estrogen activation of the unfolded protein response is linked to cell proliferation and poor survival in estrogen receptor alpha-positive breast cancer. Oncogene 2015, 34, 3760–3769. [Google Scholar] [CrossRef] [Green Version]
- Cook, K.L.; Clarke, P.A.; Parmar, J.; Hu, R.; Schwartz-Roberts, J.L.; Abu-Asab, M.; Warri, A.; Baumann, W.T.; Clarke, R. Knockdown of estrogen receptor-alpha induces autophagy and inhibits antiestrogen-mediated unfolded protein response activation, promoting ROS-induced breast cancer cell death. FASEB J. 2014, 28, 3891–3905. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, Y.; Horiuchi, Y.; Kano, F.; Murata, M. Novel prosurvival function of Yip1A in human cervical cancer cells: Constitutive activation of the IRE1 and PERK pathways of the unfolded protein response. Cell Death Dis. 2017, 8, e2718. [Google Scholar] [CrossRef] [Green Version]
- Teng, M.W.; Swann, J.B.; Koebel, C.M.; Schreiber, R.D.; Smyth, M.J. Immune-mediated dormancy: An equilibrium with cancer. J. Leukoc. Biol. 2008, 84, 988–993. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Lee, E.; Friedline, R.H.; Suk, S.; Jung, D.Y.; Dagdeviren, S.; Hu, X.; Inashima, K.; Noh, H.L.; Kwon, J.Y.; et al. Endoplasmic reticulum chaperone GRP78 regulates macrophage function and insulin resistance in diet-induced obesity. FASEB J. 2018, 32, 2292–2304. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.J.; Kusnadi, A.; Lee, E.J.; Kim, W.W.; Cho, B.C.; Lee, I.J.; Seong, J.; Ha, S.J. Tumor-infiltrating regulatory T cells delineated by upregulation of PD-1 and inhibitory receptors. Cell Immunol. 2012, 278, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Yu, X.; Wang, H.; Zuo, D.; Guo, C.; Yi, H.; Tirosh, B.; Subjeck, J.R.; Qiu, X.; Wang, X.Y. ER stress and its regulator X-box-binding protein-1 enhance polyIC-induced innate immune response in dendritic cells. Eur. J. Immunol. 2011, 41, 1086–1097. [Google Scholar] [CrossRef] [PubMed]
- Shields, J.D.; Kourtis, I.C.; Tomei, A.A.; Roberts, J.M.; Swartz, M.A. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science 2010, 328, 749–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, I.; Garrido, F.; Garcia-Lora, A.M. Metastases in immune-mediated dormancy: A new opportunity for targeting cancer. Cancer Res. 2014, 74, 6750–6757. [Google Scholar] [CrossRef] [Green Version]
- Senovilla, L.; Vacchelli, E.; Galon, J.; Adjemian, S.; Eggermont, A.; Fridman, W.H.; Sautes-Fridman, C.; Ma, Y.; Tartour, E.; Zitvogel, L.; et al. Trial watch: Prognostic and predictive value of the immune infiltrate in cancer. Oncoimmunology 2012, 1, 1323–1343. [Google Scholar] [CrossRef] [Green Version]
- Koelzer, V.H.; Canonica, K.; Dawson, H.; Sokol, L.; Karamitopoulou-Diamantis, E.; Lugli, A.; Zlobec, I. Phenotyping of tumor-associated macrophages in colorectal cancer: Impact on single cell invasion (tumor budding) and clinicopathological outcome. Oncoimmunology 2016, 5, e1106677. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; He, Y.; Sun, X.; Li, Q.; Wang, W.; Zhao, A.; Di, W. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J. Ovarian Res. 2014, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.W.; Liu, L.; Gong, C.Y.; Shi, H.S.; Zeng, Y.H.; Wang, X.Z.; Zhao, Y.W.; Wei, Y.Q. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A. Molecular pathways linking inflammation and cancer. Curr. Mol. Med. 2010, 10, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [PubMed]
- Schafer, M.; Werner, S. Cancer as an overhealing wound: An old hypothesis revisited. Nat. Rev. Mol. Cell Biol 2008, 9, 628–638. [Google Scholar] [CrossRef]
- Porta, C.; Larghi, P.; Rimoldi, M.; Totaro, M.G.; Allavena, P.; Mantovani, A.; Sica, A. Cellular and molecular pathways linking inflammation and cancer. Immunobiology 2009, 214, 761–777. [Google Scholar] [CrossRef]
- Yu, H.; Huang, X.; Liu, X.; Jin, H.; Zhang, G.; Zhang, Q.; Yu, J. Regulatory T cells and plasmacytoid dendritic cells contribute to the immune escape of papillary thyroid cancer coexisting with multinodular non-toxic goiter. Endocrine 2013, 44, 172–181. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, N.; Li, Q.; Zhang, W.; Ke, F.; Leng, Q.; Wang, H.; Chen, J.; Wang, H. Tumor-associated macrophages recruit CCR6+ regulatory T cells and promote the development of colorectal cancer via enhancing CCL20 production in mice. PLoS ONE 2011, 6, e19495. [Google Scholar] [CrossRef]
- Edin, S.; Wikberg, M.L.; Oldenborg, P.A.; Palmqvist, R. Macrophages: Good guys in colorectal cancer. Oncoimmunology 2013, 2, e23038. [Google Scholar] [CrossRef] [Green Version]
- Bogolyubova, A.V.; Belousov, P.V. Inflammatory Immune Infiltration in Human Tumors: Role in Pathogenesis and Prognostic and Diagnostic Value. Biochemistry 2016, 81, 1261–1273. [Google Scholar] [CrossRef]
- Pages, F.; Galon, J.; Dieu-Nosjean, M.C.; Tartour, E.; Sautes-Fridman, C.; Fridman, W.H. Immune infiltration in human tumors: A prognostic factor that should not be ignored. Oncogene 2010, 29, 1093–1102. [Google Scholar] [CrossRef] [Green Version]
- Ferrone, C.; Dranoff, G. Dual roles for immunity in gastrointestinal cancers. J. Clin. Oncol. 2010, 28, 4045–4051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, M.; Denardo, D.G.; Coussens, L.M. Polarized immune responses differentially regulate cancer development. Immunol. Rev. 2008, 222, 145–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiavoni, G.; Gabriele, L.; Mattei, F. The tumor microenvironment: A pitch for multiple players. Front. Oncol. 2013, 3, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marvel, D.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the tumor microenvironment: Expect the unexpected. J. Clin. Investig. 2015, 125, 3356–3364. [Google Scholar] [CrossRef]
- Preynat-Seauve, O.; Contassot, E.; Schuler, P.; French, L.E.; Huard, B. Melanoma-infiltrating dendritic cells induce protective antitumor responses mediated by T cells. Melanoma Res. 2007, 17, 169–176. [Google Scholar] [CrossRef]
- Eruslanov, E.B.; Bhojnagarwala, P.S.; Quatromoni, J.G.; Stephen, T.L.; Ranganathan, A.; Deshpande, C.; Akimova, T.; Vachani, A.; Litzky, L.; Hancock, W.W.; et al. Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. J. Clin. Investig. 2014, 124, 5466–5480. [Google Scholar] [CrossRef] [Green Version]
- De Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Mougiakakos, D.; Johansson, C.C.; Trocme, E.; All-Ericsson, C.; Economou, M.A.; Larsson, O.; Seregard, S.; Kiessling, R. Intratumoral forkhead box P3-positive regulatory T cells predict poor survival in cyclooxygenase-2-positive uveal melanoma. Cancer 2010, 116, 2224–2233. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef] [PubMed]
- Coffelt, S.B.; Lewis, C.E.; Naldini, L.; Brown, J.M.; Ferrara, N.; De Palma, M. Elusive identities and overlapping phenotypes of proangiogenic myeloid cells in tumors. Am. J. Pathol. 2010, 176, 1564–1576. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Andreu, P.; Coussens, L.M. Interactions between lymphocytes and myeloid cells regulate pro- versus anti-tumor immunity. Cancer Metastasis Rev. 2010, 29, 309–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egeblad, M.; Nakasone, E.S.; Werb, Z. Tumors as organs: Complex tissues that interface with the entire organism. Dev. Cell 2010, 18, 884–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef] [PubMed]
- De Palma, M.; Murdoch, C.; Venneri, M.A.; Naldini, L.; Lewis, C.E. Tie2-expressing monocytes: Regulation of tumor angiogenesis and therapeutic implications. Trends Immunol. 2007, 28, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Daurkin, I.; Eruslanov, E.; Stoffs, T.; Perrin, G.Q.; Algood, C.; Gilbert, S.M.; Rosser, C.J.; Su, L.M.; Vieweg, J.; Kusmartsev, S. Tumor-associated macrophages mediate immunosuppression in the renal cancer microenvironment by activating the 15-lipoxygenase-2 pathway. Cancer Res. 2011, 71, 6400–6409. [Google Scholar] [CrossRef] [Green Version]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Li, Y.; Rogoff, H.A.; Keates, S.; Gao, Y.; Murikipudi, S.; Mikule, K.; Leggett, D.; Li, W.; Pardee, A.B.; Li, C.J. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc. Natl. Acad. Sci. USA 2015, 112, 1839–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segura, E. Review of Mouse and Human Dendritic Cell Subsets. Methods Mol. Biol. 2016, 1423, 3–15. [Google Scholar] [PubMed]
- Worbs, T.; Hammerschmidt, S.I.; Forster, R. Dendritic cell migration in health and disease. Nat. Rev. Immunol. 2017, 17, 30–48. [Google Scholar] [CrossRef] [PubMed]
- Iwakoshi, N.N.; Pypaert, M.; Glimcher, L.H. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J. Exp. Med. 2007, 204, 2267–2275. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.M.; Yao, F.H.; Yao, Y.M.; Dong, N.; Yu, Y.; Sheng, Z.Y. Endoplasmic reticulum stress and its regulator XBP-1 contributes to dendritic cell maturation and activation induced by high mobility group box-1 protein. Int J. Biochem Cell Biol. 2012, 44, 1097–1105. [Google Scholar] [CrossRef]
- Goodall, J.C.; Wu, C.; Zhang, Y.; McNeill, L.; Ellis, L.; Saudek, V.; Gaston, J.S. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc. Natl. Acad. Sci. USA 2010, 107, 17698–17703. [Google Scholar] [CrossRef] [Green Version]
- Osorio, F.; Tavernier, S.J.; Hoffmann, E.; Saeys, Y.; Martens, L.; Vetters, J.; Delrue, I.; De Rycke, R.; Parthoens, E.; Pouliot, P.; et al. The unfolded-protein-response sensor IRE-1alpha regulates the function of CD8alpha+ dendritic cells. Nat. Immunol. 2014, 15, 248–257. [Google Scholar] [CrossRef]
- Shurin, G.V.; Ma, Y.; Shurin, M.R. Immunosuppressive mechanisms of regulatory dendritic cells in cancer. Cancer Microenviron. 2013, 6, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Shurin, G.V.; Gutkin, D.W.; Shurin, M.R. Tumor associated regulatory dendritic cells. Semin. Cancer Biol. 2012, 22, 298–306. [Google Scholar] [CrossRef] [Green Version]
- Shurin, M.R.; Naiditch, H.; Zhong, H.; Shurin, G.V. Regulatory dendritic cells: New targets for cancer immunotherapy. Cancer Biol. Ther. 2011, 11, 988–992. [Google Scholar] [CrossRef] [Green Version]
- Enk, A.H.; Jonuleit, H.; Saloga, J.; Knop, J. Dendritic cells as mediators of tumor-induced tolerance in metastatic melanoma. Int. J. Cancer 1997, 73, 309–316. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, C.; Sun, A.; Zheng, Y.; Wang, L.; Cao, X. Tumor-educated CD11bhighIalow regulatory dendritic cells suppress T cell response through arginase I. J. Immunol. 2009, 182, 6207–6216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Dickhout, J.G.; Lhotak, S.; Hilditch, B.A.; Basseri, S.; Colgan, S.M.; Lynn, E.G.; Carlisle, R.E.; Zhou, J.; Sood, S.K.; Ingram, A.J.; et al. Induction of the unfolded protein response after monocyte to macrophage differentiation augments cell survival in early atherosclerotic lesions. FASEB J. 2011, 25, 576–589. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.; Jung, D.Y.; Jun, J.Y.; Li, J.; Luo, S.; Ko, H.J.; Kim, J.K.; Lee, A.S. Grp78 heterozygosity promotes adaptive unfolded protein response and attenuates diet-induced obesity and insulin resistance. Diabetes 2010, 59, 6–16. [Google Scholar] [CrossRef] [Green Version]
- Tian, P.G.; Jiang, Z.X.; Li, J.H.; Zhou, Z.; Zhang, Q.H. Spliced XBP1 promotes macrophage survival and autophagy by interacting with Beclin-1. Biochem. Biophys. Res. Commun. 2015, 463, 518–523. [Google Scholar] [CrossRef]
- Qiu, Q.; Zheng, Z.; Chang, L.; Zhao, Y.S.; Tan, C.; Dandekar, A.; Zhang, Z.; Lin, Z.; Gui, M.; Li, X.; et al. Toll-like receptor-mediated IRE1alpha activation as a therapeutic target for inflammatory arthritis. EMBO J. 2013, 32, 2477–2490. [Google Scholar] [CrossRef] [Green Version]
- Guthrie, L.N.; Abiraman, K.; Plyler, E.S.; Sprenkle, N.T.; Gibson, S.A.; McFarland, B.C.; Rajbhandari, R.; Rowse, A.L.; Benveniste, E.N.; Meares, G.P. Attenuation of PKR-like ER Kinase (PERK) Signaling Selectively Controls Endoplasmic Reticulum Stress-induced Inflammation Without Compromising Immunological Responses. J. Biol. Chem. 2016, 291, 15830–15840. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, R.K.; Li, C.; Chaudhary, S.C.; Ballestas, M.E.; Elmets, C.A.; Robbins, D.J.; Matalon, S.; Deshane, J.S.; Afaq, F.; Bickers, D.R.; et al. Unfolded protein response (UPR) signaling regulates arsenic trioxide-mediated macrophage innate immune function disruption. Toxicol. Appl. Pharmacol. 2013, 272, 879–887. [Google Scholar] [CrossRef]
- Ayaub, E.A.; Kolb, P.S.; Mohammed-Ali, Z.; Tat, V.; Murphy, J.; Bellaye, P.S.; Shimbori, C.; Boivin, F.J.; Lai, R.; Lynn, E.G.; et al. GRP78 and CHOP modulate macrophage apoptosis and the development of bleomycin-induced pulmonary fibrosis. J. Pathol. 2016, 239, 411–425. [Google Scholar] [CrossRef]
- Isa, S.A.; Ruffino, J.S.; Ahluwalia, M.; Thomas, A.W.; Morris, K.; Webb, R. M2 macrophages exhibit higher sensitivity to oxLDL-induced lipotoxicity than other monocyte/macrophage subtypes. Lipids Health Dis. 2011, 10, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, B.; Wang, X.; Wu, Y.; Xu, C.; Xia, Z.; Dai, J.; Shao, M.; Zhao, F.; He, S.; Yang, L.; et al. The metabolic ER stress sensor IRE1alpha suppresses alternative activation of macrophages and impairs energy expenditure in obesity. Nat. Immunol. 2017, 18, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Seimon, T.; Timmins, J.; Li, G.; Lim, W. Macrophage apoptosis in advanced atherosclerosis. Ann. NY Acad. Sci. 2009, 1173, E40–E45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhi, H.; Kropp, E.M.; Clavo, V.F.; Kobrossi, C.R.; Han, J.; Mauer, A.S.; Yong, J.; Kaufman, R.J. C/EBP homologous protein-induced macrophage apoptosis protects mice from steatohepatitis. J. Biol. Chem. 2013, 288, 18624–18642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, A.X.; Tabas, I. The UPR in atherosclerosis. Semin. Immunopathol. 2013, 35, 321–332. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Lhotak, S.; Hilditch, B.A.; Austin, R.C. Activation of the unfolded protein response occurs at all stages of atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation 2005, 111, 1814–1821. [Google Scholar] [CrossRef] [Green Version]
- Myoishi, M.; Hao, H.; Minamino, T.; Watanabe, K.; Nishihira, K.; Hatakeyama, K.; Asada, Y.; Okada, K.; Ishibashi-Ueda, H.; Gabbiani, G.; et al. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation 2007, 116, 1226–1233. [Google Scholar] [CrossRef] [Green Version]
- Yao, S.; Yang, N.; Song, G.; Sang, H.; Tian, H.; Miao, C.; Zhang, Y.; Qin, S. Minimally modified low-density lipoprotein induces macrophage endoplasmic reticulum stress via toll-like receptor 4. Biochim. Biophys. Acta 2012, 1821, 954–963. [Google Scholar] [CrossRef]
- Yao, S.; Miao, C.; Tian, H.; Sang, H.; Yang, N.; Jiao, P.; Han, J.; Zong, C.; Qin, S. Endoplasmic reticulum stress promotes macrophage-derived foam cell formation by up-regulating cluster of differentiation 36 (CD36) expression. J. Biol. Chem. 2014, 289, 4032–4042. [Google Scholar] [CrossRef] [Green Version]
- McAlpine, C.S.; Werstuck, G.H. Protein kinase R-like endoplasmic reticulum kinase and glycogen synthase kinase-3alpha/beta regulate foam cell formation. J. Lipid Res. 2014, 55, 2320–2333. [Google Scholar] [CrossRef] [Green Version]
- Gass, J.N.; Gifford, N.M.; Brewer, J.W. Activation of an unfolded protein response during differentiation of antibody-secreting B cells. J. Biol. Chem. 2002, 277, 49047–49054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunsing, R.; Omori, S.A.; Weber, F.; Bicknell, A.; Friend, L.; Rickert, R.; Niwa, M. B- and T-cell development both involve activity of the unfolded protein response pathway. J. Biol. Chem. 2008, 283, 17954–17961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamimura, D.; Bevan, M.J. Endoplasmic Reticulum Stress Regulator XBP-1 Contributes to Effector CD8+ T Cell Differentiation during Acute Infection. J. Immunol. 2008, 181, 5433–5441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwakoshi, N.N.; Lee, A.H.; Glimcher, L.H. The X-box binding protein-1 transcription factor is required for plasma cell differentiation and the unfolded protein response. Immunol. Rev. 2003, 194, 29–38. [Google Scholar] [CrossRef]
- Reimold, A.M.; Iwakoshi, N.N.; Manis, J.; Vallabhajosyula, P.; Szomolanyi-Tsuda, E.; Gravallese, E.M.; Friend, D.; Grusby, M.J.; Alt, F.; Glimcher, L.H. Plasma cell differentiation requires the transcription factor XBP-1. Nature 2001, 412, 300–307. [Google Scholar] [CrossRef]
- Li, S.; Zhu, G.; Yang, Y.; Jian, Z.; Guo, S.; Dai, W.; Shi, Q.; Ge, R.; Ma, J.; Liu, L.; et al. Oxidative stress drives CD8(+) T-cell skin trafficking in patients with vitiligo through CXCL16 upregulation by activating the unfolded protein response in keratinocytes. J. Allergy Clin. Immunol. 2017, 140, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Catakovic, K.; Klieser, E.; Neureiter, D.; Geisberger, R. T cell exhaustion: From pathophysiological basics to tumor immunotherapy. Cell Commun. Signal. 2017, 15, 1. [Google Scholar] [CrossRef] [Green Version]
- Durward-Diioia, M.; Harms, J.; Khan, M.; Hall, C.; Smith, J.A.; Splitter, G.A. CD8+ T cell exhaustion, suppressed gamma interferon production, and delayed memory response induced by chronic Brucella melitensis infection. Infect. Immun. 2015, 83, 4759–4771. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [Green Version]
- Pauken, K.E.; Wherry, E.J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015, 36, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.C.; Xu, Y.; Huang, Z.M.; Lu, X.J. T cell exhaustion in cancer: Mechanisms and clinical implications. J. Cell Biochem. 2018, 119, 4279–4286. [Google Scholar] [CrossRef] [PubMed]
- Zarour, H.M. Reversing T-cell Dysfunction and Exhaustion in Cancer. Clin. Cancer Res. 2016, 22, 1856–1864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.H.; Walter, P.; Yen, T.S. Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol. 2008, 3, 399–425. [Google Scholar] [CrossRef] [PubMed]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef]
- Peng, Y.; Li, Z.; Li, Z. GRP78 secreted by tumor cells stimulates differentiation of bone marrow mesenchymal stem cells to cancer-associated fibroblasts. Biochem. Biophys. Res. Commun. 2013, 440, 558–563. [Google Scholar] [CrossRef]
- Yu, T.; Guo, Z.; Fan, H.; Song, J.; Liu, Y.; Gao, Z.; Wang, Q. Cancer-associated fibroblasts promote non-small cell lung cancer cell invasion by upregulation of glucose-regulated protein 78 (GRP78) expression in an integrated bionic microfluidic device. Oncotarget 2016, 7, 25593–25603. [Google Scholar] [CrossRef] [Green Version]
- Holtrup, F.; Bauer, A.; Fellenberg, K.; Hilger, R.A.; Wink, M.; Hoheisel, J.D. Microarray analysis of nemorosone-induced cytotoxic effects on pancreatic cancer cells reveals activation of the unfolded protein response (UPR). Br. J. Pharmacol. 2011, 162, 1045–1059. [Google Scholar] [CrossRef] [Green Version]
- Bruning, A.; Burger, P.; Vogel, M.; Rahmeh, M.; Gingelmaiers, A.; Friese, K.; Lenhard, M.; Burges, A. Nelfinavir induces the unfolded protein response in ovarian cancer cells, resulting in ER vacuolization, cell cycle retardation and apoptosis. Cancer Biol. Ther. 2009, 8, 226–232. [Google Scholar] [CrossRef]
- Lev, A.; Lulla, A.R.; Wagner, J.; Ralff, M.D.; Kiehl, J.B.; Zhou, Y.; Benes, C.H.; Prabhu, V.V.; Oster, W.; Astsaturov, I.; et al. Anti-pancreatic cancer activity of ONC212 involves the unfolded protein response (UPR) and is reduced by IGF1-R and GRP78/BIP. Oncotarget 2017, 8, 81776–81793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.H.; Jung Jung, Y.; Koh, D.; Lim, Y.; Lee, Y.H.; Shin, S.Y. A synthetic chalcone, 2′-hydroxy-2,3,5′-trimethoxychalcone triggers unfolded protein response-mediated apoptosis in breast cancer cells. Cancer Lett. 2016, 372, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Maurel, M.; McGrath, E.P.; Mnich, K.; Healy, S.; Chevet, E.; Samali, A. Controlling the unfolded protein response-mediated life and death decisions in cancer. Semin. Cancer Biol. 2015, 33, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Ojha, R.; Amaravadi, R.K. Targeting the unfolded protein response in cancer. Pharmacol. Res. 2017, 120, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Nagelkerke, A.; Bussink, J.; Sweep, F.C.; Span, P.N. The unfolded protein response as a target for cancer therapy. Biochim. Biophys. Acta 2014, 1846, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, K.; Sakai, C.; Kawakami, S.; Yamashita, F.; Hashida, M. Inhibition of cancer cell growth by GRP78 siRNA lipoplex via activation of unfolded protein response. Biol. Pharm. Bull. 2014, 37, 648–653. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.H.; Kim, C.G.; Lim, Y.; Shin, S.Y. Aurora kinase A inhibitor TCS7010 demonstrates pro-apoptotic effect through the unfolded protein response pathway in HCT116 colon cancer cells. Oncol. Lett. 2017, 14, 6571–6577. [Google Scholar] [CrossRef]
- Simard, J.C.; Durocher, I.; Girard, D. Silver nanoparticles induce irremediable endoplasmic reticulum stress leading to unfolded protein response dependent apoptosis in breast cancer cells. Apoptosis 2016, 21, 1279–1290. [Google Scholar] [CrossRef]
- Wang, M.; Shim, J.S.; Li, R.J.; Dang, Y.; He, Q.; Das, M.; Liu, J.O. Identification of an old antibiotic clofoctol as a novel activator of unfolded protein response pathways and an inhibitor of prostate cancer. Br. J. Pharmacol. 2014, 171, 4478–4489. [Google Scholar] [CrossRef]
- Fribley, A.M.; Miller, J.R.; Brownell, A.L.; Garshott, D.M.; Zeng, Q.; Reist, T.E.; Narula, N.; Cai, P.; Xi, Y.; Callaghan, M.U.; et al. Celastrol induces unfolded protein response-dependent cell death in head and neck cancer. Exp. Cell Res. 2015, 330, 412–422. [Google Scholar] [CrossRef] [Green Version]
- Chien, W.; Ding, L.W.; Sun, Q.Y.; Torres-Fernandez, L.A.; Tan, S.Z.; Xiao, J.; Lim, S.L.; Garg, M.; Lee, K.L.; Kitajima, S.; et al. Selective inhibition of unfolded protein response induces apoptosis in pancreatic cancer cells. Oncotarget 2014, 5, 4881–4894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddalo, D.; Neeb, A.; Jehle, K.; Schmitz, K.; Muhle-Goll, C.; Shatkina, L.; Walther, T.V.; Bruchmann, A.; Gopal, S.M.; Wenzel, W.; et al. A peptidic unconjugated GRP78/BiP ligand modulates the unfolded protein response and induces prostate cancer cell death. PLoS ONE 2012, 7, e45690. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.Y.; Lee, J.M.; Lee, M.S.; Koh, D.; Jung, H.; Lim, Y.; Lee, Y.H. Targeting cancer cells via the reactive oxygen species-mediated unfolded protein response with a novel synthetic polyphenol conjugate. Clin. Cancer Res. 2014, 20, 4302–4313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, L.J.; Rivera, M.; Hawsawi, O.; Zou, J.; Hudson, T.; Wang, G.; Zhang, Q.; Cubano, L.; Boukli, N.; Odero-Marah, V. Muscadine Grape Skin Extract Induces an Unfolded Protein Response-Mediated Autophagy in Prostate Cancer Cells: A TMT-Based Quantitative Proteomic Analysis. PLoS ONE 2016, 11, e0164115. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, A.; Miller, J.R.; Tripathi, A.; Garshott, D.M.; Brownell, A.L.; Chiego, D.J.; Arevang, C.; Zeng, Q.; Jackson, L.C.; Bechler, S.A.; et al. Borrelidin Induces the Unfolded Protein Response in Oral Cancer Cells and Chop-Dependent Apoptosis. ACS Med. Chem. Lett. 2015, 6, 1122–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Liu, H.; Liu, C.; Fan, L.; Zhang, X.; Gao, A.; Hu, X.; Zhang, K.; Cao, X.; Jiang, K.; et al. Disruption of the unfolded protein response (UPR) by lead compound selectively suppresses cancer cell growth. Cancer Lett. 2015, 360, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Mokarram, P.; Albokashy, M.; Zarghooni, M.; Moosavi, M.A.; Sepehri, Z.; Chen, Q.M.; Hudecki, A.; Sargazi, A.; Alizadeh, J.; Moghadam, A.R.; et al. New frontiers in the treatment of colorectal cancer: Autophagy and the unfolded protein response as promising targets. Autophagy 2017, 13, 781–819. [Google Scholar] [CrossRef]
- Liang, G.; Fang, X.; Yang, Y.; Song, Y. Knockdown of CEMIP suppresses proliferation and induces apoptosis in colorectal cancer cells: Downregulation of GRP78 and attenuation of unfolded protein response. Biochem. Cell Biol. 2018, 96, 332–341. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, Y.; Yao, F.; Xiao, B.; Cheng, Y.; Feng, C.; Duan, C.; Zhang, C.; Liu, Y.; Li, H.; et al. Unfolded Protein Response Promotes Doxorubicin-Induced Nonsmall Cell Lung Cancer Cells Apoptosis via the mTOR Pathway Inhibition. Cancer Biother. Radiopharm. 2016, 31, 347–351. [Google Scholar] [CrossRef]
- Zhang, L.; Hapon, M.B.; Goyeneche, A.A.; Srinivasan, R.; Gamarra-Luques, C.D.; Callegari, E.A.; Drappeau, D.D.; Terpstra, E.J.; Pan, B.; Knapp, J.R.; et al. Mifepristone increases mRNA translation rate, triggers the unfolded protein response, increases autophagic flux, and kills ovarian cancer cells in combination with proteasome or lysosome inhibitors. Mol. Oncol. 2016, 10, 1099–1117. [Google Scholar] [CrossRef] [Green Version]
- Prasad, V.; Suomalainen, M.; Pennauer, M.; Yakimovich, A.; Andriasyan, V.; Hemmi, S.; Greber, U.F. Chemical induction of unfolded protein response enhances cancer cell killing through lytic virus infection. J. Virol. 2014, 88, 13086–13098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, G.F.; Cai, Z.; Meng, X.K.; Zhang, Y.; Zhu, W.; Pang, X.Y.; Dou, L. Unfolded protein response mediated JNK/AP-1 signal transduction, a target for ovarian cancer treatment. Int. J. Clin. Exp. Pathol. 2015, 8, 6505–6511. [Google Scholar]
- Schroder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Todd, D.J.; Lee, A.H.; Glimcher, L.H. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat. Rev. Immunol. 2008, 8, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.P.; Barraclough, D.L.; Stewart, C.; Joyce, K.A.; Eccles, R.M.; Barraclough, R.; Rudland, P.S.; Sibson, D.R. Expression and splicing of the unfolded protein response gene XBP-1 are significantly associated with clinical outcome of endocrine-treated breast cancer. Int. J. Cancer 2008, 123, 85–88. [Google Scholar] [CrossRef]
- Galmiche, A.; Sauzay, C.; Chevet, E.; Pluquet, O. Role of the unfolded protein response in tumor cell characteristics and cancer outcome. Curr. Opin. Oncol. 2017, 29, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Harrington, P.E.; Biswas, K.; Malwitz, D.; Tasker, A.S.; Mohr, C.; Andrews, K.L.; Dellamaggiore, K.; Kendall, R.; Beckmann, H.; Jaeckel, P.; et al. Unfolded Protein Response in Cancer: IRE1alpha Inhibition by Selective Kinase Ligands Does Not Impair Tumor Cell Viability. ACS Med. Chem. Lett. 2015, 6, 68–72. [Google Scholar] [CrossRef] [Green Version]
- Manalo, R.V.M. Anastasis and the ER stress response: Solving the paradox of the unfolded protein response in cancer. Med. Hypotheses 2017, 109, 25–27. [Google Scholar] [CrossRef]
- Tameire, F.; Verginadis, I.I.; Koumenis, C. Cell intrinsic and extrinsic activators of the unfolded protein response in cancer: Mechanisms and targets for therapy. Semin. Cancer Biol. 2015, 33, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.X.; Liu, C.M.; Gao, A.H.; Zhou, Y.B.; Li, J. Berberine combined with 2-deoxy-d-glucose synergistically enhances cancer cell proliferation inhibition via energy depletion and unfolded protein response disruption. Biochim. Biophys. Acta 2013, 1830, 5175–5183. [Google Scholar] [CrossRef]
- Ruan, Q.; Han, S.; Jiang, W.G.; Boulton, M.E.; Chen, Z.J.; Law, B.K.; Cai, J. AlphaB-crystallin, an effector of unfolded protein response, confers anti-VEGF resistance to breast cancer via maintenance of intracrine VEGF in endothelial cells. Mol. Cancer Res. 2011, 9, 1632–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, S.; Bhanja, P.; Partanen, A.; Zhang, W.; Liu, L.; Tome, W.; Guha, C. Low intensity focused ultrasound (LOFU) modulates unfolded protein response and sensitizes prostate cancer to 17AAG. Oncoscience 2014, 1, 434–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilakivi-Clarke, L.; Warri, A.; Bouker, K.B.; Zhang, X.; Cook, K.L.; Jin, L.; Zwart, A.; Nguyen, N.; Hu, R.; Cruz, M.I.; et al. Effects of In Utero Exposure to Ethinyl Estradiol on Tamoxifen Resistance and Breast Cancer Recurrence in a Preclinical Model. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, B.; Lee, A.S. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene 2013, 32, 805–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodvold, J.J.; Mahadevan, N.R.; Zanetti, M. Immune modulation by ER stress and inflammation in the tumor microenvironment. Cancer Lett. 2016, 380, 227–236. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Immune Cell Type | Cell Sub-Type | General UPR Signaling | UPR Signaling Component | |||
|---|---|---|---|---|---|---|
| GRP78 | PERK | ATF6 | IRE-1 | |||
| DCs | DCs | Required for development and functional antigen presentation and cytokine secretion. Inhibition leads to cell death. | Increased expression during maturation downstream of HMGB1 signaling. | PERK considered to have no association; however, CHOP function required for successful IL-23 secretion. | No associations found upon testing. | Increased during maturation downstream HMGB1 signaling; Required for mature function, CD80, CD86, MHC-1, and cytokine secretion. |
| Reg DCs | Unknown. | ? | ? | ? | ? | |
| Macrophages | Macrophages | Required for trafficking, function, and M1/M2 polarization. | Increased expression in maturation; decreased expression in target cells increases macrophage efficacy. | Required for mature function; Inhibiting PERK increases M1 polarization; ATF4 function associated with M1/M2 macrophage balance; maintains function during stress signaling. | ? | XBP-1 splicing increased; function required for inflammatory response; signaling associated with survival. |
| Microglia | Required for function. | ? | Required for mature function. | ? | ? | |
| Foam cell | Induces CD36 expression, positive-feedback cycle in formation and eventual cell death. | ? | Increased function leads to GSK3a/b signaling; CHOP function induces cell death. | ? | ? | |
| T cell | T cell | Required for various stages of differentiation, maturation, activation, and cytotoxic functions; also required for trafficking and homing. Excessive function associated with T-cell exhaustion. | Increased expression during differentiation. | ? | Signaling increased during differentiation, function, and immune response. | XBP-1 splicing increased during differentiation. |
| T helper | Required for differentiation but inhibited upon maturation. | Increased expression during differentiation. | ? | Signaling increased during differentiation. | XBP-1 splicing increased during differentiation. | |
| Treg | Unknown. | ? | ? | ? | ? | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramirez, M.U.; Hernandez, S.R.; Soto-Pantoja, D.R.; Cook, K.L. Endoplasmic Reticulum Stress Pathway, the Unfolded Protein Response, Modulates Immune Function in the Tumor Microenvironment to Impact Tumor Progression and Therapeutic Response. Int. J. Mol. Sci. 2020, 21, 169. https://doi.org/10.3390/ijms21010169
Ramirez MU, Hernandez SR, Soto-Pantoja DR, Cook KL. Endoplasmic Reticulum Stress Pathway, the Unfolded Protein Response, Modulates Immune Function in the Tumor Microenvironment to Impact Tumor Progression and Therapeutic Response. International Journal of Molecular Sciences. 2020; 21(1):169. https://doi.org/10.3390/ijms21010169
Chicago/Turabian StyleRamirez, Manuel U., Salvador R. Hernandez, David R. Soto-Pantoja, and Katherine L. Cook. 2020. "Endoplasmic Reticulum Stress Pathway, the Unfolded Protein Response, Modulates Immune Function in the Tumor Microenvironment to Impact Tumor Progression and Therapeutic Response" International Journal of Molecular Sciences 21, no. 1: 169. https://doi.org/10.3390/ijms21010169