Use of QSAR Global Models and Molecular Docking for Developing New Inhibitors of c-src Tyrosine Kinase



Abstract
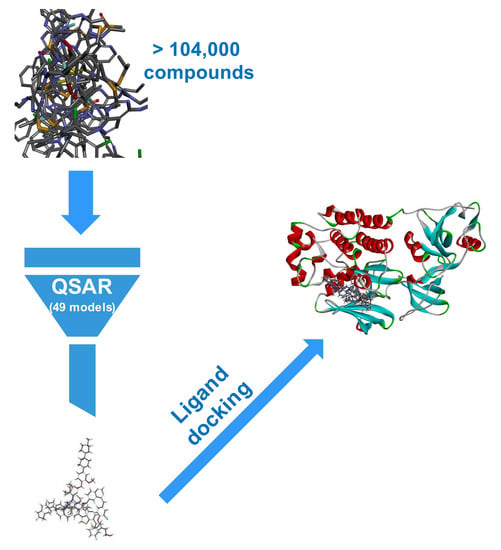
:
1. Introduction
2. Results
2.1. Dataset Analysis
2.2. Performances of Models in Nested Cross-Validation
2.3. Y-Randomization Test
2.4. Descriptors Associated with c-src Inhibitory Activity
2.5. Virtual Screening and External Validation
2.6. Applicability Domain
2.7. Molecular Docking
3. Discussion
4. Materials and Methods
4.1. Dataset
4.2. Descriptors
4.3. Feature Selection
4.4. Machine Learning Algorithms and Model Building
4.5. Performance Evaluation
4.6. Applicability Domain
4.7. Virtual Screening by QSAR
4.8. Molecular Docking Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| QSAR | Quantitative structure-activity relationship |
| AUC | Area under the ROC curve |
| PPV | Positive predictive value |
| RMSD | Root-mean-square deviation |
| BA | Balanced accuracy |
| MMCE | Mean misclassification error |
| TPR | True positive rate |
| TNR | True negative rate |
| KDEOS | Kernel Density Estimation Outlier Score |
| INFLO | Influenced Outlierness |
| MAO-A | Monoamine oxidase A |
| RF | Random forests |
| SVM | Support vector machines |
| BART | Bayesian additive regression trees |
References
- Oneyama, C.; Okada, M. MicroRNAs as the fine-tuners of Src oncogenic signalling. J. Biochem. 2015, 157, 431–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, J.T.; Parsons, S.J. Src family protein tyrosine kinases: Cooperating with growth factor and adhesion signaling pathways. Curr. Opin. Cell Biol. 1997, 9, 187–192. [Google Scholar] [CrossRef]
- Fowler, A.J.; Hebron, M.; Missner, A.A.; Wang, R.; Gao, X.; Kurd-Misto, B.T.; Liu, X.; Moussa, C.E.-H. Multikinase Abl/DDR/Src Inhibition Produces Optimal Effects for Tyrosine Kinase Inhibition in Neurodegeneration. Drugs R D 2019, 19, 149–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Gu, Y.; Luo, J.; Ye, P.; Zheng, Y.; Yu, W.; Chen, S. Inhibition of Src activation reverses pulmonary vascular remodeling in experimental pulmonary arterial hypertension via Akt/mTOR/HIF-1<alpha> signaling pathway. Exp. Cell Res. 2019, 380, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Montani, D.; Seferian, A.; Savale, L.; Simonneau, G.; Humbert, M. Drug-induced pulmonary arterial hypertension: A recent outbreak. Eur. Respir. Rev. 2013, 22, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Guignabert, C.; Phan, C.; Seferian, A.; Huertas, A.; Tu, L.; Thuillet, R.; Sattler, C.; Le Hiress, M.; Tamura, Y.; Jutant, E.-M.; et al. Dasatinib induces lung vascular toxicity and predisposes to pulmonary hypertension. J. Clin. Investig. 2016, 126, 3207–3218. [Google Scholar] [CrossRef] [PubMed]
- Özgür Yurttaş, N.; Eşkazan, A.E. Dasatinib-induced pulmonary arterial hypertension. Br. J. Clin. Pharmacol. 2018, 84, 835–845. [Google Scholar] [CrossRef]
- Broeckel, R.; Sarkar, S.; May, N.A.; Totonchy, J.; Kreklywich, C.N.; Smith, P.; Graves, L.; DeFilippis, V.R.; Heise, M.T.; Morrison, T.E.; et al. Src Family Kinase Inhibitors Block Translation of Alphavirus Subgenomic mRNAs. Antimicrob. Agents Chemother. 2019, 63, e02325. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Siemann, D. c-Src is required for hypoxia-induced metastasis-associated functions in prostate cancer cells. Onco Targets Ther. 2019, 12, 3519–3529. [Google Scholar] [CrossRef] [Green Version]
- Molinari, A.; Fallacara, A.L.; Di Maria, S.; Zamperini, C.; Poggialini, F.; Musumeci, F.; Schenone, S.; Angelucci, A.; Colapietro, A.; Crespan, E.; et al. Efficient optimization of pyrazolo [3,4-d] pyrimidines derivatives as c-Src kinase inhibitors in neuroblastoma treatment. Bioorganic Med. Chem. Lett. 2018, 28, 3454–3457. [Google Scholar] [CrossRef]
- Halaban, R.; Bacchiocchi, A.; Straub, R.; Cao, J.; Sznol, M.; Narayan, D.; Allam, A.; Krauthammer, M.; Mansour, T.S. A novel anti-melanoma SRC-family kinase inhibitor. Oncotarget 2019, 10, 2237–2251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, K.-E.; Choi, N.; Oh, S.-H.; Kim, W.-S.; Suh, W.; Sung, J.-H. Src inhibition induces melanogenesis in human G361 cells. Mol. Med. Rep. 2019, 19, 3061–3070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, Y.C.; Toro-Serra, R.; Chen, Y.; Ryu, J.; Frederick, M.J.; Zhou, G.; Gallick, G.E.; Lai, S.Y.; Clayman, G.L. Src inhibitors in suppression of papillary thyroid carcinoma growth. Head Neck 2014, 36, 375–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roelants, C.; Giacosa, S.; Pillet, C.; Bussat, R.; Champelovier, P.; Bastien, O.; Guyon, L.; Arnoux, V.; Cochet, C.; Filhol, O. Combined inhibition of PI3K and Src kinases demonstrates synergistic therapeutic efficacy in clear-cell renal carcinoma. Oncotarget 2018, 9, 30066–30078. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.; Ahn, K.; Ji, Y.G.; Cho, H.J.; Lee, D.H. Synergistic Anti-Cancer Effects of AKT and SRC Inhibition in Human Pancreatic Cancer Cells. Yonsei Med. J. 2018, 59, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Simpkins, F.; Jang, K.; Yoon, H.; Hew, K.E.; Kim, M.; Azzam, D.J.; Sun, J.; Zhao, D.; Ince, T.A.; Liu, W.; et al. Dual Src and MEK Inhibition Decreases Ovarian Cancer Growth and Targets Tumor Initiating Stem-Like Cells. Clin. Cancer Res. 2018, 24, 4874–4886. [Google Scholar] [CrossRef] [Green Version]
- PubChem Data Counts. Available online: https://pubchemdocs.ncbi.nlm.nih.gov/statistics (accessed on 17 December 2019).
- Reymond, J.-L. The Chemical Space Project. Acc. Chem. Res. 2015, 48, 722–730. [Google Scholar] [CrossRef] [Green Version]
- Polishchuk, P.G.; Madzhidov, T.I.; Varnek, A. Estimation of the size of drug-like chemical space based on GDB-17 data. J. Comput. Aided Mol. Des. 2013, 27, 675–679. [Google Scholar] [CrossRef]
- Bohacek, R.S.; McMartin, C.; Guida, W.C. The art and practice of structure-based drug design: A molecular modeling perspective. Med. Res. Rev. 1996, 16, 3–50. [Google Scholar] [CrossRef]
- Ertl, P. Cheminformatics Analysis of Organic Substituents: Identification of the Most Common Substituents, Calculation of Substituent Properties, and Automatic Identification of Drug-like Bioisosteric Groups. J. Chem. Inf. Comput. Sci. 2003, 43, 374–380. [Google Scholar] [CrossRef]
- Gini, G. QSAR: What Else? In Computational Toxicology; Nicolotti, O., Ed.; Springer: New York, NY, USA, 2018; Volume 1800, pp. 79–105. ISBN 978-1-4939-7898-4. [Google Scholar]
- Bellera, C.L.; Talevi, A. Quantitative structure–activity relationship models for compounds with anticonvulsant activity. Expert Opin. Drug Discov. 2019, 14, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Ai, S.; Lin, G.; Bai, Y.; Liu, X.; Piao, L. QSAR Classification-Based Virtual Screening Followed by Molecular Docking Identification of Potential COX-2 Inhibitors in a Natural Product Library. J. Comput. Biol. 2019, 26. [Google Scholar] [CrossRef] [PubMed]
- Allam, L.; Fatima, G.; Wiame, L.; Hamid, E.A.; Azeddine, I. Molecular screening and docking analysis of LMTK3and AKT1 combined inhibitors. Bioinformation 2018, 14, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Peng, J.; Li, P.; Du, H.; Li, Y.; Li, Y.; Zhang, L.; Sun, W.; Liu, X.; Zuo, Z. Discovery of novel indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors by virtual screening. Comput. Biol. Chem. 2019, 78, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Wang, S.; Yabes, J.G.; Chang, C.-C.H. Hybrid Density- and Partition-based Clustering Algorithm for Data with Mixed-type Variables. arXiv 2019, arXiv:1905.02257. [Google Scholar]
- Batista, J.; Vikić-Topić, D.; Lučić, B. The Difference between the Accuracy of Real and the Corresponding Random Model is a Useful Parameter for Validation of Two-State Classification Model Quality. Croat. Chem. Acta 2016, 89, 527–534. [Google Scholar] [CrossRef]
- Lučić, B.; Batista, J.; Bojović, V.; Lovrić, M.; Sović Kržić, A.; Bešlo, D.; Nadramija, D.; Vikić-Topić, D. Estimation of Random Accuracy and its Use in Validation of Predictive Quality of Classification Models within Predictive Challenges. Croat. Chem. Acta 2019, 92, P1–P13. [Google Scholar] [CrossRef] [Green Version]
- Berenger, F.; Yamanishi, Y. A Distance-Based Boolean Applicability Domain for Classification of High Throughput Screening Data. J. Chem. Inf. Model. 2019, 59, 463–476. [Google Scholar] [CrossRef]
- Sahigara, F.; Mansouri, K.; Ballabio, D.; Mauri, A.; Consonni, V.; Todeschini, R. Comparison of Different Approaches to Define the Applicability Domain of QSAR Models. Molecules 2012, 17, 4791–4810. [Google Scholar] [CrossRef] [Green Version]
- Roy, K.; Kar, S.; Ambure, P. On a simple approach for determining applicability domain of QSAR models. Chemom. Intell. Lab. Syst. 2015, 145, 22–29. [Google Scholar] [CrossRef]
- Sahigara, F.; Ballabio, D.; Todeschini, R.; Consonni, V. Defining a novel k-nearest neighbours approach to assess the applicability domain of a QSAR model for reliable predictions. J. Cheminform. 2013, 5, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Caflisch, A. Discovery of ZAP70 inhibitors by high-throughput docking into a conformation of its kinase domain generated by molecular dynamics. Bioorg. Med. Chem. Lett. 2013, 23, 5721–5726. [Google Scholar] [CrossRef]
- Chen, Y.-C. Beware of docking! Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef]
- PASS online. Available online: http://www.pharmaexpert.ru/passonline (accessed on 17 December 2019).
- Tintori, C.; Magnani, M.; Schenone, S.; Botta, M. Docking, 3D-QSAR studies and in silico ADME prediction on c-Src tyrosine kinase inhibitors. Eur. J. Med. Chem. 2009, 44, 990–1000. [Google Scholar] [CrossRef]
- Bairy, S.K.; Suneel Kumar, B.V.S.; Bhalla, J.U.T.; Pramod, A.B.; Ravikumar, M. Three-dimensional quantitative structure-activity relationship studies on c-Src inhibitors based on different docking methods. Chem. Biol. Drug Des. 2009, 73, 416–427. [Google Scholar] [CrossRef]
- Cao, R.; Mi, N.; Zhang, H. 3D-QSAR study of c-Src kinase inhibitors based on docking. J. Mol. Model. 2010, 16, 361–375. [Google Scholar] [CrossRef]
- Patil, R.; Das, S.; Stanley, A.; Yadav, L.; Sudhakar, A.; Varma, A.K. Optimized hydrophobic interactions and hydrogen bonding at the target-ligand interface leads the pathways of drug-designing. PLoS ONE 2010, 5, e12029. [Google Scholar] [CrossRef]
- Chaudhry, Q.; Piclin, N.; Cotterill, J.; Pintore, M.; Price, N.R.; Chrétien, J.R.; Roncaglioni, A. Global QSAR models of skin sensitisers for regulatory purposes. Chem. Cent. J. 2010, 4, S5. [Google Scholar] [CrossRef] [Green Version]
- Fang, D.Q.; Wu, W.J.; Zhang, R.; Zeng, G.H.; Zheng, K.C. Theoretical studies of QSAR and molecular design on a novel series of ethynyl-3-quinolinecarbonitriles as SRC inhibitors. Chem. Biol. Drug Des. 2012, 80, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Yu, X. Prediction of Depuration Rate Constants for Polychlorinated Biphenyl Congeners. ACS Omega 2019, 4, 15615–15620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, F.; Wang, Z.; Yang, X.; Shi, L.; Liu, J.; Chen, G. Development of classification models for predicting chronic toxicity of chemicals to Daphnia magna and Pseudokirchneriella subcapitata. SAR QSAR Environ. Res. 2019, 30, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Vilar, S.; Ferino, G.; Quezada, E.; Santana, L.; Friedman, C. Predicting monoamine oxidase inhibitory activity through ligand-based models. Curr. Top. Med. Chem. 2012, 12, 2258–2274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanni, R.; Garcia-Domenech, R.; Galvez-Llompart, M.; Galvez, J. Alzheimer: A Decade of Drug Design. Why Molecular Topology can be an Extra Edge? Curr. Neuropharmacol. 2018, 16, 849–864. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-H.; Xia, Z.-N.; Yan, L.; Liu, S.-S. Prediction of placental barrier permeability: A model based on partial least squares variable selection procedure. Molecules 2015, 20, 8270–8286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varmuza, K.; Filzmoser, P.; Dehmer, M. Multivariate linear QSPR/QSAR models: Rigorous evaluation of variable selection for PLS. Comput. Struct. Biotechnol. J. 2013, 5, e201302007. [Google Scholar] [CrossRef] [Green Version]
- Masand, V.H.; Mahajan, D.T.; Alafeefy, A.M.; Bukhari, S.N.A.; Elsayed, N.N. Optimization of antiproliferative activity of substituted phenyl 4-(2-oxoimidazolidin-1-yl) benzenesulfonates: QSAR and CoMFA analyses. Eur. J. Pharm. Sci. 2015, 77, 230–237. [Google Scholar] [CrossRef]
- Birck, M.G.; Campos, L.J.; Melo, E.B. Estudo computacional de 1h-imidazol-2-il-pirimidina-4, 6-diaminas para a identificação de potenciais precursores de novos agentes antimaláricosf06[C-N]. Química Nova 2016, 39, 567–574. [Google Scholar]
- Baba, H.; Takahara, J.; Yamashita, F.; Hashida, M. Modeling and Prediction of Solvent Effect on Human Skin Permeability using Support Vector Regression and Random Forest. Pharm. Res. 2015, 32, 3604–3617. [Google Scholar] [CrossRef]
- Chen, C.-H.; Tanaka, K.; Funatsu, K. Random Forest Approach to QSPR Study of Fluorescence Properties Combining Quantum Chemical Descriptors and Solvent Conditions. J. Fluoresc. 2018, 28, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Zakariazadeh, M.; Barzegar, A.; Soltani, S.; Aryapour, H. Developing 2D-QSAR models for naphthyridine derivatives against HIV-1 integrase activity. Med. Chem. Res. 2015, 24, 2485–2504. [Google Scholar] [CrossRef]
- Durgapal, J.; Bisht, N.; Alam, M.; Sharma, D.; Salman, M.; Nandi, S. QSAR and Structure-Based Docking Studies of Aryl Pyrido[2 ,3-d]pyrimidin-7(8H)-ones: An Attempt to Anticancer Drug Design. Int. J. Quant. Struct. Prop. Relatsh. 2018, 3, 43–73. [Google Scholar] [CrossRef] [Green Version]
- Evelyn, C.R.; Biesiada, J.; Duan, X.; Tang, H.; Shang, X.; Papoian, R.; Seibel, W.L.; Nelson, S.; Meller, J.; Zheng, Y. Combined Rational Design and a High Throughput Screening Platform for Identifying Chemical Inhibitors of a Ras-activating Enzyme. J. Biol. Chem. 2015, 290, 12879–12898. [Google Scholar] [CrossRef] [Green Version]
- Neves, B.J.; Braga, R.C.; Melo-Filho, C.C.; Moreira-Filho, J.T.; Muratov, E.N.; Andrade, C.H. QSAR-Based Virtual Screening: Advances and Applications in Drug Discovery. Front. Pharmacol. 2018, 9, 1275. [Google Scholar] [CrossRef] [Green Version]
- Kopetz, S. Targeting SRC and epidermal growth factor receptor in colorectal cancer: Rationale and progress into the clinic. GCR 2007, 1, S37–S41. [Google Scholar]
- Kaushansky, A.; Gordus, A.; Budnik, B.A.; Lane, W.S.; Rush, J.; MacBeath, G. System-wide investigation of ErbB4 reveals 19 sites of Tyr phosphorylation that are unusually selective in their recruitment properties. Chem. Biol. 2008, 15, 808–817. [Google Scholar] [CrossRef] [Green Version]
- Olayioye, M.A.; Beuvink, I.; Horsch, K.; Daly, J.M.; Hynes, N.E. ErbB Receptor-induced Activation of Stat Transcription Factors Is Mediated by Src Tyrosine Kinases. J. Biol. Chem. 1999, 274, 17209–17218. [Google Scholar] [CrossRef] [Green Version]
- Reactome. Search Results for SRC. Available online: https://reactome.org/content/query?q=SRC&species=Homo+sapiens&types=Reaction&types=Pathway&cluster=true (accessed on 17 December 2019).
- Araujo, J.; Logothetis, C. Dasatinib: A potent SRC inhibitor in clinical development for the treatment of solid tumors. Cancer Treat. Rev. 2010, 36, 492–500. [Google Scholar] [CrossRef] [Green Version]
- IUPHAR/BPS Guide to Pharmacology erb-b2 receptor tyrosine kinase 4. Available online: https://www.guidetopharmacology.org/GRAC/LigandInteractionsDisplayForward?ligandId=7903&species=Human (accessed on 17 December 2019).
- Lo, S.H. Focal adhesions: What’s new inside. Dev. Biol. 2006, 294, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Gene Cards. Human Gene Database KIT Gene. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=KIT (accessed on 17 December 2019).
- Amanchy, R.; Zhong, J.; Hong, R.; Kim, J.H.; Gucek, M.; Cole, R.N.; Molina, H.; Pandey, A. Identification of c-Src tyrosine kinase substrates in platelet-derived growth factor receptor signaling. Mol. Oncol. 2009, 3, 439–450. [Google Scholar] [CrossRef] [PubMed]
- McCauley, J.A.; Rudd, M.T. Hepatitis C virus NS3/4a protease inhibitors. Curr. Opin. Pharmacol. 2016, 30, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Benzine, T.; Brandt, R.; Lovell, W.C.; Yamane, D.; Neddermann, P.; De Francesco, R.; Lemon, S.M.; Perelson, A.S.; Ke, R.; McGivern, D.R. NS5A inhibitors unmask differences in functional replicase complex half-life between different hepatitis C virus strains. PLoS Pathog. 2017, 13, e1006343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watkins, W.J. Evolution of HCV NS5B Non-Nucleoside Inhibitors; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Macdonald, A. The hepatitis C virus NS5A protein binds to members of the Src family of tyrosine kinases and regulates kinase activity. J. Gen. Virol. 2004, 85, 721–729. [Google Scholar] [CrossRef]
- Klinker, S.; Stindt, S.; Gremer, L.; Bode, J.G.; Gertzen, C.G.W.; Gohlke, H.; Weiergräber, O.H.; Hoffmann, S.; Willbold, D. Phosphorylated tyrosine 93 of hepatitis C virus nonstructural protein 5A is essential for interaction with host c-Src and efficient viral replication. J. Biol. Chem. 2019, 294, 7388–7402. [Google Scholar] [CrossRef]
- Baker, M. Reproducibility crisis: Blame it on the antibodies. Nature 2015, 521, 274–276. [Google Scholar] [CrossRef] [Green Version]
- Hunter, P. The reproducibility “crisis”: Reaction to replication crisis should not stifle innovation. EMBO Rep. 2017, 18, 1493–1496. [Google Scholar] [CrossRef]
- Lagunin, A.A.; Romanova, M.A.; Zadorozhny, A.D.; Kurilenko, N.S.; Shilov, B.V.; Pogodin, P.V.; Ivanov, S.M.; Filimonov, D.A.; Poroikov, V.V. Comparison of Quantitative and Qualitative (Q)SAR Models Created for the Prediction of Ki and IC50 Values of Antitarget Inhibitors. Front. Pharmacol. 2018, 9, 1136. [Google Scholar] [CrossRef]
- Li, J.; Cheng, K.; Wang, S.; Morstatter, F.; Trevino, R.P.; Tang, J.; Liu, H. Feature Selection: A Data Perspective. ACM Comput. Surv. 2017, 50, 1–45. [Google Scholar] [CrossRef] [Green Version]
- Bischl, B.; Lang, M.; Kotthoff, L.; Schiffner, J.; Richter, J.; Studerus, E.; Casalicchio, G.; Jones, Z.M. mlr: Machine Learning in R. J. Mach. Learn. Res. 2016, 17, 1–5. [Google Scholar]
- Strobl, C.; Boulesteix, A.-L.; Kneib, T.; Augustin, T.; Zeileis, A. Conditional Variable Importance for Random Forests. BMC Bioinform. 2008, 9, 307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanski, P.; Kotthoff, L. FSelector: Selecting Attributes. Available online: https://CRAN.R-project.org/package=FSelector (accessed on 16 May 2018).
- Liaw, A.; Wiener, M. Classification and Regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Ishwaran, H.; Kogalur, U.B. Fast Unified Random Forests for Survival, Regression, and Classification (RF-SRC). Available online: https://cran.r-project.org/web/packages/randomForestSRC/index.html (accessed on 18 November 2019).
- Wright, M.N.; Ziegler, A. Ranger: A Fast Implementation of Random Forests for High Dimensional Data in C++ and R. J. Stat. Softw. 2017, 77, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Ancuceanu, R.; Dinu, M.; Neaga, I.; Laszlo, F.; Boda, D. Development of QSAR machine learning-based models to forecast the effect of substances on malignant melanoma cells. Oncol. Lett. 2019, 17, 4188–4196. [Google Scholar] [CrossRef]
- Hdoufane, I.; Bjij, I.; Soliman, M.; Tadjer, A.; Villemin, D.; Bogdanov, J.; Cherqaoui, D. In Silico SAR Studies of HIV-1 Inhibitors. Pharmaceuticals 2018, 11, 69. [Google Scholar] [CrossRef] [Green Version]
- Gadaleta, D.; Manganelli, S.; Roncaglioni, A.; Toma, C.; Benfenati, E.; Mombelli, E. QSAR Modeling of ToxCast Assays Relevant to the Molecular Initiating Events of AOPs Leading to Hepatic Steatosis. J. Chem. Inf. Model. 2018, 58, 1501–1517. [Google Scholar] [CrossRef] [Green Version]
- Hodyna, D.; Kovalishyn, V.; Semenyuta, I.; Blagodatnyi, V.; Rogalsky, S.; Metelytsia, L. Imidazolium ionic liquids as effective antiseptics and disinfectants against drug resistant S. aureus: In silico and in vitro studies. Comput. Biol. Chem. 2018, 73, 127–138. [Google Scholar] [CrossRef]
- Sun, Y.; Shi, S.; Li, Y.; Wang, Q. Development of quantitative structure-activity relationship models to predict potential nephrotoxic ingredients in traditional Chinese medicines. Food Chem. Toxicol. 2019, 128, 163–170. [Google Scholar] [CrossRef]
- Chen, H.; Chen, L. Support Vector Machine Classification of Drunk Driving Behaviour. Int. J. Environ. Res. Public Health 2017, 14, 108. [Google Scholar] [CrossRef]
- Idakwo, G.; Luttrell, J.; Chen, M.; Hong, H.; Zhou, Z.; Gong, P.; Zhang, C. A review on machine learning methods for in silico toxicity prediction. J. Environ. Sci. Health Part C 2018, 36, 169–191. [Google Scholar] [CrossRef]
- Lei, T.; Sun, H.; Kang, Y.; Zhu, F.; Liu, H.; Zhou, W.; Wang, Z.; Li, D.; Li, Y.; Hou, T. ADMET Evaluation in Drug Discovery. 18. Reliable Prediction of Chemical-Induced Urinary Tract Toxicity by Boosting Machine Learning Approaches. Mol. Pharm. 2017, 14, 3935–3953. [Google Scholar] [CrossRef]
- Feng, D.; Svetnik, V.; Liaw, A.; Pratola, M.; Sheridan, R.P. Building Quantitative Structure-Activity Relationship Models Using Bayesian Additive Regression Trees. J. Chem. Inf. Model. 2019, 59, 2642–2655. [Google Scholar] [CrossRef] [PubMed]
- Dieguez-Santana, K.; Pham-The, H.; Rivera-Borroto, O.M.; Puris, A.; Le-Thi-Thu, H.; Casanola-Martin, G.M. A Two QSAR Way for Antidiabetic Agents Targeting Using α-Amylase and α-Glucosidase Inhibitors: Model Parameters Settings in Artificial Intelligence Techniques. Lett. Drug Des. Discov. 2017, 14, 862–868. [Google Scholar] [CrossRef]
- Raevsky, O.A.; Grigorev, V.Y.; Yarkov, A.V.; Polianczyk, D.E.; Tarasov, V.V.; Bovina, E.V.; Bryzhakina, E.N.; Dearden, J.C.; Avila-Rodriguez, M.; Aliev, G. Classification (Agonist/Antagonist) and Regression “Structure-Activity” Models of Drug Interaction with 5-HT6. Cent. Nerv. Syst. Agents Med. Chem. 2018, 18, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Quinlan, R. C50: C5.0 Decision Trees and Rule-Based Models. Available online: https://CRAN.R-project.org/package=C50 (accessed on 22 May 2018).
- Bharti, D.R.; Lynn, A.M. QSAR based predictive modeling for anti-malarial molecules. Bioinformation 2017, 13, 154–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Bischl, B.; Lang, M. Parallelmap: Unified Interface to Parallelization Back-Ends. Available online: https://CRAN.R-project.org/package=parallelMap (accessed on 17 May 2019).
- Wing, M.K.C.; Weston, S.; Williams, A.; Keefer, C.; Engelhardt, A.; Cooper, T.; Mayer, Z.; Kenkel, B.; Team, R.C.; Benesty, M.; et al. Caret: Classification and Regression Training. Available online: https://CRAN.R-project.org/package=caret (accessed on 27 April 2019).
- Meyer, D.; Dimitriadou, E.; Hornik, K.; Weingessel, A.; Leisch, F. E1071: Misc Functions of the Department of Statistics, Probability Theory Group (Formerly: E1071), TU Wien. R Package Version 2019, 1, 6–8. [Google Scholar]
- Witten, I.H.; Frank, E. Data Mining: Practical Machine Learning Tools and Techniques, 2nd ed.; Morgan Kaufmann: San Francisco, CA, USA, 2005. [Google Scholar]
- Hornik, K.; Buchta, C.; Zeileis, A. Open-Source Machine Learning: R Meets Weka. Comput. Stat. 2009, 24, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Kapelner, A.; Bleich, J. bartMachine: Machine Learning with Bayesian Additive Regression Trees. J. Stat. Softw. 2016, 70, 1–40. [Google Scholar] [CrossRef] [Green Version]
- Maechler, M.; Rousseeuw, P.; Struyf, A.; Hubert, M.; Hornik, K. Cluster: Cluster Analysis Basics and Extensions. R Package Version 2012, 1, 56. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Hahsler, M.; Hornik, K.; Buchta, C. Getting things in order: An introduction to the R package seriation. J. Stat. Softw. 2008, 25, 1–34. [Google Scholar] [CrossRef] [Green Version]
- Baumann, D.; Baumann, K. Reliable estimation of prediction errors for QSAR models under model uncertainty using double cross-validation. J. Cheminform. 2014, 6, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gramatica, P. Principles of QSAR models validation: Internal and external. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Roy, K.; Ambure, P. The “double cross-validation” software tool for MLR QSAR model development. Chemom. Intell. Lab. Syst. 2016, 159, 108–126. [Google Scholar] [CrossRef]
- Tetko, I.V.; Sushko, I.; Pandey, A.K.; Zhu, H.; Tropsha, A.; Papa, E.; Oberg, T.; Todeschini, R.; Fourches, D.; Varnek, A. Critical assessment of QSAR models of environmental toxicity against Tetrahymena pyriformis: Focusing on applicability domain and overfitting by variable selection. J. Chem. Inf. Model. 2008, 48, 1733–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capuzzi, S.J.; Sun, W.; Muratov, E.N.; Martínez-Romero, C.; He, S.; Zhu, W.; Li, H.; Tawa, G.; Fisher, E.G.; Xu, M.; et al. Computer-Aided Discovery and Characterization of Novel Ebola Virus Inhibitors. J. Med. Chem. 2018, 61, 3582–3594. [Google Scholar] [CrossRef] [PubMed]
- Warnes, G.R.; Bolker, B.; Lumley, T. Gtools: Various R Programming Tools. Available online: https://CRAN.R-project.org/package=gtools (accessed on 26 June 2018).
- Yang, H.; Du, Z.; Lv, W.-J.; Zhang, X.-Y.; Zhai, H.-L. In silico toxicity evaluation of dioxins using structure–activity relationship (SAR) and two-dimensional quantitative structure–activity relationship (2D-QSAR). Arch. Toxicol. 2019, 93, 3207–3218. [Google Scholar] [CrossRef]
- Madsen, J.H. DDoutlier: Distance & Density-Based Outlier Detection. Available online: https://github.com/jhmadsen/DDoutlier (accessed on 30 May 2018).
- Schubert, E.; Zimek, A.; Kriegel, H.-P. Generalized Outlier Detection with Flexible Kernel Density Estimates. In Proceedings of the 2014 SIAM International Conference on Data Mining; Society for Industrial and Applied Mathematics: Philadelphia, PA, USA, 2014; pp. 542–550. [Google Scholar]
- Jin, W.; Tung, A.K.H.; Han, J.; Wang, W. Ranking Outliers Using Symmetric Neighborhood Relationship. In Advances in Knowledge Discovery and Data Mining; Ng, W.-K., Kitsuregawa, M., Li, J., Chang, K., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 3918, pp. 577–593. ISBN 978-3-540-33206-0. [Google Scholar]
- Levinson, N.M.; Boxer, S.G. Human Src Kinase Bound to Kinase Inhibitor Bosutinib. Available online: https://www.rcsb.org/structure/4mxo (accessed on 4 December 2013).
- Boubeva, R.; Pernot, L.; Perozzo, R.; Scapozza, L. Crystal Structure of the L317I Mutant of the C-src Tyrosine Kinase Domain Complexed with Dasatinib. Available online: http://www.rcsb.org/structure/3QLG (accessed on 8 February 2012).
- Roskoski, R. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015, 94, 9–25. [Google Scholar] [CrossRef]
- Xu, W.; Doshi, A.; Lei, M.; Eck, M.J.; Harrison, S.C. Crystal Structure of Human Tyrosine-Protein Kinase C-Src, in Complex with Amp-Pnp. Available online: http://www.rcsb.org/pdb/explore/litView.do?structureId=2SRC (accessed on 22 July 1999).
- Garcia-Sosa, A.T.; Hetenyi, C.; Maran, U. Drug efficiency indices for improvement of molecular docking scoring functions. J. Comput. Chem. 2010, 31, 174–184. [Google Scholar] [CrossRef]
- Thiele, C. cutpointr: Determine and Evaluate Optimal Cutpoints in Binary Classification Tasks. Available online: https://CRAN.R-project.org/package=cutpointr (accessed on 17 September 2019).
- Bell, E.W.; Zhang, Y. DockRMSD: An open-source tool for atom mapping and RMSD calculation of symmetric molecules through graph isomorphism. J. Cheminform. 2019, 11, 40. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model * | BA (%) | PPV (%) | MMCE (%) | AUC (%) | TPR (%) | TNR (%) | Q2 − Q2, rnd |
|---|---|---|---|---|---|---|---|
| RF_anova_23 | 70.24 | 78.26 | 18.60 | 82.56 | 45.39 | 95.08 | 21.33 |
| RF_auc_20 | 70.07 | 78.08 | 18.69 | 82.85 | 45.04 | 95.09 | 21.23 |
| RF_cforest_13 | 70.07 | 79.39 | 18.60 | 82.96 | 44.80 | 95.34 | 21.33 |
| RF_kruskal_30 | 70.52 | 77.42 | 18.60 | 82.61 | 46.35 | 94.68 | 21.33 |
| RF_RFimp_30 | 71.54 | 80.04 | 17.73 | 86.03 | 47.69 | 95.39 | 22.19 |
| RF_RF.SRCimp_20 | 71.01 | 77.44 | 18.31 | 83.76 | 47.18 | 94.83 | 21.62 |
| RF_RF.SRCvarselect_10 | 72.93 | 78.72 | 17.34 | 86.01 | 51.29 | 94.56 | 22.58 |
| RF_impurity_15 | 70.67 | 76.43 | 18.69 | 83.72 | 46.91 | 94.43 | 21.23 |
| RF_permutation_10 | 71.53 | 80.51 | 17.83 | 83.63 | 47.86 | 95.20 | 22.10 |
| RF_univariate_30 | 71.48 | 83.49 | 17.44 | 84.31 | 46.80 | 96.16 | 22.48 |
| SVM_anova_30 | 71.83 | 71.26 | 19.07 | 82.08 | 51.60 | 92.05 | 20.48 |
| SVM_auc_30 | 72.02 | 71.56 | 18.98 | 83.25 | 51.99 | 92.05 | 20.94 |
| SVM_cforest_30 | 75.11 | 74.96 | 17.05 | 85.60 | 57.65 | 92.57 | 22.87 |
| SVM_chi.sq_30 | 71.91 | 75.44 | 18.59 | 82.45 | 50.86 | 92.97 | 21.33 |
| SVM_gainratio_30 | 72.03 | 72.78 | 18.98 | 82.85 | 51.99 | 92.07 | 20.94 |
| SVM_information_30 | 72.44 | 73.34 | 18.59 | 83.91 | 52.54 | 92.35 | 21.33 |
| SVM_kruskal_20 | 72.06 | 72.29 | 18.98 | 82.06 | 52.06 | 92.05 | 20.94 |
| SVM_oneR_30 | 72.49 | 78.08 | 17.73 | 81.16 | 50.68 | 94.31 | 22.19 |
| SVM_RFimp_30 | 74.74 | 74.71 | 17.25 | 86.92 | 57.16 | 92.32 | 22.68 |
| SVM_RF.SRCimp_30 | 75.92 | 77.07 | 16.28 | 86.20 | 58.57 | 93.28 | 23.64 |
| SVM_RF.SRCvarselect_20 | 76.33 | 76.22 | 16.28 | 86.75 | 60.10 | 92.56 | 23.64 |
| SVM_impurity_30 | 73.96 | 73.86 | 17.82 | 84.27 | 55.61 | 92.30 | 22.10 |
| SVM_permutation_20 | 72.14 | 73.82 | 18.59 | 84.37 | 51.58 | 92.71 | 21.33 |
| SVM_relief_30 | 72.42 | 71.93 | 19.08 | 82.15 | 53.57 | 91.26 | 20.84 |
| SVM_sym.uncertain_20 | 71.91 | 73.31 | 18.69 | 83.33 | 50.99 | 92.84 | 21.23 |
| Adabm1_RFimp_30 | 71.06 | 73.50 | 19.08 | 83.49 | 49.11 | 93.00 | 20.84 |
| Adabm1_RF.SRCvarselect_20 | 71.15 | 70.36 | 19.56 | 81.96 | 50.36 | 91.95 | 20.36 |
| Adabm1_impurity_20 | 71.22 | 73.34 | 18.80 | 83.66 | 49.18 | 93.26 | 21.13 |
| Adabm1_univariate_30 | 70.50 | 74.30 | 19.27 | 82.36 | 47.61 | 93.39 | 20.65 |
| BartM_chi.sq_30 | 73.15 | 73.28 | 18.11 | 83.54 | 53.87 | 92.42 | 21.81 |
| BartM_gainratio_20 | 71.61 | 70.19 | 19.37 | 82.45 | 51.57 | 91.64 | 20.56 |
| BartM_information_20 | 73.56 | 73.52 | 17.92 | 84.08 | 54.68 | 92.44 | 22.00 |
| BartM_RFimp_25 | 74.24 | 71.45 | 18.02 | 85.28 | 57.13 | 91.36 | 21.90 |
| BartM_impurity_20 | 73.48 | 70.94 | 18.50 | 83.79 | 55.74 | 91.22 | 21.42 |
| BartM_permutation_22 | 74.70 | 71.64 | 17.82 | 85.04 | 58.17 | 91.23 | 22.10 |
| BartM_sym.uncertain_30 | 73.59 | 71.19 | 18.31 | 84.36 | 55.69 | 91.49 | 21.62 |
| C50_anova_30 | 75.96 | 72.56 | 17.05 | 84.73 | 60.70 | 91.23 | 22.87 |
| C50_auc_20 | 74.00 | 72.03 | 18.12 | 83.75 | 56.80 | 91.19 | 21.81 |
| C50_cforest_20 | 75.08 | 71.62 | 17.73 | 85.06 | 59.32 | 90.84 | 22.19 |
| C50_chi.sq_30 | 75.55 | 70.40 | 17.73 | 83.55 | 60.79 | 90.32 | 22.19 |
| C50_gainratio_30 | 75.26 | 70.85 | 17.82 | 84.43 | 60.08 | 90.45 | 22.10 |
| C50_kruskal_30 | 74.56 | 71.35 | 18.02 | 84.52 | 58.03 | 91.10 | 21.90 |
| C50_oneR_30 | 73.91 | 72.78 | 18.41 | 83.62 | 57.06 | 90.76 | 21.52 |
| C50_RFimp_30 | 78.56 | 75.39 | 15.32 | 87.24 | 65.23 | 91.89 | 24.60 |
| C50_RF.SRCimp_30 | 76.21 | 72.82 | 17.05 | 85.45 | 61.32 | 91.10 | 22.87 |
| C50_RF.SRCvarselect_20 | 77.64 | 72.08 | 16.76 | 87.84 | 65.43 | 89.86 | 23.16 |
| C50_impurity_20 | 76.40 | 76.14 | 16.10 | 86.70 | 60.13 | 92.66 | 23.83 |
| C50_permutation_30 | 75.93 | 72.28 | 16.96 | 86.29 | 60.51 | 91.36 | 22.96 |
| C50_univariate_30 | 75.44 | 70.55 | 17.73 | 85.47 | 60.46 | 90.43 | 22.19 |
| Name | Interpretation | Descriptor Block (Group) | Frequency Occurring among the First Five Most Important Features |
|---|---|---|---|
| SpMax4_Bh(m) | Largest eigenvalue n. 4 of Burden matrix weighted by mass | Burden eigenvalues | 14 |
| DECC | Eccentric topological index | Topological indices | 11 |
| SpMax5_Bh(m) | Largest eigenvalue n. 5 of Burden matrix weighted by mass | Burden eigenvalues | 8 |
| SpMax3_Bh(m) | Largest eigenvalue n. 3 of Burden matrix weighted by mass | Burden eigenvalues | 8 |
| J_D | Balaban-like index from topological distance matrix (Balaban distance connectivity index) | 2D matrix-based descriptors | 6 |
| F06[C–N] | Frequency of C–N at topological distance 6 | 2D Atom Pairs | 5 |
| Chi1_EA(dm) | Connectivity-like index of order 1 from edge adjacency mat. weighted by dipole moment | Edge adjacency indices | 4 |
| P_VSA_MR_6 | P_VSA-like on Molar Refractivity, bin 6 | P_VSA-like descriptors | 3 |
| SpMax6_Bh(m) | largest eigenvalue n. 6 of Burden matrix weighted by mass | Burden eigenvalues | 3 |
| N-073 | Ar2NH/Ar3N/Ar2N-Al/R..N..R | Atom-centered fragments | 2 |
| F05[C–N] | Frequency of C–N at topological distance 5 | 2D Atom Pairs | 2 |
| ZINC Code | Substance Name | Confirmation in Wet Lab Experiments * | Activity Confirmed on Other Tyrosin Kinases * | Presence in the Training Set | Energy of Binding ** |
|---|---|---|---|---|---|
| ZINC000001550477 | Lapatinib | Yes | Yes | Yes | −10.07 (0.67) |
| ZINC000034638188 | Pf-562271 | Yes | Yes | Yes | −9.3 (0.74) |
| ZINC000063298074 | Ilorasertib | Yes | Yes | Yes | −10.09 (0.66) |
| ZINC000034800096 | Gw583373a | No | Yes | No | −11.02 (1.01) |
| ZINC000027184814 | Vibriobactin | NA | No | No | −9.77 (0.74) |
| ZINC000034800093 | Gw580496a | No | Yes | No | −9.33 (1.09) |
| ZINC000150528975 | Vedroprevir | NA | No | No | −11.51 (1.04) |
| ZINC000034800112 | Gw576484x | No | Yes | No | −10.36 (0.84) |
| ZINC000072190218 | Avatrombopag | NA | No | No | −9.28 (0.43) |
| ZINC000034800091 | Gw576609a | No | Yes | No | −11.38 (0.69) |
| ZINC000044418656 | Gw784684x | No | Yes | No | −10.77 (0.93) |
| ZINC000042804069 | Gsk-182497a | No | Yes | No | −9.57 (0.37) |
| ZINC000103297739 | Defactinib | No | Yes | No | −10.23 (0.40) |
| ZINC000004215255 | Cefpimizole | NA | No | No | −10.54 (0.70) |
| ZINC000042834127 | Gsk1751853a | No | Yes | No | −10.34 (1.40) |
| ZINC000014945166 | Gw830365a | No | Yes | No | −9.53 (0.29) |
| ZINC000150339466 | Ciluprevir | NA | No | No | −10.95 (0.88) |
| ZINC000043195317 | Golvatinib | No | Yes | No | −14 (1.06) |
| ZINC000042201866 | Gw566221a | No | Yes | No | −10.06 (0.71) |
| ZINC000095615094 | Patellamide G | NA | No | No | −9.32 (0.79) |
| ZINC000003604326 | Vaneprim | NA | No | No | −11.01 (0.79) |
| ZINC000002007399 | Gw458787a | No | Yes | No | −10.95 (0.76) |
| ZINC000028639340 | Posaconazole | NA | No | No | −10.92 (1.01) |
| ZINC000072122048 | Gsk259178a | No | Yes | No | −12.44 (0.49) |
| ZINC000068204830 | Daclatasvir | NA | No | No | −10.75 (0.42) |
| ZINC000043131420 | Fostamatinib | NA | Yes | No | −10.77 (1.11) |
| ZINC000169289453 | Simeprevir | NA | No | No | −11.45 (0.88) |
| ZINC000042834162 | Gw869810x | No | Yes | No | −12.11 (0.76) |
| ZINC000049709569 | Asperazine | NA | No | No | −11.6 (0.82) |
| ZINC000096928979 | Deleobuvir | NA | No | No | −10.2 (0.68) |
| ZINC000042201868 | Gw568377a | No | No | No | −9.36 (0.60) |
| ZINC000014945147 | Gw809897x | Yes | Yes | No | −10.44 (0.71) |
| ZINC000014945171 | Gw830263a | Yes | Yes | No | −10.53 (0.57) |
| ZINC000014945045 | Gw569530a | No | Yes | No | −9.52 (0.55) |
| ZINC000003925087 | Gw806742x | Yes | Yes | No | −10.43 (0.78) |
| ZINC000095618748 | Candesartan O-Glucuronide | NA | No | No | −9.71 (0.58) |
| ZINC000098052868 | Olcegepant | NA | No | No | −9.55 (0.48) |
| ZINC000049833405 | Preulicyclamide | NA | No | No | −11.13 (0.62) |
| ZINC000034800110 | Gw574782a | No | Yes | No | −10.42 (0.60) |
| ZINC000014965596 | Gw683134a | Yes | Yes | No | −10.91 (0.80) |
| ZINC000034800112 | Gw576484x | No | Yes | No | −9.93 (0.36) |
| ZINC000019862646 | Fedratinib | Yes | Yes | No | −10.23 (0.64) |
| ZINC000150377731 | Bms-247243 | NA | No | No | −10.42 (0.83) |
| ZINC000003986669 | Bx-795 | Yes | Yes | No | −9.28 (0.69) |
| ZINC000095615898 | Tyrokeradine A | NA | No | No | −11.14 (0.76) |
| ZINC000003919988 | L-766892 | NA | No | No | −9.59 (0.67) |
| ZINC000095544067 | Ulithiacyclamide F | NA | No | No | −9.76 (0.52) |
| ZINC000049889335 | Edulirin A | NA | No | No | −11.45 (1.04) |
| ZINC000003995140 | Gw621823a | No | Yes | No | −10.63 (0.63) |
| ZINC000040379218 | Gw684626b | No | Yes | No | −10.46 (0.87) |
| ZINC000034800121 | Gw567808a | No | Yes | No | −10.42 (0.53) |
| ZINC000169306513 | Hydroxyitraconazole | NA | No | No | −9.78 (1.02) |
| ZINC000169368380 | Kni-1039 | NA | No | No | −10.13 (0.41) |
| ZINC000150601177 | Ombitasvir | NA | No | No | −10.07 (0.69) |
| ZINC000040404350 | Gsk-969786a | No | Yes | No | −10.2 (0.75) |
| ZINC000150592451 | Micromide | NA | No | No | −12.96 (1.00) |
| ZINC000028249631 | Pd-170292 | NA | No | No | −10.1 (0.73) |
| ZINC000169366333 | Porphyrin | NA | No | No | −11.05 (0.71) |
| ZINC000034800119 | Gw576924a | No | Yes | No | −10.18 (0.92) |
| ZINC000150362888 | Pyropheophytin B | NA | No | No | −10.23 (0.73) |
| ZINC000100057121 | Tegobuvir | NA | No | No | −10.55 (0.58) |
| ZINC000103213128 | Heptamethylene 1,7-Bis-Imadacloprid | NA | No | No | −9.58 (0.47) |
| ZINC000169291993 | Sansanmycin F | NA | No | No | −9.5 (0.56) |
| ZINC000230052516 | Urobilin | NA | No | No | −10.9 (0.85) |
| ZINC000003994828 | Brecanavir | NA | No | No | −10.41 (0.86) |
| ZINC000169363931 | Ansacarbamitocin C | NA | No | No | −10.56 (0.52) |
| ZINC000095535868 | Rwj-58259 | NA | No | No | −10.09 (0.77) |
| ZINC000003921862 | Tallimustine | NA | No | No | −9.76 (0.67) |
| ZINC000063933734 | Rebastinib | No | Yes | No | −9.73 (0.57) |
| ZINC000095615652 | Patellamide C | NA | No | No | −9.46 (0.73) |
| ZINC000197688172 | S-[(3e,5z)-3,5-Octadienoate | NA | No | No | −9.6 (0.67) |
| ZINC000014965588 | Gw709042a | No | Yes | No | −9.89 (0.89) |
| ZINC000085537136 | Barixibat | NA | No | No | −9.72 (0.56) |
| ZINC000169291499 | Kibdelomycin | NA | No | No | −10.99 (0.66) |
| ZINC000003946578 | Mitratapide | NA | No | No | −10.41 (0.62) |
| ZINC000001481922 | Setipafant | NA | No | No | −10.05 (0.62) |
| ZINC000072173092 | Deoxyvobstusine Lactone | NA | No | No | −9.66 (0.64) |
| ZINC000006717126 | Quarfloxin | NA | No | No | −9.85 (0.78) |
| ZINC000077301904 | Losartan N2-Glucuronide | NA | No | No | −10.86 (1.27) |
| ZINC000150609364 | Pseudoceratinazole A | NA | No | No | −11.38 (0.97) |
| ZINC000095616246 | Ulithiacyclamide E | NA | No | No | −9.35 (0.69) |
| ZINC000068151111 | Narlaprevir | NA | No | No | −9.96 (0.44) |
| ZINC000150351429 | Phytosulfokine B | NA | No | No | −9.7 (0.70) |
| ZINC000003989268 | Ceftaroline Fosamil | NA | No | No | −9.84 (0.62) |
| ZINC000008552132 | Stafac | NA | No | No | −11.01 (0.91) |
| ZINC000095618880 | Clofazimine Glucuronide | NA | No | No | −9.65 (0.58) |
| ZINC000096006065 | Xv638 | NA | No | No | −9.56 (0.57) |
| ZINC000169292535 | Rifapentine | NA | No | No | −12.81 (0.92) |
| ZINC000150341961 | Mafodotin | NA | No | No | −9.32 (0.71) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ancuceanu, R.; Tamba, B.; Stoicescu, C.S.; Dinu, M. Use of QSAR Global Models and Molecular Docking for Developing New Inhibitors of c-src Tyrosine Kinase. Int. J. Mol. Sci. 2020, 21, 19. https://doi.org/10.3390/ijms21010019
Ancuceanu R, Tamba B, Stoicescu CS, Dinu M. Use of QSAR Global Models and Molecular Docking for Developing New Inhibitors of c-src Tyrosine Kinase. International Journal of Molecular Sciences. 2020; 21(1):19. https://doi.org/10.3390/ijms21010019
Chicago/Turabian StyleAncuceanu, Robert, Bogdan Tamba, Cristina Silvia Stoicescu, and Mihaela Dinu. 2020. "Use of QSAR Global Models and Molecular Docking for Developing New Inhibitors of c-src Tyrosine Kinase" International Journal of Molecular Sciences 21, no. 1: 19. https://doi.org/10.3390/ijms21010019
APA StyleAncuceanu, R., Tamba, B., Stoicescu, C. S., & Dinu, M. (2020). Use of QSAR Global Models and Molecular Docking for Developing New Inhibitors of c-src Tyrosine Kinase. International Journal of Molecular Sciences, 21(1), 19. https://doi.org/10.3390/ijms21010019