Role of c-Jun N-Terminal Kinases (JNKs) in Epilepsy and Metabolic Cognitive Impairment

 ,
,  , , , , ,
, , , , , 
Abstract
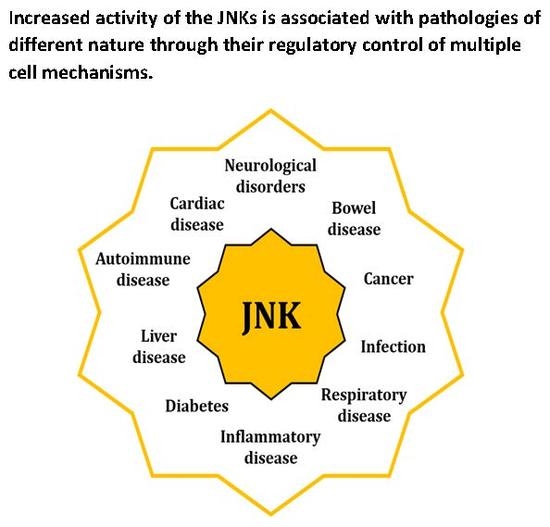
1. Introduction
2. Mitogen Activated Protein Kinases (MAPKs)
3. C-JUN N-Terminal Kinases (JNKs)
3.1. Role of JNKs in Subcellular Compartments
3.1.1. JNK Activation Alters Mitochondrial Activity
3.1.2. Endoplasmic Reticulum Stress (ERS) Activates JNK
3.2. JNKs in the Brain
4. Role of the JNKs in the Development of Disease
4.1. Temporal Lobe Epilepsy
4.2. The Metabolic-Cognitive Syndrome
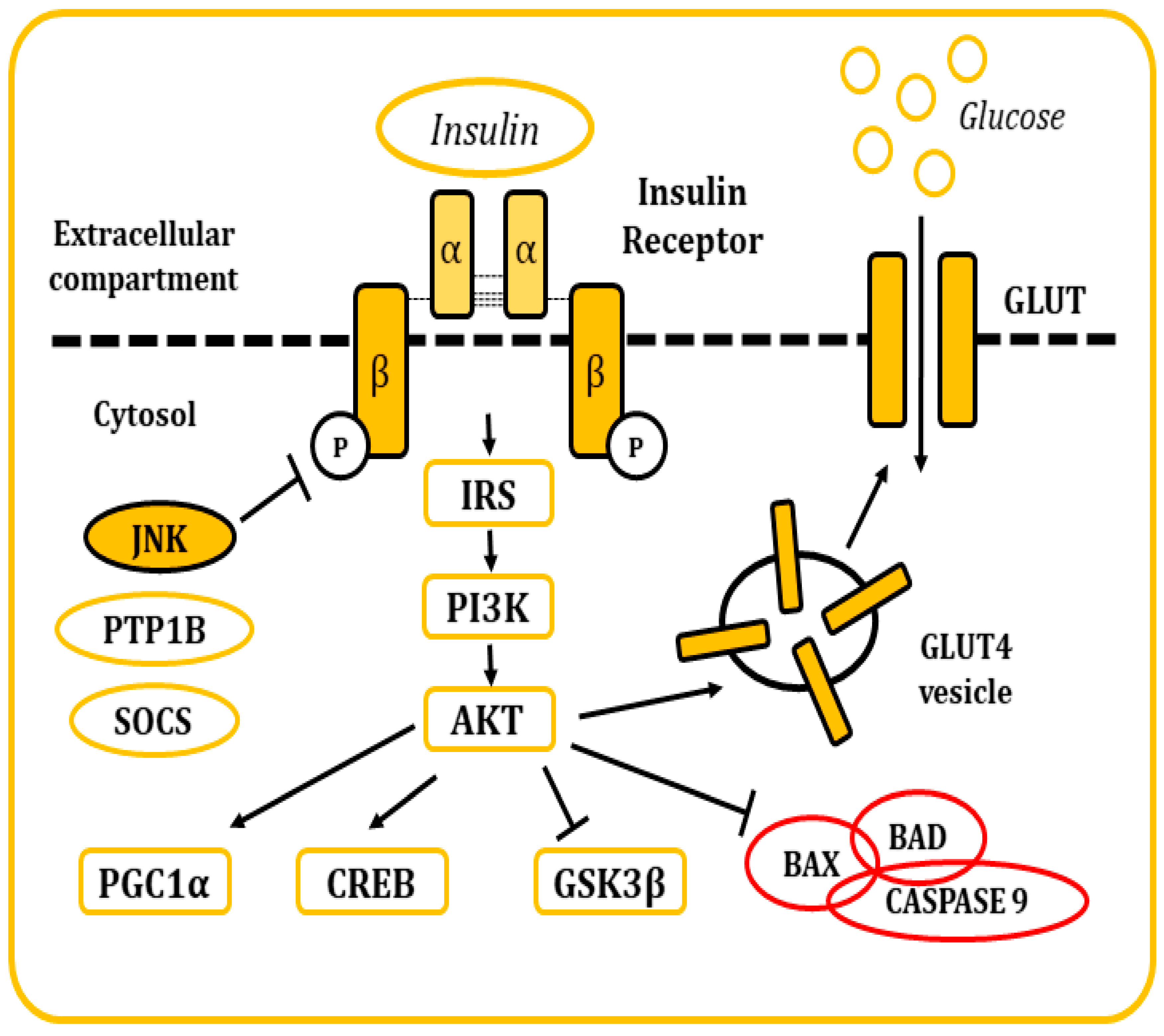
4.2.1. The Metabolism of Insulin
4.2.2. The Metabolism of Insulin in the Brain
4.2.3. Role of JNKs in Metabolic-Related Dementias
5. JNKs as Potential Therapeutic Targets
6. Concluding Remarks
Funding
Conflicts of Interest
References
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1619–1633. [Google Scholar] [CrossRef] [PubMed]
- Sabapathy, K. Role of the JNK Pathway in human diseases. In Progress in Molecular Biology and Translational Science, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2012; Volume 106, pp. 145–169. [Google Scholar]
- Sun, Y.; Liu, W.Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal Transduct. 2015, 35, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.J.; Patrick, S.L.; Huang, W.C.; Connors, B.W. Initiation, propagation, and termination of epileptiform activity in rodent neocortex in vitro involve distinct mechanisms. J. Neurosci. 2005, 25, 8131–8140. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Finegan, K.G.; Robinson, A.C.; Knowles, L.; Khosravi-Far, R.; Hinchliffe, K.A.; Boot-Handford, R.P.; Tournier, C. Activation of extracellular signal-regulated protein kinase 5 downregulates FasL upon osmotic stress. Cell Death Differ. 2006, 13, 2099. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signaling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef]
- Cui, J.; Zhang, M.; Zhang, Y.Q.; Xu, Z.H. JNK pathway: Diseases and therapeutic potential. Acta Pharmacol. Sin. 2007, 28, 601–608. [Google Scholar] [CrossRef]
- Yasuda, J.; Whitmarsh, A.J.; Cavanagh, J.; Sharma, M.; Davis, R.J. The JIP Group of Mitogen-Activated Protein Kinase Scaffold Proteins. Mol. Cell. Biol. 1999, 19, 7245–7254. [Google Scholar] [CrossRef]
- Coffey, E.T. Nuclear and cytosolic JNK signalling in neurons. Nat. Rev. Neurosci. 2014, 15, 285–299. [Google Scholar] [CrossRef]
- Borsello, T. The JNK signalling transduction pathway in the brain. Front. Biosci. 2012, 4, 2110–2120. [Google Scholar] [CrossRef]
- Yarza, R.; Vela, S.; Solas, M.; Ramirez, M.J. c-Jun N-terminal kinase (JNK) signaling as a therapeutic target for Alzheimer’s disease. Front. Pharmacol. 2016, 6, 321. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Llambi, F. Cell death signaling. Cold Spring Harb. Perspect. Biol. 2015, 7, a006080. [Google Scholar] [CrossRef]
- Ventura, J.J.; Hübner, A.; Zhang, C.; Flavell, R.A.; Shokat, K.M.; Davis, R.J. Chemical genetic analysis of the time course of signal transduction by JNK. Mol. Cell 2006, 21, 701–710. [Google Scholar] [CrossRef]
- Madhumita, D.; Sabio, G.; Jiang, F.; Rincón, M.; Flavell, R.A.; Davis, R.J. Induction of hepatitis by JNK-mediated expression of TNFα. Cell 2009, 136, 249–260. [Google Scholar]
- Koch, P.; Gehringer, M.; Laufer, S.A. Inhibitors of c-Jun N-terminal kinases: An update. J. Med. Chem. 2015, 58, 72–95. [Google Scholar] [CrossRef]
- Tournier, C.; Hess, P.; Yang, D.D.; Xu, J.; Turner, T.K.; Nimnual ABar-Sagi, D.; Jones, S.N.; Flavell, R.A.; Davis, R.J. Requirement of JNK for Stress- Induced Activation of the Cytochrome c–Mediated Death Pathway. Science 2000, 288, 870–874. [Google Scholar] [CrossRef]
- Waldbaum, S.; Patel, M. Mitochondria, oxidative stress, and temporal lobe epilepsy. Epilepsy Res. 2010, 88, 23–45. [Google Scholar] [CrossRef]
- Picard, M.; Taivassalo, T.; Gouspillou, G.; Hepple, R.T. Mitochondria: Isolation, structure and function. J. Physiol. 2011, 589, 4413–4421. [Google Scholar] [CrossRef]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, 1005–1028. [Google Scholar] [CrossRef] [PubMed]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial Metabolism of Reactive Oxygen Species. Biochemistry 2005, 70, 200–214. [Google Scholar] [CrossRef] [PubMed]
- Boveris, A. Determination of the Production of Superoxide Radicals and Hydrogen Peroxide in Mitochondria. Methods Enzymol. 1984, 105, 429–435. [Google Scholar] [PubMed]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef]
- Hanawa, N.; Shinohara, M.; Saberi, B.; Gaarde, W.A.; Han, D.; Kaplowitz, N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J. Biol. Chem. 2008, 283, 13565–13577. [Google Scholar] [CrossRef]
- Chambers, J.W.; LoGrasso, P.V. Mitochondrial c-Jun N-terminal Kinase (JNK) signaling initiates physiological changes resulting in amplification of reactive oxygen species generation. J. Biol. Chem. 2011, 286, 16052–16062. [Google Scholar] [CrossRef]
- Schönthal, A.H. Endoplasmic Reticulum Stress: Its Role in Disease and Novel Prospects for Therapy. Scientifica (Cairo) 2012, 2012, 1–26. [Google Scholar] [CrossRef]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef]
- Haas, I.; Wabl, M. Immunoglobulin heavy chain binding protein. Nature 1983, 306, 387–389. [Google Scholar] [CrossRef]
- Kaneto, H.; Matsuoka, T.A.; Nakatani, Y.; Kawamori, D.; Miyatsuka, T.; Matsuhisa, M.; Yamasaki, Y. Oxidative stress, ER stress, and the JNK pathway in type 2 diabetes. J. Mol. Med. 2005, 83, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 2007, 18, 716–731. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Fels, D.R.; Koumenis, C. The PERK/eIF2α/ATF4 module of the UPR in hypoxia resistance and tumor growth. Cancer Biol. Ther. 2006, 5, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef]
- Lin, J.H.; Walter, P.; Yen, T.S.B. Endoplasmic Reticulum Stress in Disease Pathogenesis. Annu. Rev. Pathol. 2008, 3, 399–425. [Google Scholar] [CrossRef]
- Kaneko, M.; Imaizumi, K.; Saito, A.; Kanemoto, S.; Asada, R.; Matsuhisa, K.; Ohtake, Y. ER Stress and Disease: Toward Prevention and Treatment. Biol. Pharm. Bull. 2017, 40, 1337–1343. [Google Scholar] [CrossRef]
- Ron, D.; Hubbard, S.R. How IRE1 Reacts to ER Stress. Cell 2008, 132, 24–26. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of Stress in the ER to Activation of JNK protein kinases by Protein Kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Nishitoh, H. CHOP is a multifunctional transcription factor in the ER stress response. J. Biochem. 2012, 151, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Davis, R.J. Cell signaling and stress responses. Cold Spring Harb. Perspect. Biol. 2016, 8, a006072. [Google Scholar] [CrossRef] [PubMed]
- Coffey, E.T.; Courtney, M.J. 12 Regulation of SAPKs in CNS neurons. Biochem. Soc. Trans. 1997, 25, 568. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Raber, J.; Yang, D.; Su, B.; Mucke, L. Dynamic regulation of c-Jun N-terminal kinase activity in mouse brain by environmental stimuli. Proc. Natl. Acad. Sci. USA 1997, 94, 12655–12660. [Google Scholar] [CrossRef]
- Lee, J.K.; Park, J.; Lee, Y.D.; Lee, S.H.; Han, P.L. Distinct localization of SAPK isoforms in neurons of adult mouse brain implies multiple signaling modes of SAPK pathway. Mol. Brain Res. 1999, 70, 116–124. [Google Scholar] [CrossRef]
- Mao, L.M.; Wang, J.Q. Synaptically Localized Mitogen-Activated Protein Kinases: Local Substrates and Regulation. Mol. Neurobiol. 2016, 53, 6309–6315. [Google Scholar] [CrossRef]
- Weston, C.R.; Davis, R.J. The JNK signal transduction pathway. Curr. Opin. Cell Biol. 2007, 19, 142–149. [Google Scholar] [CrossRef]
- Haeusgen, W.; Boehm, R.; Zhao, Y.; Herdegen, T.; Waetzig, V. Specific activities of individual c-Jun N-terminal kinases in the brain. Neuroscience 2009, 161, 951–959. [Google Scholar] [CrossRef]
- Kaminska, B.; Gozdz, A.; Zawadzka, M.; Ellert-Miklaszewska, A.; Lipko, M. MAPK signal transduction underlying brain inflammation and gliosis as therapeutic target. Anat. Rec. 2009, 292, 1902–1913. [Google Scholar] [CrossRef]
- Devinsky, O.; Vezzani, A.; O’Brien, T.J.; Jette, N.; Scheffer, I.E.; de Curtis, M.; Perucca, P. Epilepsy. Nat. Rev. Dis. Primers 2018, 4, 18024. [Google Scholar] [CrossRef]
- Vezzani, A.; French, J.; Bartfai, T.; Baram, T.Z. The role of inflammation in epilepsy. Nat. Rev. Neurol. 2011, 7, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Prilopko, L. Epilepsy Care in the World. In Epilepsy Health Atlas; World Health Organization: Geneva, Switzerland, 2005. [Google Scholar]
- Steinhäuser, C.; Grunnet, M.; Carmignoto, G. Crucial role of astrocytes in temporal lobe epilepsy. Neuroscience 2016, 323, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.; Löscher, W. Drug resistance in epilepsy: Putative neurobiologic and clinical mechanisms. Epilepsia 2005, 46, 858–877. [Google Scholar] [CrossRef]
- Lévesque, M.; Avoli, M. The kainic acid model of temporal lobe epilepsy. Neurosci. Biobehav. Rev. 2016, 37, 2887–2899. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Kang, H.; You, L.; Rastogi, P.; Venkatesh, D.; Chandra, M. Neuropsychological deficits in temporal lobe epilepsy: A comprehensive review. Ann. Indian Acad. Neurol. 2014, 17, 374–382. [Google Scholar] [PubMed]
- Tramoni-Negre, E.; Lambert, I.; Bartolomei, F.; Felician, O. Long-term memory deficits in temporal lobe epilepsy. Rev. Neurol. 2017, 173, 490–497. [Google Scholar] [CrossRef]
- Lévesque, M.; Avoli, M.; Bernard, C. Animal models of temporal lobe epilepsy following systemic chemoconvulsant administration. J. Neurosci. Methods 2016, 260, 45–52. [Google Scholar] [CrossRef]
- Nirwan, N.; Vyas, P.; Vohora, D. Animal models of status epilepticus and temporal lobe epilepsy: A narrative review. Rev. Neurosci. 2018, 29, 757–770. [Google Scholar] [CrossRef]
- Rana, A.; Musto, A.E. The role of inflammation in the development of epilepsy. J. Neuroinflamm. 2018, 15, 144. [Google Scholar] [CrossRef]
- Liu, G.; Guo, H.; Guo, C.; Zhao, S.; Gong, D.; Zhao, Y. Involvement of IRE1α signaling in the hippocampus in patients with mesial temporal lobe epilepsy. Brain Res. Bull. 2011, 84, 94–102. [Google Scholar] [CrossRef]
- Chatzikonstantinou, A. Epilepsy and the hippocampus. Hippocampus Clin. Neurosci. 2014, 34, 121–142. [Google Scholar]
- Puttachary, S.; Sharma, S.; Stark, S.; Thippeswamy, T. Seizure-induced oxidative stress in temporal lobe epilepsy. BioMed Res. Int. 2015, 2015, 745613. [Google Scholar] [CrossRef] [PubMed]
- Henshall, D. Apoptosis signaling pathways in seizure-induced neuronal death and epilepsy. Biochem. Soc. Trans. 2007, 35, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Tai, T.Y.; Warner, L.N.; Jones, T.D.; Jung, S.; Concepcion, F.A.; Skyrud, D.W.; Fender, J.; Liu, Y.; Williams, A.D.; Neumaier, J.F.; et al. Antiepileptic action of c-Jun N-terminal kinase (JNK) inhibition in an animal model of temporal lobe epilepsy. Neuroscience 2017, 349, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Gorter, J.A.; Iyer, A.; White, I.; Colzi, A.; van Vliet, E.A.; Sisodiya, S.; Aronica, E. Hippocampal subregion-specific microRNA expression during epileptogenesis in experimental temporal lobe epilepsy. Neurobiol. Dis. 2014, 62, 508–520. [Google Scholar] [CrossRef]
- Tsai, N.P.; Wei, L.N. RhoA/ROCK1 signaling regulates stress granule formation and apoptosis. Cell. Signal. 2010, 22, 668–675. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, Y.; Lü, Y.; Chen, X.; Cheng, L.; Mi, X.; Xu, X.; Deng, W.; Zhang, Y.; Wang, N.; et al. Effects of JIP3 on epileptic seizures: Evidence from temporal lobe epilepsy patients, kainic-induced acute seizures and pentylenetetrazole-induced kindled seizures. Neuroscience 2015, 300, 314–324. [Google Scholar] [CrossRef]
- Schauwecker, P.E. Seizure-induced neuronal death is associated with induction of c-Jun N-terminal kinase and is dependent on genetic background. Brain Res. 2000, 884, 116–128. [Google Scholar] [CrossRef]
- De Lemos, L.; Junyent, F.; Camins, A.; Castro-Torres, R.D.; Folch, J.; Olloquequi, J.; Beas-Zarate, C.; Verdaguer, E.; Auladell, C. Neuroprotective Effects of the Absence of JNK1 or JNK3 Isoforms on Kainic Acid-Induced Temporal Lobe Epilepsy-Like Symptoms. Mol. Neurobiol. 2018, 55, 4437–4452. [Google Scholar] [CrossRef]
- Castro-Torres, R.D.; Landa, J.; Rabaza, M.; Busquets, O.; Olloquequi, J.; Ettcheto, M.; Beas-Zarate, C.; Folch, J.; Camins, A.; Auladell, C.; et al. JNK Isoforms Are Involved in the Control of Adult Hippocampal Neurogenesis in Mice, Both in Physiological Conditions and in an Experimental Model of Temporal Lobe Epilepsy. Mol. Neurobiol. 2019, 56, 5856–5865. [Google Scholar] [CrossRef]
- Duque-Guimarães, D.E.; Ozanne, S.E. Nutritional programming of insulin resistance: Causes and consequences. Trends Endocrinol. Metab. 2013, 24, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Dodd, G.T.; Tiganis, T. Insulin action in the brain: Roles in energy and glucose homeostasis. J. Neuroendocrinol. 2017, 29, 1–14. [Google Scholar] [CrossRef] [PubMed]
- McCracken, E.; Monaghan, M.; Sreenivasan, S. Pathophysiology of the metabolic syndrome. Clin. Dermatol. 2018, 36, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Onyango, A.N. Cellular Stresses and Stress Responses in the Pathogenesis of Insulin Resistance. Oxidative Med. Cell. Longev. 2018, 4321714. [Google Scholar] [CrossRef]
- Onyanfar, U.; Khaliq, S.; Ahmad, H.U.; Manzoor, S.; Lone, K.P. Metabolic syndrome: An update on diagnostic criteria, pathogenesis, and genetic links. Hormones 2018, 17, 299–313. [Google Scholar]
- Henneberg, N.; Hoyer, S. Desensitization of the neuronal insulin receptor: A new approach in the etiopathogenesis of late-onset sporadic dementia of the Alzheimer type (SDAT)? Arch. Gerontol. Geriatr. 1995, 21, 63–74. [Google Scholar] [CrossRef]
- Ott, A.; Stolk, R.P.; van Harskamp, F.; Pols, H.A.P.; Hofman, A.; Breteler, M.M.B. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999, 53, 1937–1942. [Google Scholar] [CrossRef]
- De Felice, F.G.; Lourenco, M.V.; Ferreira, S.T. How does brain insulin resistance develop in Alzheimer’s disease? Alzheimers Dement 2014, 10 (Suppl. 1), S26–S32. [Google Scholar] [CrossRef]
- Lee, J.; Pilch, P.F. The insulin receptor: Structure, function, and signalling. Am. J. Physiol. Cell Physiol. 1994, 266, 319–334. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Ronald Kahn, C. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef]
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Metabolic Signalling: Biochemical and cellular properties of insulin receptor signaling. Nat. Rev. Mol. Cell Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Sah, S.P.; Singh, B.; Choudhary, S.; Kumar, A. Animal models of insulin resistance: A review. Pharmacol. Rep. 2016, 68, 1165–1177. [Google Scholar] [CrossRef] [PubMed]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Wang, H.G. The protein kinase PKB/Akt regulates cell survival and apoptosis by inhibiting Bax conformational change. Oncogene 2001, 20, 7779–7786. [Google Scholar] [CrossRef]
- Gardai, S.J.; Hildeman, D.A.; Frankel, S.K.; Whitlock, B.B.; Frasch, S.C.; Borregaard, N.; Marrack, P.; Bratton, D.L.; Henson, P.M. Phosphorylation of Bax ser184 by Akt regulates its activity and apoptosis in neutrophils. J. Biol. Chem. 2004, 279, 21085–21095. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.J.; Voeltz, G.K. Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol. 2016, 17, 69–82. [Google Scholar] [CrossRef]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Osborn, O.; Olefsky, J.M. The cellular and signaling networks linking the immune system and metabolism in disease. Nat. Med. 2012, 18, 363–374. [Google Scholar] [CrossRef]
- Saltiel, A.R. Insulin Signaling in the Control of Glucose and Lipid Homeostasis. Handb. Exp. Pharmacol. 2016, 233, 51–57. [Google Scholar]
- Emanuelli, B.; Peraldi, P.; Filloux, C.; Sawka-Verhelle, D.; Hilton, D.; Van Obberghen, E. SOCS-3 is an insulin-induced negative regulator of insulin signaling. J. Biol. Chem. 2000, 275, 15985–15991. [Google Scholar] [CrossRef]
- Després, J.P.; Lemieux, I. Abdominal obesity and metabolic syndrome. Nature 2006, 444, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Engin, A. Obesity and Lipotoxicity: The Definition and Prevalence of Obesity and Metabolic Syndrome. In Obesity and Lipotoxicity; Springer: Cham, Switzerland, 2017; Volume 960, pp. 1–17. [Google Scholar] [CrossRef]
- Hill, J.M.; Lesniak, M.A.; Pert, C.B.; Roth, J. Autoradiographic localization of insulin receptors in rat brain: Prominence in olfactory and limbic areas. Neuroscience 1986, 17, 1127–1138. [Google Scholar] [CrossRef]
- Kullmann, S.; Heni, M.; Hallschmid, M.; Fritsche, A.; Preissl, H.; Häring, H.U. Brain insulin resistance at the crossroads of metabolic and cognitive disorders in humans. Physiol. Rev. 2016, 96, 1169–1209. [Google Scholar] [CrossRef]
- Bilotta, F.; Lauretta, M.P.; Tewari, A.; Haque, M.; Hara, N.; Uchino, H.; Rosa, G. Insulin and the Brain: A Sweet Relationship with Intensive Care. J. Intensive Care Med. 2017, 32, 48–58. [Google Scholar] [CrossRef]
- Gralle, M. The neuronal insulin receptor in its environment. J. Neurochem. 2017, 140, 359–367. [Google Scholar] [CrossRef]
- Al Haj Ahmad, R.M.; Al-Domi, H.A. Thinking about brain insulin resistance. Diabetes Metab. Syndr. Clin. Res. Rev. 2018, 12, 1091–1094. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Wands, J.R. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—Is this type 3 diabetes? J. Alzheimers Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef]
- Kroner, Z. The relationship between Alzheimer’s disease and diabetes: Type 3 diabetes? Altern. Med. Rev. 2009, 14, 373–379. [Google Scholar]
- Ma, L.; Wang, J.; Li, Y. Insulin resistance and cognitive dysfunction. Clin. Chim. Acta 2015, 444, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Mittal, K.; Mani, R.J.; Katare, D.P. Type 3 Diabetes: Cross Talk between Differentially Regulated Proteins of Type 2 Diabetes Mellitus and Alzheimer’s Disease. Sci. Rep. 2016, 6, 25589. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.W.; Figlewicz, D.F.; Kahn, S.E.; Baskin, D.G.; Greenwood, M.R.C.; Porte, D. Insulin binding to brain capillaries is reduced in genetically obese, hyperinsulinemic Zucker rats. Peptides 1990, 11, 467–472. [Google Scholar] [CrossRef]
- Özcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Özdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L.H.; Hotamisligil, G.S.; et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef]
- Nakamura, T.; Furuhashi, M.; Li, P.; Cao, H.; Tuncman, G.; Sonenberg, N.; Gorgun, C.Z.; Hotamisligil, G. Double-stranded RNA-dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell 2010, 140, 338–348. [Google Scholar] [CrossRef]
- Biessels, G.J.; Reagan, L.P. Hippocampal insulin resistance and cognitive dysfunction. Nat. Rev. Neurosci. 2015, 16, 660–671. [Google Scholar] [CrossRef]
- Solinas, G.; Becattini, B. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol. Metab. 2017, 6, 174–184. [Google Scholar] [CrossRef]
- Solinas, G.; Karin, M. JNK1 and IKKβ: Molecular links between obesity and metabolic dysfunction. FASEB J. 2010, 24, 2596–2611. [Google Scholar] [CrossRef]
- Mohammad, H.; Marchisella, F.; Ortega-Martinez, S.; Hollos, P.; Eerola, K.; Komulainen, E.; Kulesskaya NFreemantle, E.; Fagerholm, V.; Savontaus, E.; Rauvala, H. JNK1 controls adult hippocampal neurogenesis and imposes cell-autonomous control of anxiety behaviour from the neurogenic niche. Mol. Psychiatry 2018, 23, 362–374. [Google Scholar] [CrossRef]
- Belgardt, B.F.; Mauer, J.; Wunderlich, F.T.; Ernst, M.B.; Pal, M.; Spohn, G.; Brönneke, H.S.; Brodesser, S.; Hampel, B.; Schauss, A.C.; et al. Hypothalamic and pituitary c-Jun N-terminal kinase 1 signaling coordinately regulates glucose metabolism. Proc. Natl. Acad. Sci. USA 2010, 107, 6028–6033. [Google Scholar] [CrossRef]
- Sabio, G.; Davis, R.J. CJun NH2-terminal kinase 1 (JNK1): Roles in metabolic regulation of insulin resistance. Trends Biochem. Sci. 2010, 35, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Vernia, S.; Morel, C.; Madara, J.C.; Cavanagh-Kyros, J.; Barrett, T.; Chase, K.; Kennedy, N.J.; Jung, D.Y.; Kim, J.K.; Aronin, N.; et al. Excitatory transmission onto AgRP neurons is regulated by cjun NH2-terminal kinase 3 in response to metabolic stress. Elife 2016, 5, e10031. [Google Scholar] [CrossRef] [PubMed]
- Racine, R.J. Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 1972, 31, 281–294. [Google Scholar] [CrossRef]
- Bogoyevitch, M.A.; Arthur, P.G. Inhibitors of c-Jun N-terminal kinases-JuNK no more? Biochim. Biophys. Acta Proteins Proteom. 2008, 1784, 76–93. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q.H.; Pei, D.S.; Liu, X.M.; Wang, X.T.; Xu, T.L.; Zhang, G.Y. Neuroprotection against ischemic brain injury by SP600125 via suppressing the extrinsic and intrinsic pathways of apoptosis. Brain Res. 2006, 1092, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, K.; Ueno, M.; Lee, S.; Nakamura, Y.; Sato, A.; Yoshimura, K.; Kishima, H.; Yoshimine, T.; Yamashita, T. C-Jun N-terminal kinase induces axonal degeneration and limits motor recovery after spinal cord injury in mice. Neurosci. Res. 2011, 71, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Murata, Y.; Fujiwara, N.; Seo, J.H.; Yan, F.; Liu, X.; Terasaki, Y.; Luo, Y.; Arai, K.; Ji, X.; Lo, E.H. Delayed Inhibition of c-Jun N-Terminal Kinase Worsens Outcomes after Focal Cerebral Ischemia. J. Neurosci. 2012, 32, 8112–8115. [Google Scholar] [CrossRef]
- Sabapathy, K.; Hochedlinger, K.; Nam, S.Y.; Bauer, A.; Karin, M.; Wagner, E.F. Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun-dependent cell proliferation. Mol. Cell 2004, 15, 713–725. [Google Scholar] [CrossRef]
- Eshraghi, A.A.; Aranke, M.; Salvi, R.; Ding, D.; Coleman, J.K.M., Jr.; Ocak, E.; Mittal, R.; Meyer, T. Preclinical and clinical otoprotective applications of cell-penetrating peptide D-JNKI-1 (AM-111). Hear Res. 2018, 368, 86–91. [Google Scholar] [CrossRef]
- Eshraghi, A.A.; Wang, J.; Adil, E.; He, J.; Zine, A.; Bublik, M.; Bonny, C.; Puel, J.L.; Balkany, T.J.; Van De Water, T.R. Blocking c-Jun-N-terminal kinase signaling can prevent hearing loss induced by both electrode insertion trauma and neomycin ototoxicity. Hear Res. 2007, 226, 168–177. [Google Scholar] [CrossRef]
- Zhao, Y.; Spigolon, G.; Bonny, C.; Culman, J.; Vercelli, A.; Herdegen, T. The JNK inhibitor D-JNKI-1 blocks apoptotic JNK signaling in brain mitochondria. Mol. Cell. Neurosci. 2012, 49, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ruel, J.; Ladrech, S.; Bonny, C.; van de Water, T.R.; Puel, J.L. Inhibition of the c-Jun N-terminal kinase-mediated mitochondrial cell death pathway restores auditory function in sound-exposed animals. Mol. Pharmacol. 2007, 71, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Howard, S.; LoGrasso, P.V. Pharmacological Inhibition of c-Jun N-terminal Kinase Reduces Food Intake and Sensitizes Leptin’s Anorectic Signaling Actions. Sci. Rep. 2017, 7, 41795. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Yu, L.R.; Abdelmegeed, M.A.; Gao, Y.; Banerjee, A.; Song, B.J. Critical role of c-jun N-terminal protein kinase in promoting mitochondrial dysfunction and acute liver injury. Redox Biol. 2015, 6, 552–564. [Google Scholar] [CrossRef]
- Plotnikov, M.B.; Chernysheva, G.A.; Aliev, O.I.; Smol’iakova, V.I.; Fomina, T.I.; Osipenko, A.N.; Rydchenko, V.S.; Anfinogenova, Y.J.; Khlebnikov, A.I.; Schepetkin, I.A.; et al. Protective Effects of a New C-Jun N-terminal Kinase Inhibitor in the Model of Global Cerebral Ischemia in Rats. Molecules 2019, 24, 1722. [Google Scholar] [CrossRef]
- Graczyk, P. JNK inhibitors as anti-inflammatory and neuroprotective agents. Future Med. Chem. 2013, 5, 539–551. [Google Scholar] [CrossRef]
- Solinas, G.; Vilcu, C.; Neels, J.G.; Bandyopadhyay, G.K.; Luo, J.L.; Naugler, W.; Grivennikov, S.; Wynshaw-Boris, A.; Scadeng, M.; Olefsky, J.M.; et al. JNK1 in Hematopoietically Derived Cells Contributes to Diet-Induced Inflammation and Insulin Resistance without Affecting Obesity. Cell Metab. 2007, 6, 386–397. [Google Scholar] [CrossRef]
- Guma, M.; Ronacher, L.M.; Firestein, G.S.; Karin, M.; Corr, M. JNK-1 deficiency limits macrophage-mediated antigen-induced arthritis. Arthritis Rheum. 2011, 63, 1603–1612. [Google Scholar] [CrossRef]
- Kamenecka, T.; Jiang, R.; Song, X.; Duckett, D.; Chen, W.; Ling, Y.Y.; Habel, J.; Laughlin, J.D.; Chambers, J.; Figuera-Losada, M.; et al. Synthesis, biological evaluation, X-ray structure, and pharmacokinetics of aminopyrimidine c-jun-N-terminal kinase (JNK) inhibitors. J. Med. Chem. 2010, 53, 419–431. [Google Scholar] [CrossRef]
- Rochfort, S.; Towerzey, L.; Carroll, A.; King, G.; Michael, A.; Pierens, G.; Rali, T.; Redburn, J.; Whitmore, J.; Quinn, R.J. Latifolians A and B, novel JNK3 kinase inhibitors from the Papua New Guinean plant Gnetum latifolium. J. Nat. Prod. 2005, 68, 1080–1082. [Google Scholar] [CrossRef]
- Yao, K.; Chen, H.; Lee, M.H.; Li, H.; Ma, W.; Peng, C.; Song, N.R.; Lee, K.W.; Bode, A.M.; Dong, Z.; et al. Licochalcone A, a natural inhibitor of c-Jun N-terminal kinase 1. Cancer Prev. Res. 2014, 7, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Liou, C.J.; Lee, Y.K.; Ting, N.C.; Chen, Y.L.; Shen, S.C.; Wu, S.J.; Huang, W.C. Protective Effects of Licochalcone A Ameliorates Obesity and Non-Alcoholic Fatty Liver Disease Via Promotion of the Sirt-1/AMPK Pathway in Mice Fed a High-Fat Diet. Cells 2019, 8, 447. [Google Scholar] [CrossRef] [PubMed]
- Raman, M.; Chen, W.; Cobb, M.H. Differential regulation and properties of MAPKs. Oncogene 2007, 26, 3100–3112. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.W.; Pachori, A.; Howard, S.; Ganno, M.; Hansen, D., Jr.; Kamenecka, T.; Song, X.; Duckett, D.; Chen, W.; Ling, Y.Y.; et al. Small Molecule c-jun-N-Terminal Kinase Inhibitors protect Dopaminergic Neurons in a Model of Parkinson’s Disease. ACS Chem. Neurosci. 2011, 2, 198–206. [Google Scholar] [CrossRef]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef]
- Ip, Y.T.; Davis, R.J. Signal transduction by the c-Jun N-terminal kinase (JNK)—From inflammation to development. Curr. Opin. Cell Biol. 1998, 10, 205–219. [Google Scholar] [CrossRef]
- Cardoso, S.; Moreira, P.I. Diabesity and brain disturbances: A metabolic perspective. Mol. Asp. Med. 2019, 66, 71–79. [Google Scholar] [CrossRef]
- Grillo, C.A.; Tamashiro, K.L.; Piroli, G.G.; Melhorn, S.; Gass, J.T.; Newsom, R.J.; Reznikov, L.R.; Smith, A.; Wilson, S.P.; Sakai, R.R.; et al. Lentivirus-mediated downregulation of hypothalamic insulin receptor expression. Physiol. Behav. 2007, 92, 691–701. [Google Scholar] [CrossRef]
- Liu, Z.; Patil, I.Y.; Jiang, T.; Sancheti, H.; Walsh, J.P.; Stiles, B.L.; Yin, F.; Cadenas, E. High-fat diet induces hepatic insulin resistance and impairment of synaptic plasticity. PLoS ONE 2015, 10, e0128274. [Google Scholar] [CrossRef]
- Mendelson, K.G.; Contois, L.R.; Tevosian, S.G.; Davis, R.J.; Paulson, K.E. Independent regulation of JNK/p38 mitogen-activated protein kinases by metabolic oxidative stress in the liver. Proc. Natl. Acad. Sci. USA 1996, 93, 12908–12913. [Google Scholar] [CrossRef]
- Muresan, Z.; Muresan, V. C-Jun NH2-terminal kinase-interactin g613protein-3 facilitates phosphorylation and controls localization o f614amyloid-beta precursor protein. J. Neurosci. 2005, 25, 3741–3751. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, D.; Picone, P.; Baldassano, S.; Caruana, L.; Messina, E.; Gammazza, A.; Cappello, F.; Mulè, F.; Di Carlo, M. Insulin Resistance as Common Molecular Denominator Linking Obesity to Alzheimer’s Disease. Curr. Alzheimer Res. 2015, 12, 723–735. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, S.; O’Driscoll, L. Metabolic syndrome: A closer look at the growing epidemic and its associated pathologies. Obes. Rev. 2015, 16, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Petrov, D.; Pedrós, I.; Artiach, G.; Sureda, F.X.; Barroso, E.; Pallàs, M.; Casadesús, G.; Beas-Zarate, C.; Carro, E.; Ferrer, I.; et al. High-fat diet-induced deregulation of hippocampal insulin signaling and mitochondrial homeostasis deficiences contribute to Alzheimer disease pathology in rodents. Biochim. Biophys. Acta Mol. Basis. Dis. 2015, 1852, 1687–1699. [Google Scholar] [CrossRef]
- Raciti, M.; Lotti, L.V.; Valia, S.; Pulcinelli, F.M.; Di Renzo, L. JNK2 is activated during ER stress and promotes cell survival. Cell Death Dis. 2012, 3, e429. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Phase | CT Number | Disease |
|---|---|---|---|
| D-JNKI-1 | III | NCT02809118 | Idiopathic sudden sensorineural hearing loss |
| D-JNKI-1 | III | NCT02235272 | Post-cataract Surgery Intraocular Inflammation |
| D-JNKI-1 | I | NCT01570205 | Safety, Tolerability and PK of a Single iv Infusion of 10, 40, and 80 µg/kg XG-102 Administered to Healthy Volunteers |
| D-JNKI-1 | III | NCT02561091 | Acute Inner Ear Hearing Loss |
| D-JNKI-1 | III | NCT02508337 | Reduction of Post-cataract Surgery Intraocular Inflammation and Pain |
| D-JNKI-1 | II | NCT00802425 | Acute Sensorineural Hearing Loss |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Busquets, O.; Ettcheto, M.; Cano, A.; R. Manzine, P.; Sánchez-Lopez, E.; Espinosa-Jiménez, T.; Verdaguer, E.; Dario Castro-Torres, R.; Beas-Zarate, C.; X. Sureda, F.; et al. Role of c-Jun N-Terminal Kinases (JNKs) in Epilepsy and Metabolic Cognitive Impairment. Int. J. Mol. Sci. 2020, 21, 255. https://doi.org/10.3390/ijms21010255
Busquets O, Ettcheto M, Cano A, R. Manzine P, Sánchez-Lopez E, Espinosa-Jiménez T, Verdaguer E, Dario Castro-Torres R, Beas-Zarate C, X. Sureda F, et al. Role of c-Jun N-Terminal Kinases (JNKs) in Epilepsy and Metabolic Cognitive Impairment. International Journal of Molecular Sciences. 2020; 21(1):255. https://doi.org/10.3390/ijms21010255
Chicago/Turabian StyleBusquets, Oriol, Miren Ettcheto, Amanda Cano, Patricia R. Manzine, Elena Sánchez-Lopez, Triana Espinosa-Jiménez, Ester Verdaguer, Rubén Dario Castro-Torres, Carlos Beas-Zarate, Francesc X. Sureda, and et al. 2020. "Role of c-Jun N-Terminal Kinases (JNKs) in Epilepsy and Metabolic Cognitive Impairment" International Journal of Molecular Sciences 21, no. 1: 255. https://doi.org/10.3390/ijms21010255
APA StyleBusquets, O., Ettcheto, M., Cano, A., R. Manzine, P., Sánchez-Lopez, E., Espinosa-Jiménez, T., Verdaguer, E., Dario Castro-Torres, R., Beas-Zarate, C., X. Sureda, F., Olloquequi, J., Auladell, C., Folch, J., & Camins, A. (2020). Role of c-Jun N-Terminal Kinases (JNKs) in Epilepsy and Metabolic Cognitive Impairment. International Journal of Molecular Sciences, 21(1), 255. https://doi.org/10.3390/ijms21010255